
ABSTRACT
Epigenetic chemical probes are potent, cell-active, small molecule inhibitors or antagonists of specific domains in a protein; they have been indispensable for studying bromodomains and protein methyltransferases. The Structural Genomics Consortium (SGC), comprising scientists from academic and pharmaceutical laboratories, has generated most of the current epigenetic chemical probes. Moreover, the SGC has shared about 4 thousand aliquots of these probes, which have been used primarily for phenotypic profiling or to validate targets in cell lines or primary patient samples cultured in vitro. Epigenetic chemical probes have been critical tools in oncology research and have uncovered mechanistic insights into well-established targets, as well as identify new therapeutic starting points. Indeed, the literature primarily links epigenetic proteins to oncology, but applications in inflammation, viral, metabolic and neurodegenerative diseases are now being reported. We summarize the literature of these emerging applications and provide examples where existing probes might be used.
Abbreviations
| BAZ2A/2B | = | Bromodomain adjacent to zinc finger domain protein 2A/2B |
| BET | = | Bromo and extra-terminal domain |
| BRD | = | Bromodomain |
| BRD2/3/4 | = | Bromodomain-containing protein 2/3/4 |
| BRD7 | = | Bromodomain-containing protein 7 |
| BRD9 | = | Bromodomain-containing protein 9 |
| BRDT | = | Bromodomain testis-specific protein |
| BRPF1/2/3 | = | Bromodomain and PHD finger containing 1/2/3 |
| CECR2 | = | Cat eye syndrome critical region protein 2 |
| CREBBP | = | CREB-binding protein |
| CXCL11 | = | C-X-C motif chemokine 11 |
| DOT1L | = | Disruptor of Telomere Silencing 1-like |
| EED | = | Embryonic Ectoderm Development |
| EP300 | = | Histone acetyltransferase p300 |
| EZH2/H1 | = | Enhancer of Zeste Homolog 2/1 |
| G9a/GLP | = | Histone-lysine N-methyltransferase EHMT2/EHMT1 |
| HAT | = | Histone acetyltransferase |
| IDH1 mutant | = | Isocitrate dehydrogenase 1 |
| JMJD3/UTX | = | Lysine-specific demethylase 6B/6A |
| L3MBTL3 | = | Lethal(3)malignant brain tumor-like protein 3 |
| LSD1 | = | Lysine-specific histone demethylase 1A |
| PAD4 | = | Protein arginine deiminase |
| PB1 | = | Protein polybromo-1 |
| PMT | = | Protein methyltransferase |
| PRMT | = | Protein arginine methyltransferase |
| SETD7 | = | SET domain-containing protein 7 |
| SMARCA2/4 | = | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2/4 |
| SMYD2 | = | SET and MYND domain-containing protein 2 |
| SUV420H1/H2 | = | Suppressor of variegation 4–20 homolog 1/2 |
| TAF1 | = | TBP-associated factor |
| WDR5 | = | WD repeat-containing protein 5. |
Introduction
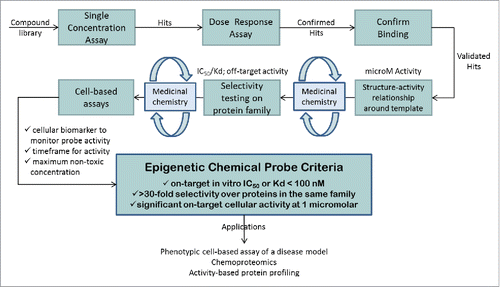
Chemical probes are cell-active small molecules that are used to explore the biology and therapeutic potential of their targets, for example, proteins or DNA. Well-characterized chemical probes,Citation1-5 in addition to relevant controls, are critical complements of genetic knockdown approaches. While knockdown experiments are well established in many laboratories, new methods using CRISPR-based technologies are now gaining on the more traditional shRNA and siRNA based approaches. These systems even allow dynamic spatial and temporal control of gene regulation, but are mostly used to prevent protein expression rather than edit a protein function as a chemical inhibitor does.Citation6 Information regarding the scaffolding functions of a protein or the function of a specific protein domain are therefore not easily obtained and require more intricate studies using complicated genetic and biochemical experiments, unless a specific inhibitor is available to modulate the activity of the domain of interest. Moreover, inhibitors can be useful for cell types that are not easily amenable to knockdown experiments and may additionally form the basis for drug development programs. More recent approaches include inhibitors linked to molecules that target the bound protein for proteolytic degradation, which often results in more dramatic and prolonged effects than those seen with the inhibitor alone.Citation7,8,9 However, for closely related protein targets, it may sometimes be difficult to generate a specific inhibitor, in which case knockdown methods are a useful complementation to link the observed phenotype to a specific target. Together with several pharmaceutical companies and academic collaborators, the Structural Genomics Consortium (SGC) has established a chemical probe program in which potential probes undergo rigorous potency and selectivity triage as shown in .
Figure 1. Schematic representation of the workflow used to generate epigenetic chemical probes.
Two families of epigenetic targets have been particularly amenable to small molecule intervention: bromodomains (BRDs) and protein methyltransferases (PMTs).Citation2 The bromodomain family comprises 42 members containing 61 different bromodomains.Citation10 Most bromodomains bind to acetylated lysine residues but certain family members also recognize other modifications, such as butyrylation or crotonylation, and in some cases a single bromodomain can bind to more than one acetylation mark, making these reader domains versatile interpreters of the histone code.Citation11 Additionally, bromodomains have also been shown to interact with other non-histone acetylated proteins, in particular transcription factors, modulating their transcriptional activity.Citation10 Developing antagonists for the bromo and extra terminal (BET) family has been particularly successful. This family consists of BRD2, BRD3, BRD4, and BRDT, each containing 2 N-terminal bromodomains and an extra terminal (ET) protein interaction domain. BET family members play central roles in a variety of physiologic and pathophysiological states. The impact of BET antagonists in cancer research has been extensively reviewed.Citation12-21 Erasers, enzymes that remove a histone posttranslational modification (PTM), and writers, which add a histone PTM, do not feature as broad a literature base as the BET family. Nevertheless, these too have important roles in cancer.Citation22-24
Human PMTs are composed of 50 SET domain lysine methyltransferases and 13 known Rossmann fold enzymes. PMTs transfer a methyl group from the cofactor S-adenosyl-L-methionine (SAM) to the side-chain nitrogen atom of lysine (K) and the terminal nitrogen atoms on the guanidinium moiety of arginine (R) residues. Lysine residues can be mono-, di-, or tri-methylated while arginine residues may be mono- or di-methylated. The latter can be symmetric, where one methyl group is added to each of the terminal nitrogen atoms or asymmetric, where both methyl groups are added to the same terminal nitrogen atom. Histones were originally thought to be the primary targets of PMTs but it soon became clear that they have other substrates. Methyl marks can be removed by another group of epigenetic proteins, lysine demethylases (KDMs), of which there are 2 groups: the FAD dependent enzymes and the iron- and 2-oxoglutarate-dependent oxygenases of the Jumonji C (JmjC) subfamily.Citation2,5
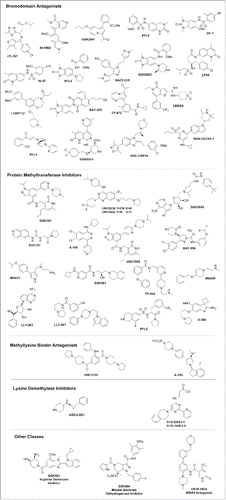
Chemical probes have enabled many biologic and clinical findings in cancer. These, in turn, have been the basis for proprietary programs and the first clinical trials in the area of specific epigenetic target classes, for example, bromodomains (BET), demethylase (LSD1), isocitrate dehydrogenase (mutant IDH1), and methyltransferases (EZH2/H1, DOT1L, PRMT5). For this review, however, we turn the spotlight on emerging applications of chemical probes including inflammation, viral, metabolic, and neurodegenerative diseases. Some of the research cited in the review has used chemical probes or tool compounds. In some cases, chemical probes are not yet available for the target described, but the examples are cited as a means to highlight the interest in developing a chemical probe for that target. The SGC has successfully generated and released 42 () highly characterized chemical probes,Citation13,25 which are accessible to the scientific community as a library. A current and updated list of epigenetic chemical probes developed by the Consortium is in and at this web page: http://www.thesgc.org/chemical-probes/epigenetics.
Table 1. Epigenetic probes generated by SGC.
Figure 2. Chemical structure of Epigenetic inhibitors available by the SGC.
Inflammation
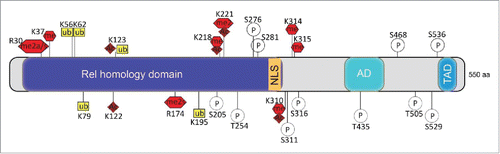
The central transcription factor in inflammation is the nuclear factor κB (NF-κB). NF-κB is a dimer of different Rel family members with the prototype of NF-κB being the heterodimer of p65 (NF-κB) and p50 subunits. The dimer resides in the cytoplasm in an inactive form, complexed with the inhibitory protein IκB. Activation by IKK (IκB kinase) results in dissociation of IκB from NF-κB, polyubiquitination, and proteasomal degradation. As a consequence, NF-κB translocates into the nucleus, where it activates transcription of its cognate genesCitation26 Incorrect regulation of inflammatory mechanisms is detrimental and is the underlying cause for widespread chronic diseases, such as asthma, arthritis, psoriasis, atopic eczema, periodontitis, and inflammatory bowel disease (IBD). The NF-κB pathway is intertwined into many biologic processes. As such, it is not surprising that it can be activated and deactivated by multiple mechanisms and that some include epigenetic modifications. Indeed, NF-κB can be extensively posttranslationally modified and phosphorylation, methylation, acetylation, and ubiquitination of the p65 subunit have been describedCitation26 ().
Figure 3. Posttranslational modifications of p65. Schematic representation of p65 subunit of NF-κB primary structure: Rel homology domain, nuclear localization signal (NLS), activation domain (AD), transactivation domain (TAD). Amino acid residues known to be posttranslationally modified are shown; ac (acetylation), ub (ubiquitination), p (phosphorylation), me (methylation), s (symmetric methylation), a (asymmetric methylation).
BET bromodomains
BET antagonists have shown positive effects in a variety of inflammatory models. As current BET antagonists do not distinguish between the different BET family members,Citation13 many of the studies do not provide unambiguous results regarding the biologic functions of BRD4 vs. BRD2 or BRD3 (BRDT is a testis specific protein). However, studies using BET antagonists combined with knockdown technologies now discriminate between the effects of the different BET family members and differential roles of specific BET family members are beginning to emerge as outlined below. There is for example now growing evidence that, apart from BRD4, BRD2 also plays important roles in inflammation.Citation27
In addition, different isoforms of BET proteins exist. For example, BRD4 exists in 2 different isoforms generated by alternative splicing that—in oncology—exert opposite effects. Mass spectrometry revealed that the short isoform is pro-metastatic and also has a broader acetylated histone binding pattern relative to the long isoform.Citation28 The role of the different isoforms in inflammation has however not yet been explored. BRD4 has also been found to be posttranslationally modified. Phosphorylation of BRD4 by CDK2 regulates its chromatin binding, and hyper-phosphorylation of BRD has been reported to be related to a resistance mechanism in triple negative breast cancer (TNBC) caused by the decreased activity of PP2a phosphatase.Citation29 Further studies are needed to explore the importance of posttranslational modifications of BET family members in other diseases.
A complex signaling event leads to co-activation of inflammatory genes by the BET family member BRD4 and NF-κB. Activation of the NF-κB pathway by LPS results first in phosphorylation of p65, which in turn recruits histone acetyl transferases (HATs) like CPB/p300 and PCAF acetylating p65 on lysine 310. BRD4 binds to acetylated p65 via its BRDs, which causes increased transcriptional activity through targeting P-TEFb to NF-κB-responsive inflammatory genes (see below). Binding of BRD4 to acetylated p65 keeps NF-κB constitutively active, at least in cancer cells.Citation30 At the same time the HATs also acetylate histones, which results in the opening of chromatin and BRD4 recruitment, which further increases transcriptional activity ().
Figure 4. Epigenetic regulation of the NF-κB pathway.
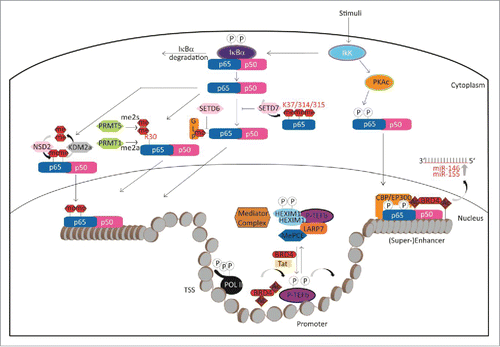
Both BRD4 and NF-κB can also co-localize on inflammation-related super enhancers co-activating inflammatory genes. Inhibition of BRD4 recruitment by BET antagonists leads to suppression of inflammation in endothelial cells and degradation of the constitutively active nuclear form of p65.Citation31,32 Additionally, the transcription of pro-inflammatory microRNAs, namely miR-146a and miR-155, is regulated by the combined action of NF-κB, BRD4 and formation of the super enhancers that drive miR-146a and miR-155 transcription. These microRNAs can then downregulate inflammatory gene expression by targeting their mediatorsCitation33 ().
The mechanism of BRD4 transcriptional activation has been extensively studied in the context of oncology, but many mechanisms may also apply for inflammatory or other diseases. BRD4 is a main regulator of transcription and does this at several different levels. First, it acts on the level of transcriptional elongation by recruiting the active positive transcriptional elongation factor b (P-TEFb) composed of cyclin-dependent kinase CDK9 and its activator cyclin T to phosphorylate RNA polymerase II (RNAP II), overcoming pausing of RNAP II and thereby activating transcription. P-TEFb is normally kept in an inactive state by association with ribonucleoprotein complex composed of snRNA 7SK and the inhibitory factor HEXIM. BRD4 binding to P-TEFb competes with this assembly recruiting P-TEFb to acetylated chromatin.Citation34 Secondly, BRD4 further acts on the level of enhancers or super enhancers, where it interacts with the mediator complex to activate transcription of specific genes.Citation35 A third way of influencing transcription is by the direct interaction with transcription factors. BRD4 has been found to interact directly with a large number of transcriptional regulators like p53, YY1, AP2, c-Jun, c-Myc/Max, C/EBPα, C/EBPβ, Acf1, and G9a.Citation29 Interaction of BRD4 with the transcription factor AIRE, which is dependent on posttranslational modifications of AIRE's caspase activation and recruitment domain, controls immunologic tolerance by influencing P-TEFb mediated transcriptional elongation.Citation36 BRD4 also binds to acetylated E2F-1 transcription factor. This association is regulated by the peptidyl arginine deiminase PAD4, which citrullinates E2F-1 in inflammatory cells increasing chromatin association of E2F-1 to cytokine genesCitation37 ().
One of the 2 key publications describing the first BET antagonists reported that the BET inhibitor I-BET exhibits potent anti-inflammatory effects. In in vitro as well as in vivo models, BET antagonism led to reduced cytokine production by LPS-induced macrophages and prevented lethal septic shock in an LPS-induced mouse model of sepsis.Citation38 Subsequent reports showed that BET antagonists specifically suppress a particular subset of LPS-induced ‘primed genes’ whose expression is independent of BRD4 activation.Citation39 The BET antagonist (+)-JQ1 has been successfully used in in vitro and mouse models of collagen-induced arthritis. (+)-JQ1 acts by reducing levels of the pro-inflammatory cytokines interleukin (IL)-1β, IL-6, IL-17, and IL-18 as shown in cultural supernatants from tumor necrosis factor (TNF)-α-stimulated human rheumatoid arthritis fibroblast-like synoviocytes, confirming results of BRD4 siRNA experiments, and reduced the inflammatory response, autoantibody production, and joint damage, respectively.Citation40 A recent study examining the role of BET proteins in synovial inflammation of RA confirmed that in particular BRD4 and BRD2 levels were elevated in the synovial fluid of RA patients; treatment with (+)-JQ1, Brd2 shRNA or Brd4 shRNA reduced levels of pro-inflammatory cytokines as well as matrix metalloproteases by inhibiting NF-κB transcriptional activation.Citation41 BET antagonism by (+)-JQ1 led to reduced pro-inflammatory cytokine production and transcription factor expression in a mouse model of periodontitis, where the number of bone resorbing osteoclasts was reduced and, therefore, bone reduction diminished.Citation42 Similarly, in a model of osteoporosis it was shown that BET antagonism by (+)-JQ1 reduced the number of osteoclasts by interfering with osteoclast differentiation while at the same time activating the bone regenerating osteoblast activity. Experiments using shRNAs specific for BRD2, BRD3, or BRD4 demonstrated a critical role of each of these BET family members in osteoclastogenesis. Together, these effects could provide a starting point for the treatment of osteoporosis.Citation43
In inflammatory renal diseases, (+)-JQ1 inhibited inflammatory responses as assessed by downregulation of pro-inflammatory genes. The BET antagonist (+)-JQ1 has been shown to lead to chromatin remodeling in promoter regions of specific genes, blockade of NF-κB pathway activation, and modulation of the Th17 immune response in human renal tubular epithelial cells stimulated with TNFα and in murine models of unilateral ureteral obstruction, anti-membrane basal GN, and infusion of Angiotensin II.Citation31 Chromatin immunoprecipitation assays demonstrated that (+)-JQ1 interfered directly with the association of BRD4 to the promoters of the pro-inflammatory genes IL-6, CCL-2, and CCL-5.Citation31
In other models of inflammation, BET antagonism has been shown to not only affect cytokine production, but also influence development of different immune cells. BET antagonists like (+)-JQ1 and I-BET-762 not only suppress expression of cytokines on the transcriptional level, but also modulate early T cell differentiation and control differentiation of Th17 cells, which play fundamental roles in autoimmune diseases.Citation44,45 Differentiation of cytotoxic CD8+ cells used for adoptive transfer in immune oncology is also affected by treatment with (+)-JQ1. T cells treated with (+)-JQ1 maintained features that more resembled those of stem-cells and T memory cells as assessed by surface markers and cytokines produced, which resulted in increased in vivo persistence and superior antitumor efficacy in several cancer immunotherapy models.Citation46 (+)-JQ1 also impairs maturation of LPS-induced monocyte derived dendritic cells (Mo-DC) by inhibiting the activity of the transcription factor STAT5. Signaling of STAT5 is important to provide the stimulation required for the complete maturation of Mo-DCs.Citation47 The interaction of BRD4 with acetylated p65 also seems to play a role for the positive effect of BET inhibitors I-BET151 and (+)-JQ1 to prevent graft vs. host disease in bone marrow transplantation. This led to an altered cytokine expression from dendritic cells via reduction of surface molecules and inhibiting T cell expansion.Citation48
Additionally, BET antagonists have been proven to be beneficial in inflammatory diseases of the lung. In airway smooth muscle cells from asthmatic patients, elevated levels of the chemokine CXCL8 contribute to the inflammatory phenotype. The increased expression level of CXCL8 could be explained by increased acetylation (H3K18) and subsequent binding of BET proteins BRD3 and BRD4 to its promoter. Treatment of cells from asthmatic patients as well as healthy individuals with BET antagonists PFI-1, (+)-JQ1, and I-BET significantly reduced expression of CXCL8 by disrupting the binding of BRD4 and RNAP II to CXCL8 promoter.Citation49 Interestingly, in another cellular model of lung inflammation, using LPS stimulated A549 lung cell line, a model is proposed in which BET antagonism, and in particular antagonism of BRD2, indirectly reduces inflammation by influencing the expression levels of the histone deacetylase sirtuin 1 (SIRT1).Citation50 Also in chronic lung disease idiopathic pulmonary fibrosis, BET antagonism showed positive response attenuating migration, proliferation, and IL-6 release from lung fibroblasts of patients as well as reducing infiltration of pro-inflammatory cells and reducing fibrosis as assessed by histology in a mouse model of lung fibrosis.Citation51
BET antagonism also influences IL17-producing T helper cells (Th17), a subset of T helper cells implicated in autoimmune disorders as well as in the defense against fungal and bacterial infections. BET family members were shown to influence differentiation of CD4+ T cells to Th17 cells, but not other T cell lineages, as well as activation of Th17 cells through downregulation of cytokine expression necessary for Th17 differentiation and activation (e.g., IL-17, IL-21, GM-CSF, IL-22, and IL-23R). Knockdown experiments revealed non-redundant functions of BRD2 and BRD4 in Th17 cells, as knocking down either transcript individually was sufficient to recapitulate the phenotype observed with (+)-JQ1 or with double RNA interference. Studies in mice further demonstrated that both Brd2 and Brd4 are associated with chromatin at the Il17a control region.Citation45 Moreover, mice could be significantly protected from experimentally-induced models of autoimmune diseases, collagen-induced arthritis, and experimental autoimmune encephalomyelitis (EAE), by treatment with (+)-JQ1, which was critically dependent on generation and/or function of Th17 cells.Citation45
The cytokine IL-17A together with IL-22 and IL-23 are also key cytokines in the pathogenesis of psoriasis, and (+)-JQ1 demonstrated beneficial effects in a mouse model of IMQ-induced skin inflammation, as reflected by a decrease in ear thickness/myeloperoxidase activity, and RORC/IL-17A/IL-22 expression.Citation52 Th17 cells also play a role in cystic fibrosis (CF) and elevated levels of the Th17 effector cytokine IL-17 have been reported in the sputum of CF patients and shown to contribute to IL-17–mediated immunity in CF lungs. The BET antagonist CPI-203, derived from (+)-JQ1, was able to suppress IL-17 production in a mouse model of P. aeruginosa lung infection as well as suppress Th17 response in explanted CF patient tissue.Citation53 A different approach has been reported recently in a study of the autoimmune disease juvenile idiopathic arthritis. Epigenetic profiling, using primary patient-derived CD4+ memory/effector T cells from the site of inflammation revealed the presence of a disease specific super enhancer characterized by the active H3K27ac mark. Treatment with (+)-JQ1 resulted in preferential downregulation of genes associated with the disease and under control of the disease specific super enhancer, suggesting that regulation of disease associated genes can be inhibited by BET antagonists.Citation54
In another autoimmune disease, systemic lupus erythematosus (SLE), which is a systemic autoimmune disease characterized by autoantibodies against nuclear factors, the BET antagonist (+)-JQ1 has also shown promise in a mouse model of lupus (MRL-lpr mice) by reducing levels of pro-inflammatory B cell activating factors, while increasing IL-10. The latter inhibits production of several pro-inflammatory mediators. Treatment of mice also resulted in reduction of serum anti-dsDNA antibody and associated deposition of immune complexes in the kidney, which led to overall increased survival rate of the treated animals.Citation55 Although the authors show that CD4+ BRD4 levels are elevated in this mouse model, which can be reduced by treatment with (+)-JQ1, BRD2 has also been linked to autoimmune diseases, and SNPs in the BRD2 locus have been associated with autoimmune and inflammatory processes like rheumatoid arthritis.Citation56 Indeed, studies using a genetic model of brd2 lo mice, expressing about half of the normal amount of Brd2, led to reduced expression of pro-inflammatory cytokines by regulating NF-κB transcription.Citation57
Promising results have been reported on Helicobacter pylori infection, which is characterized by chronic gastritis and peptic ulceration. The disease is associated with increased inflammatory gene expression, of which about half can be downregulated by treatment with (+)-JQ1. Mechanistically, (+)-JQ1 thereby acts on both mRNA as well as enhancer RNA (eRNA) level, inhibiting H. pylori-induced interaction between Brd4 and p65 and the recruitment of Brd4 and RNAP II to the promoters and enhancers of inflammatory genes. Knockdown experiments identified Brd4 as the most relevant BET protein as only depletion of Brd4 also significantly reduced H. pylori–induced IL1A and IL1B mRNA expression, whereas depletion of Brd2 or Brd3 had little effect on the expression of IL1A but still impaired H. pylori–induced IL1B gene expression. Validity of the approach was demonstrated in H. pylori-infected mice, which showed significantly attenuated inflammation with reduced number of infiltrated immune cells and decreased inflammation score.Citation58
Other bromodomains
Antagonists of bromodomains other than BETs have also revealed a therapeutic potential for their respective targets in inflammation. CBP and EP300 are general transcriptional co-activators harboring a catalytic histone acetyltransferase (HAT) domain, in addition to the bromodomain, which catalyzes the transfer of acetyl moieties to lysine residues on histones, in particular H3K56, and non-histone proteins. The mechanism of the anti-inflammatory effect of the CBP/EP300 bromodomain antagonists is less well studied. Studies using in vitro FRAP assays suggest that targeting the BRD of CBP does not displace this protein completely from chromatin. However, displacement from specific loci may still happen.Citation59 The selective CBP/EP300 bromodomain antagonist SGC-CBP30 has shown potential in autoimmune disease. Profiling the biologic activity of SGC-CBP30 on primary human cell types by BioMAP® analysis revealed that SGC-CBP30 reduced immune cell production of IL-17A and other pro-inflammatory cytokines. Moreover, SGC-CBP30 inhibited IL-17A secretion from CD4+ T cells purified from healthy donors as well as from patients with ankylosing spondylitis and psoriatic arthritis. Transcriptional profiling of human T cells treated with SGC-CBP30 showed a much more restricted effect on gene expression than that observed with (+)-JQ1,Citation60 but further studies will be necessary to fully explore the potential of CBP antagonism in autoimmune diseases. Independently, Genentech (GNE) and Constellation Pharmaceuticals (CPI) found a role for CBP inhibitors in the biology of regulatory T cells, which play important roles in cancer immunotherapy.Citation61 The p300/CBP associated factor, PCAF, another bromodomaining containing HAT has also been implicated in inflammatory diseases, and specific antagonists to the bromodomain of this protein have just been reported.Citation62,63
BRD9 and BRD7 are part of the SWI/SNF remodeling complexes BAF and PBAF, large multisubunit complexes harboring several bromodomain-containing proteins including the mutually exclusive catalytically active components SMARCA2/BRM and SMARCA4/BRG1. BRD9 is associated with the BAF complex, while BRD7 is part of the PBAF complex, which also harbors the 6-bromodomain containing protein polybromo 1 (PB1 or PBRM1) anchoring the complex to chromatin.Citation2 Antagonism of BRD7/9 by the chemical probe LP-99 was found to inhibit the secretion of IL-6 from cultured THP-1 cells and the selective BRD9 antagonist I-BRD9 was found to regulate genes involved in inflammatory pathways when Kasumi leukemia cells were treated with the inhibitor.Citation64 Interestingly, a recent analysis of BRD7 knockout mice showed that by the age of 6–12 months these animals were more prone to inflammation. Mouse embryonic fibroblasts (MEFs) from these animals stimulated with LPS demonstrated increased pro-inflammatory cytokine response as compared with wild type MEFs, accompanied by an increase of translocation of p65 to the nucleus at early time points (1 h). BRD7 deficiency also promoted early acute colitis induced by dextran sodium sulfate.Citation65 As BRD9 and BRD7 have been proposed to be associated to different SWI/SNF complexes, they may play opposing roles in inflammation.
Lysine demethylases
Very few high quality demethylase probes are available to date.Citation66 GSK-J1 and its cell active pro-drug GSK-J4 have been used to elucidate the role of the KDM6 family member JMJD3/KDM6B in inflammation. GSK-J1/J4 inhibit JMJD3/KDM6B as well as the related UTX/KDM6A. The histone demethylases of the KDM6 family remove the repressive H3K27me3/2 marks, allowing for permissive chromatin. These repressive marks are added by the methyltransferase activity of the PRC2 complex, and inhibitors against the EZH catalytic subunit have been described below. Both KDM6A/UTX as well as KDM6B/JMJD3 can overcome this repressive chromatin state to achieve normal gene expression. Activation of specific genes is particularly required during cell lineage commitment and KDM6 not only initiates gene expression by removing the repressive mark, but also promotes transcription elongation by forming a complex with factors bound to the elongating form of RNAP II.Citation67
Differentiation of macrophages in response to pro-inflammatory stimuli is one such event regulated by KDM6. KDM6B has been shown to bind PcG target genes regulating their H3K27me3 levels and transcriptional activity.Citation67 The inhibitor of KDM6, GSK-J4, has accordingly shown potential in rheumatoid arthritis (RA) reducing production of the pro-inflammatory cytokine TNF-α from macrophages derived from RA patients.Citation68 Interestingly, like studies shown on the H3K27me3 methyltransferase EZH2, GSK-J4 as well as genetic depletion of KDM6B in CD4+ T cells has been shown to suppress differentiation of Th17 cells, suggesting a potential role for this target in autoimmune responses.Citation69 Surprisingly, T cells isolated from mice in which Ezh2 has been deleted in CD4+ T cells also show a reduced differentiation to Th17 cells.Citation70 These apparent contradictory results could be addressed using inhibitors against both enzymes in parallel to elucidate the precise mechanism and compare the effects observed inhibiting the catalytic activity compared with the effect in the knockout cells, which are also missing the enzyme scaffolding functions.
GSK-J4 showed promising results in reducing expression of pro-inflammatory cytokines in a model for the autoimmune disease Type 1 diabetes mellitus. In this disease, the insulin-producing pancreatic β-cells are selectively destroyed by the host immune system. Pancreatic islets exposed to the cytokines IFNγ, Il-1β, and TNFα overcame H3K27me3 mediated repression and induced rapid chemokine gene activation, which persisted for several days. Treatment of cytokine-exposed islets with GSK-J4 led to significant reduction of chemokines and cytokines. However, GSK-J4 exposure also induced significant β-cell dysfunction and islet cell apoptosis.Citation71 In another model, experimental autoimmune encephalomyelitis (EAE), GSK-J4 showed promising results in vivo. Treatment of mice with GSK-J4 inhibited the development of EAE. This effect was mediated by dendritic cells, which adopted a more tolerogenic phenotype accompanied by changes in the ratio of H3K27me3 and H3K4me3 on specific promoters. The reduction of H3K27me3 increased H3K4me3 levels on the IL-6 and TGF-β1 promoters, moreover indicating that the effect is mediated by KDM6 inhibition. Promotion of a more tolerant profile was characterized by reduced expression of co-stimulatory molecules CD80/CD86, an increased expression of tolerogenic molecules CD103 and TGF-β1, and reduced secretion of pro-inflammatory cytokines IL-6, IFN-γ, and TNFα. The effect was accompanied by increased generation, stability, and activity of Treg cells but not Th1 or Th17 cells.Citation72 Inhibition of JMJD3 may also be beneficial in the treatment of osteoarthritis. Treatment of human mesenchymal stem cells with GSK-J4 inhibited collagen production. This occurred at the level of gene expression, as key chondrogenic genes, including SOX9 and COL2A1, were suppressed, resulting in disrupted glycosaminoglycan and collagen synthesis. JMJD3, and not UTX, whose activity is also inhibited by GSK-J4, was identified by knockdown studies as the key enzyme in chondrogenesis and osteoarthritis.Citation73 Suppression of inflammatory cytokines by treatment with GSK-J4 and knockdown studies has also been observed in a model of keratinocyte wound healing. However, treatment or JMJD3 depletion resulted in significantly delayed wound closure, most likely as a result of suppression of MMP and growth factor gene activation. The study also confirms the association and cooperation of JMJD3 and p65, but did not analyze in detail the effect of GSK-J4 on this interaction.Citation74 It is important to notice that GSK-J1/J4 also inhibits, albeit to a lesser extent, demethylases of the KDM5 subfamily, namely KDM5B (JARID1B) and KDM5C (JARID1C).Citation75,76 It is therefore important to complement studies using these inhibitors by alternative methods, e.g., specific knockdowns, able to distinguish between the targets involved.
Few specific inhibitors for the other JmjC family members have been reported to have an effect outside oncology.Citation77 In particular, the related enzymes KDM2A (also called F-box and leucine-rich repeat protein 11, FBXL11) and PHD Finger Protein 8 (PHF8) could play potential roles in inflammatory diseases, and chemical probes would help to manifest their therapeutic potential. FBXL11 has been reported to demethylate the p65 subunit of NF-κB at lysine residues K218 and K221, and the authors show that in mouse embryo fibroblasts, the activation of most p65-dependent genes relied on K218/K221 methylation. In addition, expression of the KDM2A gene is driven by NF-κB, resulting in a negative regulatory feedback loop. Additional regulation occurs at the level of microRNA. The microRNA miR-19b downregulates expression of KDM2A resulting in a positive effect on NF-κB signaling.Citation78 On the other hand, PHF8 has been identified as a pro-inflammatory regulator. In an LPS-induced acute-inflammation macrophage model, knockdown of PHF8 led to a reduction of pro-inflammatory cytokines accompanied by an increase in the repressive H3K9me2 mark. Additionally, PHF8 promoted T cell activation and proliferation, providing the first link between the epigenetic regulation of inflammation and adaptive immunity.Citation79
Deiminases
A less explored histone modification is citrullination or deamination. PAD4 is a calcium-dependent enzyme, which catalyzes the transformation of protein arginine residues into citrulline. There is a strong association of PAD4 to rheumatoid arthritis and antibodies against citrullinated epitopes as well as against PAD4 itself represent diagnostic hallmarks of the disease. In addition, PAD4 is known to promote profound chromatin decondensation during the innate immune response to infection in neutrophils by mediating formation of neutrophil extracellular traps (NETs). NETs were originally described in the context of trapping pathogens for host-defense purposes, but findings have now been extended to demonstrate that unrestrained NETosis may be crucial for pathological deep venous thrombosis ischemia/reperfusion injury, systemic lupus erythematosis, and small vessel vasculitis as well as rheumatoid arthritis.Citation80 The PAD4 inhibitor GSK484 specifically binds to the low-calcium form of PAD4 and fully inhibited mouse NETosis, while NET-like structures in human neutrophils were only partially inhibited, the reason for which is still unclear.
Protein methyltransferases
Lysine,8182 and arginineCitation83 methyltransferases, such as, SETD784, GLP,Citation85 NSD1 and NSD2,Citation86 G9a,Citation87 SETD6,Citation87 SUV420,Citation88 PRMT5,Citation89-91 PRMT1,Citation92-97 PRMT6,Citation98 SMYD2,Citation99 EZH2,Citation100 and SETDB1,Citation101 also modulate the NF-κB pathway. Recent reviews outline the known R and K methylation sites of p65,Citation82 and summarize the known roles of some PRMTs in inflammationCitation83 (). Chemical probes are available for all targets described in this section except NSD1, NSD2, SETDB1, and SETD6.
Under conditions of NF-κb activation by TNF-α, p65 is modified by PRMT1 to form the asymmetric dimethyl mark on R30 (p65R30me2a) and the mono-methyl mark at R236 (p65R236me).Citation96 Interestingly, transcription of NF-κB target genes is reduced, which means that p65R30me2a in conjunction with p65R236me triggers a repressive state. The impact of p65R236me may be a result of its proximity to the C terminus of the Rel homology domain (RHD) and, thus, to the (inhibitory) IκBα-interacting and dimerization interfaces. The pivotal role of the conserved residue R30 is illustrated by its symmetric dimethylation by PRMT5 (p65R30me2s) in response to IL-1β and subsequent activation of the NF-κB pathwayCitation89 ().
In another example, endothelial cells co-stimulated with TNF-α and IFN-γ produce the chemokine CXCL11. The mechanism involves PRMT5 catalyzed symmetric dimethylation of p65 at R174 (to form p65R174me2s) and subsequent binding of p65 to the CXCL11 promoter.Citation90 In a knockdown of PRMT5, p65 is unable to associate with the promoter and CXCL11 mRNA and protein are reduced (in co-stimulated cells). When endogenous p65 is depleted and then reconstituted with R174K mutants, the potency of CXCL11 production by co-stimulated cells is reduced. These data illustrate how a single epigenetic modification in different contexts results in different phenotypes. Also in endothelial cells, excess S-adenosylhomocysteine (SAH) activates conserved and non-traditional NF-κB pathways leading to reduction of EZH2 and H3K27me3.Citation100 This results in secretion of the pro-inflammatory cytokine IL-1β. These effects were mimicked more closely by inhibition of SAH hydrolase relative to knockdown of EZH2. Nevertheless, this non-canonical pathway may represent yet another alternative route to modulating the NF-κB response.
Toll-like receptors (TLRs) play an important role in the innate immune response, particularly in the initial interaction between foreign material and macrophages. SMYD2 is an H3K4 and H3K36 methyltransferase that plays a critical role in TLR4-induced macrophage activation and pro-inflammatory cytokine secretion.Citation99 The H3K36me2 mark suppresses TNF-α and IL-6 expression but does not appear to alter the expression of other genes characteristic of M1 macrophages, such as IL-1β and IL-12. IL-6 is important for Th17 differentiation and the reciprocal differentiation of Treg relative to Th17 cells. As such, overexpressed SYMD2 in macrophages promotes Treg cell differentiation via a mechanism that inhibits the NF-κB and ERK signaling pathways.
Systemic inflammatory response syndrome (SIRS) or sepsis can lead to complete organ failure.Citation102 NF-κB transcription factor RelB is induced by TLR4 stimulation and is able to silence transcription of TNF-α and IL-1β in SIRS.Citation85 The data implicates a complex between RelB, G9a, and HP1 at the promoter of IL-1β, which results in a switch to heterochromatin. Although the nature of the molecular interaction is unknown, it was shown that RelB co-immunoprecipitated predominately at a binding site within the first 500 amino acids of G9a. Further, formation of heterochromatin is favored when G9a is knocked down. RelB still binds to the IL-1β promoter but G9a and HP1 do not. In macrophages, SETDB1 (another H3K9 methyltransferase) suppresses TLR-4 mediated expression of cytokines such as IL-6.Citation101 Loss of SETDB1 results in an increase in recruitment of NF-κB to the proximal promoter of IL-6, thus increasing IL-6 production. Unfortunately, there are no published chemical probes for SETDB1.
Fibroblasts are the main source of the extracellular matrix and contribute to the inflammatory changes in pulmonary inflammation and related diseases. Three studies describe the role of PRMT1 in pulmonary inflammation.Citation94,95,97 Stimulation by IL-4 (one of the most important pro-inflammatory cytokines driving airway inflammation) produced TGF-β and other inflammatory cytokines, which encouraged fibroblast proliferation and PRMT1 expression in sub-epithelial fibroblasts.Citation94 Upregulated PRMT1 (in turn) activates fibroblast production via COX2 and VEGF. However, blocking TGF-β did not abolish PRMT1 expression and implicated involvement of other cytokines. A follow-up studyCitation97 identified platelet derived growth factor B homodimer (PDGF-BB) but not TNF-α or TGF-β1 as the strongest inducer of PRMT1 expression. The mechanism presumably involves a cascade such as this: “PDGF-BB → ERK1/2 (MAPK) → STAT1 → PRMT1.” In line with this, PDGF-BB exposure caused fibroblasts to produce more collagen-1A1 and fibronectin (which are found in the extracellular matrix). Inhibition of PRMT1 with AMI-1 (a type 1 PRMT inhibitor) at 10 micromolar considerably reduced all pro-fibrotic activities of lung fibroblasts via COX2 (and in the same way as the previous study). MS023,Citation103 a more potent type 1 PRMT inhibitor that has an IC50 = 9 ± 0.2 nM for reduction of H4R3me2a in cells, may help to corroborate the findings. Finally, the NF-κB pathway was identified as the mediator for IL-1β stimulated fibroblasts, while the STAT6 pathway was identified as the mediator for IL-4 stimulated epithelial cells.Citation97 PRMT1 expression was induced in both acute and chronic asthma.
Neuroinflammation
The interest in exploring the role of epigenetics in neurologic disorders was catalyzed by 3 main publications. These publications highlighted the role of HDAC inhibitors in stopping progressive neurodegeneration in a polyglutamine-repeat Drosophila model,Citation104 and the role of mutations in proteins that regulate DNA or histone methylation.Citation105,106 Since then, key roles for epigenetic proteins have been implicated in Alzheimer disease (AD), polyglutamine repeat disorders, Parkinson disease (PD), and fused in sarcoma (FUS) accumulation.
AD is the most prevalent cause of dementia and is characterized by progressive cognitive and functional impairment and memory loss. Well-defined neuropathological brain changes include amyloid plaques, which are extracellular deposits of β-amyloid (Aβ), and neurofibrillary tangles, which are primarily composed of hyper-phosphorylated tau. The disease is accompanied by chronic neuro-inflammation. Although the pathophysiology of AD is still unknown, much evidence indicates that Aβ and tau species make important contributions to disease progression. Recent developments show that the inflammatory disease is not a mere consequence of disease onset, but that immune system-mediated actions contribute to and drive AD pathogenesis. Both innate immune cells like microglia as well as macrophages are the prime targets for modulating neuroinflammation.Citation107
Bromodomains
In a mouse model of AD, treatment with the brain penetrant BET bromodomain antagonist (+)-JQ1 resulted in reduced neuroinflammation assessed by expression of pro-inflammatory modulators. In addition, (+)-JQ1-treated mice showed a reduction of tau phosphorylation at Ser396 in the hippocampus and frontal cortex, while total levels of tau remained unaffected. However, no improved learning or memory deficits were observed in the (+)-JQ1-treated mice.
Likewise, studies in mouse primary astrocytes stimulated with LPS showed involvement of the bromodomain containing protein Brd2, which was increased both at the mRNA and protein levels upon LPS stimulation. Knockdown of Brd2 mRNA by small interfering RNA or treatment with (+)-JQ1 in cultured mouse primary astrocytes specifically inhibited LPS-induced mRNA expression and secretion of plasminogen activator inhibitor-1 (PAI-1) but not that of other pro-inflammatory cytokines measured, providing evidence for the involvement of Brd2 in neuroinflammation.Citation27
Protein methyltransferases
Polyglutamine diseases all share an expansion of the CAG repeat in specific genes; however, the impact of each mutation is uniquely manifested. Huntington's disease (HD) is by far the best known of these diseases, whereas spinal and bulbar muscular atrophy (SBMA), for example, is less so. ChIP analysis identified fewer peaks with the activating mark H3K4me3 at transcription start sites (TSS) in HD subjects relative to controls.Citation108 Also, an unexpected number of H3K4me3 peaks were located at sites downstream of the TSS including about 1/3 at known enhancer and chromatin remodeling sites, such as EZH2 and SUZ12. Although the latter were differentially enriched in the distal H3K4me3 peaks from HD subjects, PRC2 was reduced. Thus, in addition to showing discordant transcriptional regulation in HD subjects, there may be a PRC2-independent role for EZH2 in HD. This is a typical example where available chemical probes selective for EZH2, such as GSK343109 (or GSK126110), EPZ005687,Citation111 in parallel with the EED antagonist, A-395 [http://www.thesgc.org/chemical-probes/A-395], may be used to further explore the findings.
SBMA is an X-linked recessive disorder caused by a mutation in the androgen receptor (AR) gene resulting in a CAG expansion. PRMT6 is a co-activator of both wild type and mutant AR but co-activation of mutant AR is enhanced in SBMA.Citation112 Knockdown of PRMT6 suppressed the toxicity of polyQ-AR and, at least in a fly model, was able to rescue the phenotype associated with overexpression of normal AR. It is proposed that in the absence of protective phosphorylation of polyQ-AR at the Akt consensus RXRXXS, PRMT6 methylation at this site results in binding to testosterone and toxicity. There are 3 chemical probes, EPZ020411,Citation8 MS023,Citation103 and MS049,Citation66 which inhibit PRMT6. Unfortunately, none is sufficiently selective to be used alone. However, if used in parallel, EPZ0204118 and MS023103 (at 20 nM or less), may help corroborate the involvement of PRMT6 in the fly model of SBMA.
RNA transcriptome profiling from CSF of 27 subjects with PD and 30 age- and sex-matched controls identified diverse fragmented RNA including some that are dysregulated in PD.Citation113 Protein-coding RNA fragments comprised 80% while noncoding RNAs comprised 13% of the total transcripts. However, of the differentially expressed transcripts, noncoding RNAs are more abundant than protein-coding RNAs. The upregulated transcripts mostly included chromatin remodeling while the downregulated transcripts were mainly represented by protein-tyrosine phosphatases, endoplasmic reticulum membrane proteins, and genes that regulate phosphorylation, de-phosphorylation and phospholipase activity. Of the chromatin remodeling transcripts, EZH2 was upregulated and DNMT1 downregulated, thus aligning with ChIP analysis of HD neuronal tissue.Citation108
One of the roles of the endoplasmic reticulum (ER) is synthesis and folding of secreted and transmembrane proteins. When misfolded and unfolded proteins accumulate in the lumen of the ER, the unfolded protein response (UPR) pathway, also recently reviewed,Citation114 is triggered. Persistent over-activation of the ER stress response pathway has been implicated in neurodegenerative diseases,Citation115 immune response,Citation116 and inflammation.Citation117 Two proteins that are central to the ER stress response are cyclic AMP-dependent transcription factor (Atf4) and DNA damage-inducible transcript 3 (Ddit3). DOT1L is currently the only published H3K79 methyltransferase in mammals. When DOT1L is overexpressed, it is known to disrupt telomere silencing. Inhibition of DOT1L by SGC0946118 or EPZ00567675 in neural stem cells (NSCs) results in an increase in transcription of Atf4 and Ddit3; this was confirmed by a reduction of H3K79me2 at their promoters.Citation119 However, in 2 unrelated cell types, mouse embryonic stem cells and a leukemia cell line, the ER stress response genes were not induced after DOT1L inhibition. Thus, this is another example of a context-specific role of a methyltransferase.
Amyotrophic lateral sclerosis (ALS) and fronto-temporal lobar degeneration (FTLD) are neurodegenerative diseases that are characterized by deposits of the fused in sarcoma (FUS) DNA/RNA-binding protein. In ALS, PRMT1 asymmetrically dimethylates FUS—while in FTLD, PRMT1 monomethylates FUSCitation120 even though unmethylated FUS is the predominant form. In ALS, the nuclear function of PRMT1 appears to play a role in the toxicity of mutant FUSCitation121 because its re-distribution to the cytoplasm associates with cytoplasmic accumulation of FUS and changes in repressive histone marks.
M-channels (which are encoded by Kv7/KCNQ genes) are K+ channel proteins that modulate neuronal excitability. Dysregulation of the KCNQ genes is one underlying cause of epilepsy. In a mouse model, PRMT1 heterozygotes have impulsive seizures and their brains feature lower expression of asymmetrically dimethylated KCNQ2 relative to wild types.Citation122 Further, methylated KCNQ had higher affinity for their binding partner phosphatidylinositol-4,5-bisphosphate (PIP2). In vitro experiments identified 4 modified arginine residues, R333, R345, R353, and R435, on KCNQ2 that also reside in the PIP2-binding region. These modifications require PRMT1 but do not appear to need its close relative PRMT8.Citation122 PIP2 could not fully rescue M-currents in neurons of PRMT1 heterozygous mice; this points to the involvement or redundancy of other PRMTs.
Immune response
Protein methyltransferases
Class II major histocompatibility complex (MHC II) transcription is activated by IFN-γ via the class II transactivator (CITTA), which in turn interacts with epigenetic factors, including protein methyl- and acetyl-transferases.Citation123 The mechanism of activation by IFN-γ appears to involve recruitment of PRMT5 by CITTA to the promoter of MHC II and is evidenced by the appearance of H3R2me2s (and CITTA at the promoter).Citation91 The transactivator may also play a role in promoting communication between the H3K4 methyltransferase complex and PRMT5 on the MHC II promoter. Since PRMT5 depletion downregulates, but does not remove, the expression of MHC II in macrophages, there may be other PRMTs involved in modulating MHC II transcription. Chemical probes that inhibit the type 1 PRMT family and that are selective for PRMT3, PRMT4, and PRMT5 would be very useful for follow-up studies.
IBD is a chronic inflammatory disease of the gastrointestinal tract of which Crohn's disease (CD) and ulcerative colitis (UC)Citation124 are the 2 main types. There is evidence for both genetic and epigenetic influences in IBD. Two recent publications investigate the role of T cell activation and function in intestinal inflammation in the context of epigenetic modulation by G9aCitation125 and EZH2126. G9a, which dimethylates H3K9, is involved in regulating gene expression during lineage commitment in adult CD4+ T cells.Citation127 Antignano et al.Citation125 show that H3K9me2 (a mark that is associated with repression of lineage-promiscuous genes) controls access to chromatin and regulates responsiveness to TGF-β1 (a lineage-specifying cytokine) in Th17 and Treg cells. H3K9me2 also contributes to T cell mediated colitis by limiting differentiation to Th17 and Treg cells. In naïve T cells, G9a maintains H3K9me2 at high levels, which are removed upon activation. In the absence of G9a-dependent H3K9me2, ∼20 times less TGF-β1 is able to induce differentiation to Th17 and Treg cells. Thus, the lack of this mark reduces the amount of stimulation required for differentiation into the protective state. The authors surmised that G9a-dependent H3K9me2 modulates chromatin accessibility because chromatin rich in the H3K9me2 mark is located near the nuclear lamina where accessibility and gene expression is reduced.Citation128,129 Chemical inhibition of G9a methyltransferase activity in wild type and G9a deficient T cells in the absence and presence of UNC0638130 and BIX01294,Citation131 and in Treg promoting conditions show that G9a methyltransferase activity in naïve T cells is a negative regulator of Th17 and Treg differentiation.
Paneth cells, a specialized component of intestinal epithelial cells, release peptides that protect the intestines against inflammation and intestinal pathogens. Abnormal function of Paneth cells has been linked to IBD.Citation132 GSEA analysis identified upregulation of “SUZ12_TARGETS” and ChIP analysis of PrkciIEC-KO (knockout) and Prkcifl/fl mice showed EZH2 occupying regulatory regions of Atoh1 and Gfi1 (which are key transcription factors in Paneth cell differentiation). Similar results were obtained when PKCλ/ι was knocked out with CRISPR/Cas9 gene editing. Further, an in vitro kinase experiment illustrated that PKCλ/ι predominately phosphorylates EZH2 at T487 and to lesser extents at S690 and T345. These threonine phosphorylation sites are implicated in protein degradation.Citation133 In line with this, the EZH2 inhibitor GSK126109 resulted in upregulation of Paneth cells in PrkciIEC-KO organoids. The authors also showed that the expression of PKCλ/ι decreases with the progression of Crohn's disease.
EZH2 suppresses CD4+ T cell differentiation into Th1, Th2, and Th17 cells in vitro but is required to generate the best conditions for inducing Treg cell production in vivo.Citation69 The effect of EZH2 deletion in a mouse model resulted in reduced Foxp3+ T cells in the spleen and lymph nodes with no measurable change in the thymus or lamina propria. Also, production of Foxp3 was attenuated in the surviving Treg cells. The EZH2-deficient effect on Foxp3 production was due to aberrant overproduction of effector cytokines IFN-γ and IL-4. This observation was recapitulated in a transfer colitis model but when mice were infected with T. gondii, there was an insufficient effector cytokine response and the mice succumbed to the infection. These observations may explain the number of seemingly contradicting reports on the role of EZH2 in CD4+ T cell differentiation. In a very recent study using a mouse model of intestine specific deletion of EZH2 or EED,Citation134 it is clear that EZH2 is dispensable for wnt signaling and proliferation but not differentiation. However, deletion of EED resulted in loss of differentiation and stem cell maintenance (including wnt signaling and differentiation) in intestines. Here again is another opportunity to use the available chemical probes for EZH2 and EED.
Viral diseases
Human immunodeficiency virus
Several epigenetic chemical probes have been explored in the context of HIV treatment. Effective antiretroviral therapy targeting key viral enzymes is now available resulting in undetectable HIV RNA in the plasma of HIV patients. However, affected individuals are not cured as the virus persists in long-lived cells like memory T cells and re-emerges upon cessation of the treatment. This dormant status of the virus, termed latency, has long been thought not to be therapeutically accessible. The viral genome is integrated in the genome of host cells, which presents in resting T cells in a repressive state preventing transcription of viral genes. Among other factors, latency can be affected by the levels of cellular transcription factors, including P-TEFb and NF-κB, both key targets of epigenetic proteins like BRD4, as described above. Also, changes in chromatin can affect transcription of viral genes. Reactivation of HIV in resting cells and subsequent targeting has been shown to be a useful strategy to eliminate the reactivated cells, an approach referred to as ‘shock and kill’.Citation135,136 However, unwanted reactivation of an undiagnosed viral disease by treatment with epigenetic inhibitors for a different application may result in deleterious effects ().
Figure 5. Schematic representation of epigenetic proteins influencing HIV latency.
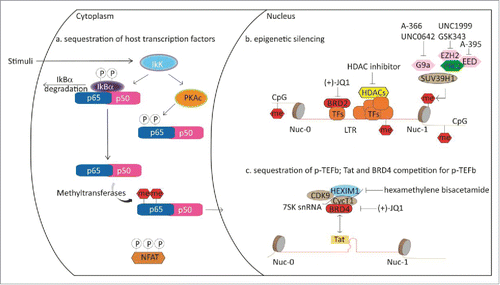
Bromodomains
Viral transcription requires recruitment of P-TEFb to the viral promoter as well as overcoming transcriptional pausing leading to elongation of the transcript. Much progress has been made in elucidating the mechanism in detail.Citation135 Prior to production of the HIV transactivator protein Tat, BRD4 recruits P-TEFb to the HIV promoter upon cellular stimulation. Upon synthesis of Tat, Tat and BRD4 compete for P-TEFb binding, making BRD4 a negative regulator of Tat transactivation. Consequently, HIV expression can be induced by treatment with BET antagonists. Several reports describe that treatment of HIV infected cells with (+)-JQ1 or other BET antagonists reactivates the virus.Citation137,138,139 However, there seems to be a fair amount of variability in the response to (+)-JQ1 treatment and some patient cells do not respond at all. The best response seems to be achieved upon co-treatment with other latency reversing compounds like protein kinase C (PKC) agonists prostratin, bryostatin-1, and ingenol-B, which work by increasing PKC-dependent phosphorylation of IκB and ultimately by activation of NF-κB-dependent genes. BET antagonists may thereby not only act on the level of P-TEFb, but also via NFκB, although the precise mechanism is not yet clear.Citation140 The PKC agonist, PEP005, an ingenol-3-angelate, also showed synergistic effect with (+)-JQ1 in HIV reactivation. In the study, induction of the pS643/S676-PKCδ/θ-IκBα/ϵ- NF-κB signaling pathway was investigated in detail.Citation139 Contrary to results observed with ingenol-B levels of NF-κB were not affected in this study. PEP005 also induced expression of HIV RNAs in primary CD4+ T cells from HIV infected individuals receiving suppressive antiretroviral therapy.Citation138 Similar positive results of HIV reactivation in T cells from patients receiving suppressive antiretroviral therapy were recently published with the PKC agonist prostratin in combination with the BET antagonist OTX015, which has entered phase Ib clinical trials for advanced hematologic malignancies. The mechanism is Tat dependent as OTX015 significantly increased expression of a HIV-1 LTR driven luciferase reporter in the presence, compared with the absence of Tat.Citation141
A different Tat-independent mechanism has been described for BRD2. Knockdown experiments revealed that absence of BRD2 resulted in a robust activation of the HIV LTR, which was only slightly enhanced in response to (+)-JQ1 and is thought to be mediated by mechanisms involving repressive chromatin marks mediated by HDACs and interaction with the transcription factor E2F, which together with NF-κB, binds to the HIV enhancer to repress HIV transcription ().Citation142,13
Lysine demethylases
UTX1, but not JMJD3, leads to induction of genes during Tat-mediated HIV-1 transactivation by removal of the repressive H3K27me3 mark. In a cellular model, Zhang et al. found Tat-induced HIV-1 transactivation is dependent on UTX, but not JMJD3. Treatment with GSK-J4, as well as knockdown of UTX, led to inhibition of Tat-mediated transactivation, indicating that the catalytic activity is responsible for the effect. Additionally, ectopic expression of UTX, but not of an inactive mutant corroborated this finding. Downregulation of UTX was accompanied by downregulation of H3K27 trimethylation and upregulation of H3K4 methylation on the specific regions of HIV-1 LTR as well as translocation of p65 to the nucleus and binding of NFκB to the LTR promoterCitation143 In a similar manner, the genome of the latent herpes simplex virus (HSV) is associated with repressive chromatin marks H3K27me3 and H3K9me2/me3. Treatment of HSV-infected neuronal cells with GSK-J4 reactivated the virus. This study, however, did not investigate the specific role of UTX or JMJD3.Citation144
Protein methyltransferases
A number of protein methyltransferases (including G9a, SUV39H2, and EZH2) are involved in maintaining HIV silencing.Citation145,146 Recently, it was shown that chemical inhibition of EZH2 plays a role in reactivating the latent HIV long-terminal repeat (LTR).Citation110,147 Pre-treating a primary resting T cell model of HIV with the EZH2 inhibitor GSK343109 [at a concentration that reduced methylation of H3K27 at the HIV LTR nucleosome 1 (Nuc-1) site] is associated directly with sensitization of the HIV LTR to subsequent exposure to vorinostat (an HDAC inhibitor and latency reversing agent in its own right). ChIP showed that GSK343 treatment reduced methylation at Nuc-1 (near the LTR transcription start site) and Nuc-0 (in the enhancer region). This led to enhanced pro-viral expression and latency reversal beyond that achieved by vorinostat alone. Using a dual-color reporter virus to monitor LTR kinetics, histone covalent modifications, particularly polycomb repressive complex 2 (PRC2)-mediated H3K27 trimethylation, appeared to dominate viral transcription at the early phase. PRC2 also contributes to time-dependent LTR dormancy in the chronic phase of the infection. Significant differences in sensitivity against several physiologic and pharmacological (GSK126) stimuli were observed between these 2 distinct populations ().
HIV-1 Tat protein can control different cellular processes but this activity is dependent on its nuclear/nucleolar localization. Tat enters the nucleus via its nuclear localization sequence (NLS) to allow transcriptional elongation of the HIV-1 genomeCitation148 through recruitment of positive elongation factor b (P-TEFb) complex, as well as chromatin remodeling factors.Citation149 Tat protein undergoes considerable posttranslational modification to augment cofactor recruitment including methylation by PRMT6150,151 on R52 and R53.Citation152 More recently, it was shown that ectopically expressed PRMT6 results in exclusion of GFP-tagged Tat from the nucleolus; in the presence of PRMT6, the fractional recovery was reduced but the rate of recovery remained the same. Tat-GFP in the presence of inactive full-length PRMT6 or Tat R52/53A mutants did not affect entry to the nucleolus. Therefore, it is unlikely that PRMT6 influences the rate of entry to the nucleolus.
Other viral diseases
Bromodomains
Other viruses make use of BRD4 to hitchhike the host transcriptional machinery. Raccoon polyomavirus (RacPyV) is a virus that has been detected in every neuroglial tumor diagnosed in free-ranging raccoons in the western United States. The virus was distributed throughout the body but accumulated in neuroglial tumor tissue. A recent study demonstrated that the tumors most likely originate from neural stem cells, based on the identified markers. ChIP experiments moreover demonstrated a tethering of BRD4 to the viral genome, which is thought to be critical for maintenance of the viral genome and oncogenic transformation. BET antagonists like (+)-JQ1 and IBET-151 (GSK1210151A) reduced the presence of RacPyV genome within cells as well as viral gene transcripts LT and VP1.Citation153 The precise mechanism of gene activation remains to be elucidated. Contrary to human polyomavirus JC (JCV), the mechanism of the positive effects seen upon treatment with BET antagonists has been elucidated and linked to the NF-κB pathway. Human neurotropic JCV replicates in glial cells causing cytolytic death of oligodendrocytes and giving rise to expanding multifocal lesions of myelin loss. The disease, called progressive multifocal leukoencephalopathy (PML), is often fatal. Expression of viral genes from this double-stranded DNA virus is regulated by a noncoding control region (NCCR) containing promoter/enhancer elements for viral gene expression including a binding site for NF-κB. Similar to the HIV virus, pro-inflammatory cytokines including TNF-α are thought to lead to reactivation of the virus and progressive multifocal leukoencephalopathy (PML). Treatment with a BET antagonist showed that JCV replication was increased by BRD4 expression and decreased by BRD4 antagonist treatment in part though inhibition of JCV early promoter activity. In addition, mutational studies demonstrated that both BRD4 and p65 acetylated at K218 and K221 are necessary for p65 nuclear localization, suggesting that the direct interaction of BRD4 and p65 is critical for p65 binding to chromatin and viral transcription.Citation154 Epstein-Barr virus (EBV) is a key etiologic agent in the development of numerous cancers including infectious mononucleosis, Burkitt's lymphoma, Hodgkin's lymphoma, HIV-related lymphomas, post-transplant lymphoproliferative diseases (PTLDs), nasopharyngeal carcinoma, and some gastric cancers. Primary infection with the virus resides in the B cell compartment, and latently-infected B cells are also the reservoir from which the virus establishes lifelong infection. One of the genes through which the early viral genes exert transcriptional activation is MYC.Citation155 Using ENCODE data, Zhou et al. found that the early viral proteins EBNA2, EBNALP, EBNA3A, EBNA3C co-localized with members of the NF-κB family on H3K27ac marked EBV super enhancers to activate expression of B cell relevant transcription factors, including the oncogene MYC. (+)-JQ1 treatment reduced MYC expression and also halted growth of lymphoblastic cell lines.Citation155 Earlier studies have shown that (+)-JQ1 also acts on the viral C promoter Cp by interfering with P-TEFb recruitment via BRD4, which is a crucial step in cellular immortalization.Citation156
In human papilloma virus (HPV), associated with cervical and anal cancer, the E2 protein is the master regulator of viral early gene transcription and also critical for persistence of the viral genome in host cells. The E2 protein itself is composed of an N-terminal transactivation domain and a C-terminal dimerization domain connected by a flexible hinge region.Citation157 The C-terminal portion of BRD4 has been shown to directly interact with E2, stimulating E2-mediated transcription. Similarly, in reporter assays, both BRD4 knockdown and disruption of the E2-BRD4 interaction reduce E2 ability to repress expression of the reporter gene. The transcriptional activity is mediated by recruitment of P-TEFb to the virus genome, as shown by ChIP experiments. Treatment of papilloma-infected cells released BRD4 and E2 from chromatin and inhibited E2-mediated transcriptional activation.Citation158 Phosphorylation of E2 at specific Serine residues located in the hinge region has been shown to be important for the interaction of BRD4 with chromatin as well as transcriptional activation.Citation157 The interaction of E2 with BRD4 is also critical for viral replication of HPV.Citation159
Regulation of c-MYC, a key target of BRD4-mediated transcription, has been shown to be important for the replication of hepatitis C virus (HCV) using c-Myc, HIF-1α, and glycolytic enzymes, which were found to be significantly higher in HCV-infected human liver and hepatocytes than in uninfected controls. This upregulation of c-Myc may play a role in HCV-associated hepatocellular carcinoma. Metabolic adaptation of the host cell is required for efficient virus replication. Interestingly, the metabolism of infected cells resembles that of cancer cells. Concomitant overexpression of MYC, HIF-1 α, and glycolytic enzymes also point to a combined regulation of these proteins. Treatment of HCV infected cells with (+)-JQ1 not only downregulated expression of Myc, but also transcription of glycolytic enzymes and inhibited viral replication.Citation160
The Merkel cell virus (MCV) recruits BRD4 to the replication origin complex in the nucleus via direct interaction with the large T antigen, a protein critical for viral replication, which in turn recruits replication factor C to the viral replication sites. Displacement of BRD4 from chromatin via (+)-JQ1 increased freely available BRD4, which enhanced MCV DNA replication.Citation161 Through interaction of BRD4 with the retroviral integrase of murine leukemia virus (MLV), this protein is recruited to the transcriptional start sites of genes. Displacement of BRD4 from chromatin by treatment of cells with BET antagonists (+)-JQ1 and I-BET released BET proteins from the transcriptional start site, thus decreasing MLV integration.Citation162 The viral integrase has been shown to interact with BRD2 and BRD3 as well as BRD4 in vitro via the C-terminal ET domain. The ET domain was found to be sufficient to stimulate integration in vitro and in vivo.Citation163
Protein methyltransferases
Human cytomegalovirus (HCMV) is an omnipresent herpes virus that leads to a life-long persistence. The regulatory protein pUL69 of HCMV acts as a viral RNA export factor. Among the 8 PRMTs (1–8), only PRMT2 and PRMT6 co-localized with pUL69 in the nucleus.Citation164 PRMT6 methylates pUL69 within the functionally important arginine rich R1 box. Although this study did not present the exact role of methylation, it did highlight the possibilities. Mutation studies identified R22, R23, and R25-R28 as being important for RNA export of pUL69. However, R27 and R28 are not required for binding PRMT6 or the RNA export factor UAP56, and are likely the putative sites of methylation. Once methylated, R27 and R28 may act as a hub for methyl-arginine binding proteins.
West Nile virus (WNV) belongs to the Flaviviridae family and has been connected to non- and neuro-invasive diseases. Members of this family feature single-stranded, type I capped, positive-strand RNA, which are limited in their coding capacity. As such, they depend on various host-cell factors. In the case of WNV, AU-rich element binding protein 1 (AUF1) isoform p45 (AUF1-p45) is essential for viral replication. As a further level of modulation, posttranslational modification by PRMT1 to generate AUF1-p45me2a, increases the affinity for WNV (at the RNA termini) and results in increased viral replication.Citation165
SET7 expression was upregulated in hepatitis C infection of a tumor cell line and was found to be overexpressed in sera, PBMC, and liver tissue of patients with HCV.Citation166 An increase in SET7 expression resulted in suppression of IFN-related signaling, specifically IFN-α, via the NF-κB pathway. The methyltransferase activity of SET7 is deemed important because a non-specific methyltransferase inhibitor reversed suppression of IFN signaling and a SET7 mutant lacking methyltransferase activity had no effect on IFN signaling pathway. In a related manner, long, noncoding RNA from CD244 mediates EZH2 recruitment to IFN-γ and TNF-α promoters in active tuberculosis infection.Citation167 Although there is no direct evidence presented for modulation of the H3K27me3 mark after EZH2 docking at these promoters, there is a concomitant reduction in IFN-γ and TNF-α.
Metabolic diseases
Epigenetic proteins are involved in diseases caused by metabolic dysregulation.Citation166,168-180 Cellular metabolism as well as nutritional behavior is known to be linked to epigenetic regulation, and histone modifications are known to be intricately linked to the maintenance of glucose homeostasis and the development of metabolic disease. Defects in metabolic pathways are implicated in diabetes, which is accompanied by inflammatory cytokine expression and changes of NF-κB levels.Citation181
Diabetes tends to enhance susceptibility to other indications, such as heart and kidney disease. SET7 is reported to have multiple roles in diabetes and related diseases.Citation168-172,180 Perhaps one of the underlying mechanisms is due to SET7 methylation of HIF-1α at K32 and HIF-2α at K29.Citation180 These modifications inhibit transcription of HIF-1α/-2α target genes. ShRNA-mediated knockdown and chemical inhibition of SET7 with (R)-PFI-2182 promoted transcription of HIF-1α/-2α target genes and increased glucose uptake.
SET7 is upregulated in subjects with type 2 diabetes relative to controlsCitation171 resulting in increased H3K4me at the promoter of NF-κB p65, which led to a boost in the expression of transcription factors and oxidant inflammatory genes. Knockdown of SET7 in a hyperglycemia model reduced the induction of oxidant and inflammatory pathways. In a related publication on Prep1, a gene exerting major effects on the insulin sensitivity of glucose transport, it was shown that high glucose (HG) exposure induces NF-κB recruitment and histone modification at the Prep1 5′ region.Citation168 HG exposure also resulted in the recruitment of EP300 and SET7 (which was confirmed by an increase in H3K9/14 acetylation and H3K4me/me2) at the Prep1 promoter. The posttranslational modifications were reduced in the presence of chemical inhibitors of NF-κB, thus indicating the leading role that NF-κB recruitment plays in driving the action of epigenetic enzymes at the Prep1 5′ regulatory region.
Pdx1 is a transcription factor that is important to the function of islet β cells; these cells are the main producer of circulating insulin. Conditional knockout of Set7 in islet β cells in a mouse model resulted in similar glucose intolerance as Pdx1-deficient mice.Citation172 Furthermore, the islets isolated from the Set7 knockout mice featured reduced expression of Pdx1 related genes. The authors identified 2 Set7 catalyzed methylation sites on Pdx1 at residues K123 and K131. Residue K131 is more important for supplementing Pdx1 (basal) activity but it does not improve binding to DNA in an in vitro model. As such, the authors suggest that methylation may play a role in post-DNA binding events.Citation172
Two studies in diabetic nephropathy outline roles for SET7. Animal studies show that H2AK119Ub and H2B120Ub play a role in renal fibrosis via regulation of H3K4me2 and H3K9me marks.Citation169 These marks are regulated by controlling the expression of SET7 and SUV39H1. Dsylipidemia is also a contributing factor to diabetic nephropathy. 12/15-lipoxygenase enhances the expression of oxidized lipids such as 12(S)-hydroxyeicosatetraenoic (12(S)-HETE). The latter promotes several processes leading to diabetic nephropathy. 12(S)-HETE increase was linked to increased levels of SET7 protein, which was subsequently translocated to the nucleus and gravitated to the profibrotic gene promoters. Genetic silencing of SET7 reduced 12(S)-HETE-induced gene expression of profibrotic genes.
PRMTs are also involved in glucose dysregulation and some of their roles has been reviewed.Citation183 Since then, Kim et al. defined a role for SIRT1, PRMT1, and PRMT4 in diabetic retinopathy.Citation177 SIRT1, a protein deacetylase, is involved in the onset of metabolic diseases. Its expression decreases under conditions (of oxidative stress) that damage retinal pigment epithelial (RPE) cells while PRMT1 and PRMT4 increase. However, PRMT1, but not PRMT4, induced damage to RPE cells via SIRT1. This highlights an example of the redundancy of PRMT1 and PRMT4 depending on the expression of SIRT1.
A recent study investigated the potential of BET antagonism for the treatment of insulin resistance and diabetes. Pancreatic β cells treated with (+)-JQ1 showed an increase in insulin content as well as secretion from islet cells. Higher concentrations of (+)-JQ1 also decreased intracellular triglyceride stores in INS-1 cells as a result of increased fatty acid oxidation. Interestingly, si-RNA studies revealed that although both antagonism of BRD4 and BRD2 could increase insulin transcription, only knockdown of Brd2 increased fatty acid oxidation.Citation184 These data are in line with the studies in mice where knockdown of Brd2 caused severe obesity with enhanced levels of pro-inflammatory cytokines, but prevented obesity-induced inflammatory responses as well as protected animals from insulin resistance, glucose intolerance, and pancreatic β cell dysfunction.Citation178,179
Hematologic diseases
Sickle-cell disease (SCD) is a group of genetically inherited blood disorders due to aberrant β-globin gene expression. A critical distal regulatory element directing expression within the gene cluster is the locus control region (LCR). The globin genes compete for occupation on the LCR,Citation185 via the LDB1 complex, as part of developmental regulation. Fetal hemoglobin (HbF, α2γ2) is used to treat SCD because it inhibits polymerization of sickle hemoglobin under deoxygenated conditions.Citation186 Silencing of HbF in adults is also mediated by epigenetic changes including DNA methyltransferases,Citation187 HDACs,Citation188 PRMT5,Citation189 and LSD1,Citation190 and it is likely that other epigenetic proteins are involved. Two independent studies reported that UNC0638,Citation130 a chemical probe for G9a and GLP, increased the expression of γ-globin (HBG1/2) genes during erythropoiesis in CD34+ HSPC-derived erythroid precursors, which was corroborated with gene knockdown methods.Citation191,192 They also reported that UNC0638 increased the transcript level of Hbb-εy and, to a lesser extent, Hbb-βh1 in a dose-dependent manner in murine erythroleukemia (MEL) cells commonly used to study globin switching. Renneville et al.Citation192 used CRISPR/Cas9 gene editing in MEL cells to generate clones with bi-allelic inactivation of G9a. These knockout clones featured a measurable increase in Hbb-εy and Hbb-βh1 relative to parental cells and phenocopied treatment with 1 µM UNC0638. (Unfortunately, the corresponding knockout clone of GLP could not be generated.) ChIP-seq showed that UNC0638 reduced the repressive H3K9me2 mark genome wide, at the γ-globin genes and the LCR, while the competing H3K9ac mark noticeably increased at γ-globin genes.Citation191 H3K9me3 was also reduced at the loci of γ-globin repressors BCL11A and MYB; however, H3K9Ac only increased slightly. Globally, 387 genes were upregulated and 252 were downregulated by at least a factor of 2. HBG1 and HBG2 were among the significantly upregulated genes while HBB and HBD were significantly decreased. The major erythroid transcription factors GATA1, KLF1, and NFE2 and the γ-globin repressors BCL11A and MYB were not significantly affected by UNC0638. Finally, a cumulative effect was observed for the combination of UNC0638 and HDAC (entinostat) or DNMT (decitabine) inhibitors.Citation192 However, knockdown of G9a or GLP plus either entinostat or decitabine also had additive effects on the induction of γ-globin gene expression. This indicates that γ-globin expression through either inhibition of EHMT1/2 and HDAC or DNA methyltransferase occurs via different mechanisms.
Probing the biological role of epigenetic proteins
A large body of literature is focused on understanding the therapeutic potential of epigenetic proteins. Earlier studies used gene knockdown and/or knockouts while more recent research combines these genetic approaches with chemical probes. The latter, on their own, have been used to gain deeper insight into the biologic role of their targets and in particular to dissect the function of specific domains in a protein. Here we summarize some of these examples.
SMARCA2/BRM and SMARCA4/BRG1 are mutually exclusive catalytic subunits of the BAF and PBAF complex, respectively. Both proteins have been implicated in a variety of diseases ranging from inflammation to several different cancers.Citation193 SMARCA2/4 proteins contain a bromodomain apart from their catalytic chromatin remodeling ATPase domain. A highly specific BRD antagonist, PFI-3, has been developed and also shown to displace ectopically expressed SMARCA2 and SMARCA4 from chromatin.Citation59,194 However, no anti-proliferative effect could be observed in cell lines from several tumor types tested indicating that the ATPase domain may be more relevant for tumorigenesis,Citation194,195 nor was an effect seen in a series of primary human cells assaying inflammatory cytokine release.Citation196 However, studies using PFI-3 revealed the crucial role of the bromodomain in several differentiation processes.Citation194,196 During embryogenesis, among the earliest cell types are the embryonic stem (ES) cells, which develop into the embryo, as well as the trophectoderm, which gives rise to the embryonic part of the placenta. Treatment of ES cells with PFI-3 resulted in differentiation of ES cells accompanied with an increased H3K27me3 mark at common Brg1 and Stat3 binding sites at promoters and at transcription start sites. Also, trophoblast stem cells showed an increase of differentiation markers as assessed by RNA sequencing. The effect could be shown to be at least in part persistent as demonstrated by washout experiments.Citation194 Contrary, differentiation of myoblast to myotubes was inhibited by PFI-3, as was differentiation of adipocytes with severely decreased lipid accumulation in the adipocytes.Citation196 Also, BRD4 plays a role in early embryogenesis. In a study using quantitative mass spectrometry during ES cells differentiation into neuronal cells, levels of chromatin acetylation were found to dramatically decrease concomitantly with a decrease in expression of BRD4. Treatment of ES cells with (+)-JQ1 led to differentiation of ES cells and changes of the epigenetic landscape with altered histone modifications, the reason of which needs further elucidation.Citation197
Members of the KDM6 family also play crucial roles during early development in both a demethylase-dependent as well as -independent manner. Treatment of mouse differentiating (but not undifferentiated) ESCs with GSK-J4 was accompanied by cell cycle arrest of the embryonic bodies demonstrating that this process is dependent on KDM6A and KDM6B enzymatic activities. DNA damage was identified as the underlying cause for the cell cycle disturbance, which could be replicated at least partially by the combined knockdown of KDM6A and KDM6B.Citation198
A particular case of repressive chromatin occurs during X-chromosome inactivation. One of the 2 female X chromosomes, inactivated in somatic cells, is covered with the repressive H3K27me3 mark. UTX has been implicated in the process and is part of a large complex that also harbors Swi/Snf members. The process of X chromosome inactivation is regulated by 2 long noncoding RNAs: the silencing RNA Xist and the antisense counterpart of it, Tsix. Treatment of female undifferentiated ES cells with GSK-J4 resulted in upregulation of Xist and reduced Tsix expression accompanied by increased H3K27me3 at their respective transcriptional start sites. ChIP experiments identified UTX to be the responsible demethylase in this process. Genes found to be regulated upon treatment could mostly be attributed to UTX inhibition, but some genes, including Xist, Nodal, and HoxC13, which were upregulated and accompanied by increased H3K4me3 levels, may originate from concomitant inhibition of KDM5 demethylases.Citation199 Similarly, in a retinoic acid induced differentiation model of human carcinoma cells, GSK-J4 induced differentiation due to reduction of H3K27me3 at differentiation genes. Although not further investigated all observed changes in gene expression could not be attributed to JMJD3 or UTX inhibition.Citation200
Bone morphogenesis is regulated by a delicate balance of bone formation, which occurs through differentiation of osteoblasts into osteocytes and the deposition of inorganic salts, and the activity of bone-resorbing osteoclasts that differentiate from resident monocytes upon stimulation of specific factors like RANKL, which triggers expression of the key transcription factor for osteoclastogenesis, NFATc. Additionally, the BET antagonist (+)-JQ1 has been shown to inhibit both expression of NFATc and the transcription factor of osteoblast Runx2 suppressing osteoclast differentiation. Knockdown experiments demonstrated that BRD4 prevented osteoclast differentiation but strongly activates osteoblast mineralization activity. These findings in basic biology may have implications for bone-related disorders such as osteoporosis. Indeed, (+)-JQ1 rescued pathologic bone loss in a post-ovariectomy osteoporosis model, increasing the trabecular bone volume and restoring mechanical properties.Citation43
Learning and memory formation requires rapid activation of immediate early genes in response to stimulation. BRD4 is expressed throughout the brain and, as in the periphery, necessary for P-TEFb mediated transcription. Recruitment of BRD4 to target promoters was found to be dependent on CK2-dependent phosphorylation of Brd4 on serine 492 in response to neuronal activity. Blocking BRD4 binding to chromatin by (+)-JQ1 decreased transcription of immediate early genes and downregulated expression of neuronal receptors. Mouse studies further demonstrated that treatment with (+)-JQ1 blocked memory formation. However, a positive effect of decreased seizure susceptibility was observed in models, opening potential new avenues for the treatment of epilepsy.Citation201 Also BRD7 has been shown to have important functions in the brain, although it has a more restricted expression. Knockout mice studies demonstrated that disruption of BRD7 was not toxic to neurons. Still, mice showed cognitive deficiency, accompanied by downregulation of synaptic related proteins like PSD95; however, antagonists have not been used to further elucidate the role of BRD7 bromodomain in the brain.Citation202
Conclusions
In the last 10 years, >50 high-quality probe compounds for epigenetic targets have become available, most of them through the SGC. Usually, the initial application of these probes has been in the oncology area, but, increasingly, links to other indications, in particular inflammatory disorders, are being discovered and the developed probes are used to explore the underlying mechanisms giving rise to these diseases. Not surprisingly the transcription factor NF-κB plays a crucial role by providing a central effector for many epigenetic regulators. The mechanistic findings on the inflammation system facilitated by chemical probes may also be useful for related emerging areas as immuno-oncology. We hope that chemical probes will be tested in the future in more complex cellular systems, such as co-culture systems or patient-derived primary tissues, which will provide better models for human disease. There is a rich literature linking other epigenetic targets like KDMs and KMTs to inflammatory and viral diseases. Probes are now becoming available to confirm the role of these targets in different diseases and the prospect of pharmacological targeting. Given the biologic functions of these epigenetic modulators in normal biology, ranging from embryonic development, cell differentiation processes to memory formation and other functions in the brain, it is however currently not clear if inhibitors against these targets will be suitable for long-term systemic treatment of inflammatory diseases and the development of specific drug targeting systems may be necessary for safety in therapeutic areas outside oncology.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We apologise for the omission of many important research contributions due to space constraints in this review.
Funding
The authors gratefully receive funding from the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer-Ingelheim, Canada 1440 Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust [106169/ZZ14/Z]. 1445 SMK is supported by the DFG-funded Cluster of Excellence Macromolecular Complexes (EXC 115).
References
- Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, et al. The promise and peril of chemical probes. Nat Chem Biol 2015; 11:536-41; PMID:26196764; https://doi.org/10.1038/nchembio.1867
- Brown PJ, Muller S. Open access chemical probes for epigenetic targets. Future Med Chem 2015; 7:1901-17; PMID:26397018; https://doi.org/10.4155/fmc.15.127
- Bunnage ME. Getting pharmaceutical R&D back on target. Nat Chem Biol 2011; 7:335-9; PMID:21587251; https://doi.org/10.1038/nchembio.581
- Frye SV. The art of the chemical probe. Nat Chem Biol 2010; 6:159-61; PMID:20154659; https://doi.org/10.1038/nchembio.296
- Muller S, Brown PJ. Epigenetic chemical probes. Clin Pharmacol Therap 2012; 92:689-93; PMID:23093316; https://doi.org/10.1038/clpt.2012.154
- Bamborough P, Chung CW, Demont EH, Furze RC, Bannister AJ, Che KH, Diallo H, Douault C, Grandi P, Kouzarides T, et al. A chemical probe for the ATAD2 bromodomain. Angewandte Chemie 2016; 55:11382-6; PMID:27530368; https://doi.org/10.1002/anie.201603928
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. Hijacking the E3 Ubiquitin Ligase cereblon to efficiently target BRD4. Chem Biol 2015; 22:755-63; PMID:26051217; https://doi.org/10.1016/j.chembiol.2015.05.009
- Mitchell LH, Drew AE, Ribich SA, Rioux N, Swinger KK, Jacques SL, Lingaraj T, Boriack-Sjodin PA, Waters NJ, Wigle TJ, et al. Aryl pyrazoles as potent inhibitors of arginine Methyltransferases: identification of the first PRMT6 tool compound. Acs Med Chem Lett 2015; 6:655-9; PMID:26101569; https://doi.org/10.1021/acsmedchemlett.5b00071
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015; 348:1376-81; PMID:25999370; https://doi.org/10.1126/science.aab1433
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012; 149:214-31; PMID:22464331; https://doi.org/10.1016/j.cell.2012.02.013
- Flynn EM, Huang OW, Poy F, Oppikofer M, Bellon SF, Tang Y, Cochran AG. A Subset of Human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure 2015; 23:1801-14; PMID:26365797; https://doi.org/10.1016/j.str.2015.08.004
- Di Costanzo A, Del Gaudio N, Migliaccio A, Altucci L. Epigenetic drugs against cancer: an evolving landscape. Arch Toxicol 2014; 88:1651-68; PMID:25085708; https://doi.org/10.1007/s00204-014-1315-6
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Dis 2014; 13:337-56; PMID:24751816; https://doi.org/10.1038/nrd4286
- Wadhwa E, Nicolaides T. Bromodomain Inhibitor Review: Bromodomain and Extra-terminal family protein inhibitors as a potential new therapy in central nervous system tumors. Cureus 2016; 8:e620; PMID:27382528; https://doi.org/10.7759/cureus.620
- Ghoshal A, Yugandhar D, Srivastava AK. BET inhibitors in cancer therapeutics: a patent review. Exp Opin Therap Patents 2016; 26:505-22; PMID:26924192; https://doi.org/10.1517/13543776.2016.1159299
- Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Therap Adv Hematol 2015; 6:128-41; PMID:26137204; https://doi.org/10.1177/2040620715576662
- Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015; 7:487-501; PMID:26077433; https://doi.org/10.2217/epi.14.91
- Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L, Zhang Y, Liu B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015; 6:5501-16; PMID:25849938; https://doi.org/10.18632/oncotarget.3551
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 2014; 54:728-36; PMID:24905006; https://doi.org/10.1016/j.molcel.2014.05.016
- Lochrin SE, Price DK, Figg WD. BET bromodomain inhibitors–a novel epigenetic approach in castration-resistant prostate cancer. Cancer Biol Ther 2014; 15:1583-5; PMID:25535892; https://doi.org/10.4161/15384047.2014.962297
- Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG. Targeting bet bromodomain proteins in solid tumors. Oncotarget 2016; 7(33):53997-4009; PMID:27283767; https://doi.org/10.18632/oncotarget.9804
- Hamamoto R, Nakamura Y. Dysregulation of protein methyltransferases in human cancer: An emerging target class for anticancer therapy. Cancer Sci 2016; 107:377-84; PMID:26751963; https://doi.org/10.1111/cas.12884
- Hirsch CL, Wrana JL, Dent SY. KATapulting toward pluripotency and cancer. J Mol Biol 2016; PMID:27720985; https://doi.org/10.1016/j.jmb.2016.09.023
- Arcipowski KM, Martinez CA, Ntziachristos P. Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX. Curr Opin Genet Dev 2016; 36:59-67; PMID:27151432; https://doi.org/10.1016/j.gde.2016.03.010
- Muller S. Targeting the Bromome: are we there yet? Future Med Chem 2016; 8:1529-32; PMID:27578167; https://doi.org/10.4155/fmc-2016-0136
- Diamant G, Dikstein R. Transcriptional control by NF-kappaB: elongation in focus. Biochim Et Biophys Acta 2013; 1829:937-45; PMID:23624258; https://doi.org/10.1016/j.bbagrm.2013.04.007
- Choi CS, Hong SH, Sim S, Cho KS, Kim JW, Yang SM, Jeon SJ, You JS, Shin CY. The epigenetic reader BRD2 as a specific modulator of PAI-1 expression in lipopolysaccharide-stimulated mouse primary astrocytes. Neurochem Res 2015; 40:2211-9; PMID:26349765; https://doi.org/10.1007/s11064-015-1710-2
- Alsarraj J, Faraji F, Geiger TR, Mattaini KR, Williams M, Wu J, Ha NH, Merlino T, Walker RC, Bosley AD, et al. BRD4 short isoform interacts with RRP1B, SIPA1 and components of the LINC complex at the inner face of the nuclear membrane. PloS One 2013; 8:e80746; PMID:24260471; https://doi.org/10.1371/journal.pone.0080746
- Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 2013; 49:843-57; PMID:23317504; https://doi.org/10.1016/j.molcel.2012.12.006
- Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, Nair S, Chen LF. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene 2014; 33:2395-404; PMID:23686307; https://doi.org/10.1038/onc.2013.179
- Suarez-Alvarez B, Morgado-Pascual JL, Rayego-Mateos S, Rodriguez RM, Rodrigues-Diez R, Cannata-Ortiz P, Sanz AB, Egido J, Tharaux PL, Ortiz A, et al. Inhibition of bromodomain and extraterminal domain family proteins ameliorates experimental renal damage. J Am Soc Nephrol 2016; PMID:27436852; https://doi.org/10.1681/ASN.2015080910
- Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 2014; 56:219-31; PMID:25263595; https://doi.org/10.1016/j.molcel.2014.08.024
- Duan Q, Mao X, Xiao Y, Liu Z, Wang Y, Zhou H, Zhou Z, Cai J, Xia K, Zhu Q, et al. Super enhancers at the miR-146a and miR-155 genes contribute to self-regulation of inflammation. Biochim Et Biophy Acta 2016; 1859:564-71; PMID:26855180; https://doi.org/10.1016/j.bbagrm.2016.02.004
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005; 19:535-45; PMID:16109377; https://doi.org/10.1016/j.molcel.2005.06.029
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013; 153:320-34; PMID:23582323; https://doi.org/10.1016/j.cell.2013.03.036
- Yoshida H, Bansal K, Schaefer U, Chapman T, Rioja I, Proekt I, Anderson MS, Prinjha RK, Tarakhovsky A, Benoist C, et al. Brd4 bridges the transcriptional regulators, Aire and P-TEFb, to promote elongation of peripheral-tissue antigen transcripts in thymic stromal cells. Proc Nat Acad Sci U S A 2015; 112:E4448-57; PMID:26216992; https://doi.org/10.1073/pnas.1512081112
- Ghari F, Quirke AM, Munro S, Kawalkowska J, Picaud S, McGouran J, Subramanian V, Muth A, Williams R, Kessler B, et al. Citrullination-acetylation interplay guides E2F-1 activity during the inflammatory response. Sci Adv 2016; 2:e1501257; PMID:26989780
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010; 468:1119-23; PMID:21068722; https://doi.org/10.1038/nature09589
- Ferri E, Petosa C, McKenna CE. Bromodomains: Structure, function and pharmacology of inhibition. Biochem Pharmacol 2016; 106:1-18; PMID:26707800; https://doi.org/10.1016/j.bcp.2015.12.005
- Zhang QG, Qian J, Zhu YC. Targeting bromodomain-containing protein 4 (BRD4) benefits rheumatoid arthritis. Immunol Lett 2015; 166:103-8; PMID:26093279; https://doi.org/10.1016/j.imlet.2015.05.016
- Xiao Y, Liang L, Huang M, Qiu Q, Zeng S, Shi M, Zou Y, Ye Y, Yang X, Xu H. Bromodomain and extra-terminal domain bromodomain inhibition prevents synovial inflammation via blocking IkappaB kinase-dependent NF-kappaB activation in rheumatoid fibroblast-like synoviocytes. Rheumatology 2016; 55:173-84; PMID:26324948; https://doi.org/10.1093/rheumatology/kev312
- Meng S, Zhang L, Tang Y, Tu Q, Zheng L, Yu L, Murray D, Cheng J, Kim SH, Zhou X, et al. BET inhibitor JQ1 blocks inflammation and bone destruction. J Dental Res 2014; 93:657-62; PMID:24799421; https://doi.org/10.1177/0022034514534261
- Baud'huin M, Lamoureux F, Jacques C, Rodriguez Calleja L, Quillard T, Charrier C, Amiaud J, Berreur M, Brounais-LeRoyer B, Owen R, et al. Inhibition of BET proteins and epigenetic signaling as a potential treatment for osteoporosis. Bone 2016; 94:10-21; PMID:27669656; https://doi.org/10.1016/j.bone.2016.09.020
- Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, Molesworth AM, Smithers N, Lee K, Witherington J, et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci USA 2012; 109:14532-7; PMID:22912406; https://doi.org/10.1073/pnas.1212264109
- Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM, Lora JM. BET bromodomain inhibition suppresses TH17-mediated pathology. J Exp Med 2013; 210:2181-90; PMID:24101376; https://doi.org/10.1084/jem.20130376
- Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, Saso K, Butler MO, Arrowsmith CH, Hirano N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest 2016; 126:3479-94; PMID:27548527; https://doi.org/10.1172/JCI86437
- Toniolo PA, Liu S, Yeh JE, Moraes-Vieira PM, Walker SR, Vafaizadeh V, Barbuto JA, Frank DA. Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation. J Immunol 2015; 194:3180-90; PMID:25725100; https://doi.org/10.4049/jimmunol.1401635
- Sun Y, Wang Y, Toubai T, Oravecz-Wilson K, Liu C, Mathewson N, Wu J, Rossi C, Cummings E, Wu D, et al. BET bromodomain inhibition suppresses graft-versus-host disease after allogeneic bone marrow transplantation in mice. Blood 2015; 125:2724-8; PMID:25778533; https://doi.org/10.1182/blood-2014-08-598037
- Clifford RL, Patel JK, John AE, Tatler AL, Mazengarb L, Brightling CE, Knox AJ. CXCL8 histone H3 acetylation is dysfunctional in airway smooth muscle in asthma: regulation by BET. Am J Physiol Lung Cell Mol Physiol 2015; 308:L962-72; PMID:25713319; https://doi.org/10.1152/ajplung.00021.2015
- Kokkola T, Suuronen T, Pesonen M, Filippakopoulos P, Salminen A, Jarho EM, Lahtela-Kakkonen M. BET inhibition upregulates SIRT1 and Alleviates inflammatory responses. Chembioch Eur J Chem Biol 2015; 16:1997-2001; PMID:26212199; https://doi.org/10.1002/cbic.201500272
- Tang X, Peng R, Phillips JE, Deguzman J, Ren Y, Apparsundaram S, Luo Q, Bauer CM, Fuentes ME, DeMartino JA, et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am J Pathol 2013; 183:470-79; PMID:23759512; https://doi.org/10.1016/j.ajpath.2013.04.020
- Nadeem A, Al-Harbi NO, Al-Harbi MM, El-Sherbeeny AM, Ahmad SF, Siddiqui N, Ansari MA, Zoheir KM, Attia SM, Al-Hosaini KA, et al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation. Pharmacological Research 2015; 99:248-57; PMID:26149470; https://doi.org/10.1016/j.phrs.2015.06.001
- Chen K, Campfield BT, Wenzel SE, McAleer JP, Kreindler JL, Kurland G, Gopal R, Wang T, Chen W, Eddens T, et al. Antiinflammatory effects of bromodomain and extraterminal domain inhibition in cystic fibrosis lung inflammation. JCI insight 2016; 1(11)e87168; PMID:27517095; https://doi.org/10.1172/jci.insight.87168
- Peeters JG, Vervoort SJ, Tan SC, Mijnheer G, de Roock S, Vastert SJ, Nieuwenhuis EE, van Wijk F, Prakken BJ, Creyghton MP, et al. Inhibition of super-enhancer activity in autoinflammatory site-derived T Cells reduces disease-associated gene expression. Cell Rep 2015; 12:1986-96; PMID:26387944; https://doi.org/10.1016/j.celrep.2015.08.046
- Wei S, Sun Y, Sha H. Therapeutic targeting of BET protein BRD4 delays murine lupus. Inter Immunopharmacol 2015; 29:314-9; PMID:26590112; https://doi.org/10.1016/j.intimp.2015.10.036
- Mahdi H, Fisher BA, Källberg H, Plant D, Malmström V, Rönnelid J, Charles P, Ding B, Alfredsson L, Padyukov L, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Gen 2009; 41:1319-24; PMID:19898480; https://doi.org/10.1038/ng.480
- Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol 2013; 190:3670-8; PMID:23420887; https://doi.org/10.4049/jimmunol.1202838
- Chen J, Wang Z, Hu X, Chen R, Romero-Gallo J, Peek RM, Jr, Chen LF. BET inhibition attenuates helicobacter pylori-induced inflammatory response by suppressing inflammatory gene transcription and enhancer activation. J Immunol 2016; 196:4132-42; PMID:27084101; https://doi.org/10.4049/jimmunol.1502261
- Philpott M, Yang J, Tumber T, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Keates T, Felletar I, Ciulli A, et al. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol Biosys 2011; 7:2899-908; PMID:21804994; https://doi.org/10.1039/c1mb05099k
- Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA, Martinez FO, Al-Mossawi MH, de Wit J, Vecellio M, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Nat Acad Sci U S A 2015; 112:10768-73; PMID:26261308; https://doi.org/10.1073/pnas.1501956112
- Ghosh S, Taylor A, Chin M, Huang HR, Conery AR, Mertz JA, Salmeron A, Dakle PJ, Mele D, Cote A, et al. Regulatory T cell modulation by CBP/EP300 bromodomain inhibition. J Biol Chem 2016; 291:13014-27; PMID:27056325; https://doi.org/10.1074/jbc.M115.708560
- Humphreys PG, Bamborough P, Chung CW, Craggs PD, Gordon L, Grandi P, Hayhow TG, Hussain J, Jones KL, Lindon M, et al. Discovery of a potent, cell penetrant and selective p300/CBP-associated factor (PCAF)/General Control Non-Derepressible 5 (GCN5) Bromodomain chemical probe. J Med Chem 2016; 60(2):695-709; PMID:28002667; https://doi.org/10.1021/acs.jmedchem.6b01566
- Moustakim M, Clark PG, Trulli L, Fuentes de Arriba AL, Ehebauer MT, Chaikuad A, Murphy EJ, Mendez-Johnson J, Daniels D, Hou CD, et al. Discovery of a PCAF Bromodomain chemical probe. Angewandte Chemie 2017; 56(3):827-31; PMID:27966810; https://doi.org/10.1002/anie.201610816
- Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, Chung CW, Dittmann A, Drewes G, Drewry DH, et al. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J Med Chem 2016; 59:1425-39; PMID:25856009; https://doi.org/10.1021/acs.jmedchem.5b00256
- Zhao R, Liu Y, Wang H, Yang J, Niu W, Fan S, Xiong W, Ma J, Li X, Phillips JB, et al. BRD7 plays an anti-inflammatory role during early acute inflammation by inhibiting activation of the NF-small ka, CyrillicB signaling pathway. Cell Mol Immunol 2016; 13:1-12; PMID:27374794; https://doi.org/10.1038/cmi.2016.31
- Shen Y, Szewczyk MM, Eram MS, Smil D, Kaniskan HÜ, Ferreira de Freitas R, Senisterra G, Li F, Schapira M, Brown PJ, et al. Discovery of a potent, selective, and cell-active dual inhibitor of Protein Arginine Methyltransferase 4 and protein arginine Methyltransferase 6. J Med Chem 2016; 59:9124-39; PMID:27584694; https://doi.org/10.1021/acs.jmedchem.6b01033
- Dimitrova E, Turberfield AH, Klose RJ. Histone demethylases in chromatin biology and beyond. EMBO Rep 2015; 16:1620-39; PMID:26564907; https://doi.org/10.15252/embr.201541113
- Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012; 488:404-8; PMID:22842901; https://doi.org/10.1038/nature11262
- Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, Bonelli M, Sun HW, Jankovic D, Kanno Y, et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci Rep 2015; 5:10643; PMID:26090605; https://doi.org/10.1038/srep10643
- Liu Z, Cao W, Xu L, Chen X, Zhan Y, Yang Q, Liu S, Chen P, Jiang Y, Sun X, et al. The histone H3 lysine-27 demethylase Jmjd3 plays a critical role in specific regulation of Th17 cell differentiation. J Mol Cell Biol 2015; 7:505-16; PMID:25840993; https://doi.org/10.1093/jmcb/mjv022
- Tennant BR, Hurley P, Dhillon J, Gill A, Whiting C, Hoffman BG. The TrxG complex mediates cytokine induced De Novo enhancer formation in Islets. PloS One 2015; 10:e0141470; PMID:; PMID:26505193; https://doi.org/10.1371/journal.pone.0141470
- Donas C, Carrasco M, Fritz M, Prado C, Tejón G, Osorio-Barrios F, Manríquez V, Reyes P, Pacheco R, Bono MR, et al. The histone demethylase inhibitor GSK-J4 limits inflammation through the induction of a tolerogenic phenotype on DCs. J Autoimmun 2016; 75:105-17; PMID:27528513; https://doi.org/10.1016/j.jaut.2016.07.011
- Yapp C, Carr AJ, Price A, Oppermann U, Snelling SJ. H3K27me3 demethylases regulate in vitro chondrogenesis and chondrocyte activity in osteoarthritis. Arthritis Res Ther 2016; 18:158; PMID:27388528; https://doi.org/10.1186/s13075-016-1053-7
- Na J, Lee K, Na W, Shin JY, Lee MJ, Yune TY, Lee HK, Jung HS, Kim WS, Ju BG. Histone H3K27 demethylase JMJD3 in cooperation with NF-kappaB regulates keratinocyte wound healing. J Invest Dermatol 2016; 136:847-58; PMID:26802933; https://doi.org/10.1016/j.jid.2015.11.029
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013; 122:1017-25; PMID:23801631; https://doi.org/10.1182/blood-2013-04-497644
- Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al. Kruidenier et al. reply. Nature 2014; 514:E2; PMID:25279927; https://doi.org/10.1038/nature13689
- McAllister TE, England KS, Hopkinson RJ, Brennan PE, Kawamura A, Schofield CJ. Recent progress in Histone Demethylase Inhibitors. J Med Chem 2016; 59:1308-29; PMID:26710088; https://doi.org/10.1021/acs.jmedchem.5b01758
- Gantier MP, Stunden HJ, McCoy CE, Behlke MA, Wang D, Kaparakis-Liaskos M, Sarvestani ST, Yang YH, Xu D, Corr SC, et al. A miR-19 regulon that controls NF-kappaB signaling. Nucl Acids Res 2012; 40:8048-58; PMID:22684508; https://doi.org/10.1093/nar/gks521
- Erdogan O, Xie L, Wang L, Wu B, Kong Q, Wan Y, Chen X. Proteomic dissection of LPS-inducible, PHF8-dependent secretome reveals novel roles of PHF8 in TLR4-induced acute inflammation and T cell proliferation. Sci Rep 2016; 6:24833; PMID:27112199; https://doi.org/10.1038/srep24833
- Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol 2015; 11:189; PMID:25622091; https://doi.org/10.1038/nchembio.1735
- Alam H, Gu B, Lee MG. Histone methylation modifiers in cellular signaling pathways. Cell Mol Life Sci 2015; 72:4577-92; PMID:26305020; https://doi.org/10.1007/s00018-015-2023-y
- Lu T, Stark GR. Using sequential immunoprecipitation and mass spectrometry to identify methylation of NF-kappaB. Methods Mol Biol 2015; 1280:383-93; PMID:25736762; https://doi.org/10.1007/978-1-4939-2422-6_23
- Kim JH, Yoo BC, Yang WS, Kim E, Hong S, Cho JY. The role of protein arginine methyltransferases in inflammatory responses. Mediators Inflamm 2016; 2016:4028353; PMID:27041824; https://doi.org/10.1155/2016/4028353
- Li Y, Reddy MA, Miao F, Shanmugam N, Yee JK, Hawkins D, Ren B, Natarajan R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem 2008; 283:26771-81; PMID:18650421; https://doi.org/10.1074/jbc.M802800200
- Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem 2009; 284:27857-65; PMID:19690169; https://doi.org/10.1074/jbc.M109.000950
- Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, Gudkov AV, Stark GR. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci U S A 2010; 107:46-51; PMID:20080798; https://doi.org/10.1073/pnas.0912493107
- Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, Espejo A, Zee BM, Liu CL, Tangsombatvisit S, et al. Lysine methylation of the NF-kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-kappaB signaling. Nat Immunol 2011; 12:29-36; PMID:21131967; https://doi.org/10.1038/ni.1968
- Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell 2012; 48:28-38; PMID:22921934; https://doi.org/10.1016/j.molcel.2012.07.020
- Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, Ghosh G, Stark GR, Lu T. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci U S A 2013; 110:13516-21; PMID:23904475; https://doi.org/10.1073/pnas.1311784110
- Harris DP, Chandrasekharan UM, Bandyopadhyay S, Willard B, DiCorleto PE. PRMT5-Mediated Methylation of NF-kappaB p65 at Arg174 Is Required for Endothelial CXCL11 Gene Induction in Response to TNF-alpha and IFN-gamma Costimulation. PloS One 2016; 11:e0148905; PMID:26901772; https://doi.org/10.1371/journal.pone.0148905
- Fan Z, Kong X, Xia J, Wu X, Li H, Xu H, Fang M, Xu Y. The arginine methyltransferase PRMT5 regulates CIITA-dependent MHC II transcription. Biochim Et Biophys Acta 2016; 1859:687-96; PMID:26972221; https://doi.org/10.1016/j.bbagrm.2016.03.004
- Sun Q, Yang X, Zhong B, Jiao F, Li C, Li D, Lan X, Sun J, Lu S. Upregulated protein arginine methyltransferase 1 by IL-4 increases eotaxin-1 expression in airway epithelial cells and participates in antigen-induced pulmonary inflammation in rats. J Immunol 2012; 188:3506-12; PMID:22387551; https://doi.org/10.4049/jimmunol.1102635
- Park MJ, Kim DI, Lim SK, Choi JH, Kim JC, Yoon KC, Lee JB, Lee JH, Han HJ, Choi IP, et al. Thioredoxin-interacting protein mediates hepatic lipogenesis and inflammation via PRMT1 and PGC-1alpha regulation in vitro and in vivo. J Hepatol 2014; 61:1151-7; PMID:25003952; https://doi.org/10.1016/j.jhep.2014.06.032
- Sun Q, Liu L, Roth M, Tian J, He Q, Zhong B, Bao R, Lan X, Jiang C, Sun J, et al. PRMT1 upregulated by epithelial proinflammatory cytokines participates in COX2 expression in fibroblasts and chronic antigen-induced pulmonary inflammation. J Immunol 2015; 195:298-306; PMID:26026059; https://doi.org/10.4049/jimmunol.1402465
- Liu L, Sun Q, Bao R, Roth M, Zhong B, Lan X, Tian J, He Q, Li D, Sun J, et al. Specific regulation of PRMT1 expression by PIAS1 and RKIP in BEAS-2B epithelia cells and HFL-1 fibroblasts in lung inflammation. Sc Rep 2016; 6:21810; PMID:26911452; https://doi.org/10.1038/srep21810
- Reintjes A, Fuchs JE, Kremser L, Lindner HH, Liedl KR, Huber LA, Valovka T. Asymmetric arginine dimethylation of RelA provides a repressive mark to modulate TNFalpha/NF-kappaB response. Proc Natl Acad Sci U S A 2016; 113:4326-31; PMID:27051065; https://doi.org/10.1073/pnas.1522372113
- Sun Q, Liu L, Mandal J, Molino A, Stolz D, Tamm M, Lu S, Roth M. PDGF-BB induces PRMT1 expression through ERK1/2 dependent STAT1 activation and regulates remodeling in primary human lung fibroblasts. Cell Signal 2016; 28:307-15; PMID:26795953; https://doi.org/10.1016/j.cellsig.2016.01.004
- Di Lorenzo A, Yang Y, Macaluso M, Bedford MT. A gain-of-function mouse model identifies PRMT6 as a NF-kappaB coactivator. Nucleic Acid Res 2014; 42:8297-309; PMID:24939901; https://doi.org/10.1093/nar/gku530
- Xu G, Liu G, Xiong S, Liu H, Chen X, Zheng B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J Biol Chem 2015; 290:5414-23; PMID:25583990; https://doi.org/10.1074/jbc.M114.610345
- Barroso M, Kao D, Blom HJ, Tavares de Almeida I, Castro R, Loscalzo J, Handy DE. S-adenosylhomocysteine induces inflammation through NFkB: A possible role for EZH2 in endothelial cell activation. Biochim Et Biophy Acta 2016; 1862:82-92; PMID:26506125; https://doi.org/10.1016/j.bbadis.2015.10.019
- Hachiya R, Shiihashi T, Shirakawa I, Iwasaki Y, Matsumura Y, Oishi Y, Nakayama Y, Miyamoto Y, Manabe I, Ochi K, et al. The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci Rep 2016; 6:28845; PMID:27349785; https://doi.org/10.1038/srep28845
- Comstedt P, Storgaard M, Lassen AT. The Systemic Inflammatory Response Syndrome (SIRS) in acutely hospitalised medical patients: a cohort study. Scand J Trauma Resus Emerg Med 2009; 17:67; PMID:20035633; https://doi.org/10.1186/1757-7241-17-67
- Eram MS, Shen Y, Szewczyk MM, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HÜ, Speed BA, dela Seña C, et al. A potent, selective, and cell-active inhibitor of human Type I protein arginine methyltransferases. ACS Chem Biol 2016; 11:772-81; PMID:26598975; https://doi.org/10.1021/acschembio.5b00839
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001; 413:739-43; PMID:11607033; https://doi.org/10.1038/35099568
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Gen 2011; 43:595-600; PMID:21532572; https://doi.org/10.1038/ng.830
- Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, Melberg A, Cornelio F, Urban AE, Pizza F, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Gen 2012; 21:2205-10; PMID:22328086; https://doi.org/10.1093/hmg/dds035
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015; 16:358-72; PMID:25991443; https://doi.org/10.1038/nrn3880
- Dong X, Tsuji J, Labadorf A, Roussos P, Chen JF, Myers RH, Akbarian S, Weng Z. The role of H3K4me3 in transcriptional regulation is altered in Huntington's disease. PloS One 2015; 10:e0144398; PMID:26636336; https://doi.org/10.1371/journal.pone.0144398
- Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine Methyltransferase EZH2. ACS Med Chem Lett 2012; 3:1091-6; PMID:24900432; https://doi.org/10.1021/ml3003346
- Tripathy MK, McManamy ME, Burch BD, Archin NM, Margolis DM. H3K27 demethylation at the proviral promoter sensitizes latent HIV to the effects of vorinostat in Ex vivo cultures of resting CD4+ T Cells. J Virol 2015; 89:8392-405; PMID:26041287; https://doi.org/10.1128/JVI.00572-15
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012; 8:890-6; PMID:23023262; https://doi.org/10.1038/nchembio.1084
- Scaramuzzino C, Casci I, Parodi S, Lievens PM, Polanco MJ, Milioto C, Chivet M, Monaghan J, Mishra A, Badders N, et al. Protein arginine methyltransferase 6 enhances polyglutamine-expanded androgen receptor function and toxicity in spinal and bulbar muscular atrophy. Neuron 2015; 85:88-100; PMID:25569348; https://doi.org/10.1016/j.neuron.2014.12.031
- Hossein-Nezhad A, Fatemi RP, Ahmad R, Peskind ER, Zabetian CP, Hu SC, Shi M, Wahlestedt C, Zhang J, Faghihi MA. Transcriptomic profiling of extracellular RNAs present in cerebrospinal fluid identifies differentially expressed transcripts in Parkinson's disease. J Parkinson's Dis 2016; 6:109-17; PMID:26889637; https://doi.org/10.3233/JPD-150737
- Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol 2016; 16:469-84; PMID:27346803; https://doi.org/10.1038/nri.2016.62
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 2013; 5:206ra138; PMID:24107777; https://doi.org/10.1126/scitranslmed.300676
- Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 2008; 8:663-74; PMID:18670423; https://doi.org/10.1038/nri2359
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008; 454:455-62; PMID:18650916; https://doi.org/10.1038/nature07203
- Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Communicat 2012; 3:1288; PMID:23250418; https://doi.org/10.1038/ncomms2304
- Roidl D, Hellbach N, Bovio PP, Villarreal A, Heidrich S, Nestel S, Grüning BA, Boenisch U, Vogel T. DOT1L Activity promotes proliferation and protects cortical neural stem cells from activation of ATF4-DDIT3-Mediated ER stress In vitro. Stem Cells 2016; 34:233-45; PMID:26299268; https://doi.org/10.1002/stem.2187
- Suarez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Göbl C, Resch M, et al. Monomethylated and unmethylated FUS exhibit increased binding to Transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathologica 2016; 131:587-604; PMID:26895297; https://doi.org/10.1007/s00401-016-1544-2
- Tibshirani M, Tradewell ML, Mattina KR, Minotti S, Yang W, Zhou H, Strong MJ, Hayward LJ, Durham HD. Cytoplasmic sequestration of FUS/TLS associated with ALS alters histone marks through loss of nuclear protein arginine methyltransferase 1. Hum Mol Gen 2015; 24:773-86; PMID:25274782; https://doi.org/10.1093/hmg/ddu494
- Kim HJ, Jeong MH, Kim KR, Jung CY, Lee SY, Kim H, Koh J, Vuong TA, Jung S, Yang H, et al. Protein arginine methylation facilitates KCNQ channel-PIP2 interaction leading to seizure suppression. eLife 2016; 5:e17159; PMID:27466704; https://doi.org/10.7554/eLife.17159
- Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trend Immunol 2006; 27:405-12; PMID:16870508; https://doi.org/10.1016/j.it.2006.07.007
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007; 448:427-34; PMID:17653185; https://doi.org/10.1038/nature06005
- Antignano F, Burrows K, Hughes MR, Han JM, Kron KJ, Penrod NM, Oudhoff MJ, Wang SK, Min PH, Gold MJ, et al. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J Clin Invest 2014; 124:1945-55; PMID:24667637; https://doi.org/10.1172/JCI69592
- Nakanishi Y, Reina-Campos M, Nakanishi N, Llado V, Elmen L, Peterson S, Campos A, De SK, Leitges M, Ikeuchi H, et al. Control of Paneth cell fate, intestinal inflammation, and tumorigenesis by PKClambda/iota. Cell Reports 2016; 16:3297-310; PMID:27653691; https://doi.org/10.1016/j.celrep.2016.08.054
- Lehnertz B, Northrop JP, Antignano F, Burrows K, Hadidi S, Mullaly SC, Rossi FM, Zaph C. Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J Exp Med 2010; 207:915-22; PMID:20421388; https://doi.org/10.1084/jem.20100363
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013; 153:178-92; PMID:23523135; https://doi.org/10.1016/j.cell.2013.02.028
- Montes de Oca R, Andreassen PR, Wilson KL. Barrier-to-Autointegration Factor influences specific histone modifications. Nucleus 2011; 2:580-90; PMID:22127260; https://doi.org/10.4161/nucl.2.6.17960
- Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol 2011; 7:566-74; PMID:21743462; https://doi.org/10.1038/nchembio.599
- Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 2007; 25:473-81; PMID:17289593; https://doi.org/10.1016/j.molcel.2007.01.017
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nat 2008; 456:259-63; PMID:18849966; https://doi.org/10.1038/nature07416
- Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem 2011; 286:28511-9; PMID:21659531; https://doi.org/10.1074/jbc.M111.240515
- Koppens MA, Bounova G, Gargiulo G, Tanger E, Janssen H, Cornelissen-Steijger P, Blom M, Song JY, Wessels LF, van Lohuizen M. Deletion of polycomb repressive Complex 2 from mouse intestine causes loss of stem cells. Gastroenterol 2016; 151:684-97 e612; PMID:27342214; https://doi.org/10.1053/j.gastro.2016.06.020
- Taube R, Peterlin M. Lost in transcription: molecular mechanisms that control HIV latency. Viruses 2013; 5:902-27; PMID:23518577; https://doi.org/10.3390/v5030902
- Polizzotto MN, Chen G, Tressler RL, Godfrey C. Leveraging Cancer Therapeutics for the HIV Cure Agenda: current status and future directions. Drugs 2015; 75:1447-59; PMID:26224205; https://doi.org/10.1007/s40265-015-0426-6
- Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukocyte Biol 2012; 92:1147-54; PMID:22802445; https://doi.org/10.1189/jlb.0312165
- Jiang G, Mendes EA, Kaiser P, Sankaran-Walters S, Tang Y, Weber MG, Melcher GP, Thompson GR 3rd, Tanuri A, Pianowski LF, et al. Reactivation of HIV latency by a newly modified Ingenol derivative via protein kinase Cdelta-NF-kappaB signaling. Aids 2014; 28:1555-66; PMID:24804860; https://doi.org/10.1097/QAD.0000000000000289
- Jiang G, Mendes EA, Kaiser P, Wong DP, Tang Y, Cai I, Fenton A, Melcher GP, Hildreth JE, Thompson GR, et al. Synergistic reactivation of latent HIV expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB signaling in combination with JQ1 Induced p-TEFb activation. PLoS Pathogens 2015; 11:e1005066; PMID:26225771; https://doi.org/10.1371/journal.ppat.1005066
- Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, Delacourt N, Melard A, Kabeya K, Vanhulle C, et al. An In-Depth comparison of latency-reversing agent combinations in various in Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathogens 2015; 11:e1005063; PMID:26225566; https://doi.org/10.1371/journal.ppat.1005063
- Lu P, Qu X, Shen Y, Jiang Z, Wang P, Zeng H, Ji H, Deng J, Yang X, Li X, et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci Rep 2016; 6:24100; PMID:27067814; https://doi.org/10.1038/srep24100
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013; 12:452-62; PMID:23255218; https://doi.org/10.4161/cc.23309
- Zhang HS, Du GY, Liu Y, Zhang ZG, Zhou Z, Li H, Dai KQ, Yu XY, Gou XM. UTX-1 regulates Tat-induced HIV-1 transactivation via changing the methylated status of histone H3. Inter J Biochem Cell Biol 2016; 80:51-56; PMID:27671333; https://doi.org/10.1016/j.biocel.2016.09.016
- Messer HG, Jacobs D, Dhummakupt A, Bloom DC. Inhibition of H3K27me3-specific histone demethylases JMJD3 and UTX blocks reactivation of herpes simplex virus 1 in trigeminal ganglion neurons. J Virol 2015; 89:3417-20; PMID:25552720; https://doi.org/10.1128/JVI.03052-14
- Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem 2010; 285:16538-45; PMID:20335163; https://doi.org/10.1074/jbc.M110.103531
- Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J Virol 2011; 85:9078-89; PMID:21715480; https://doi.org/10.1128/JVI.00836-11
- Matsuda Y, Kobayashi-Ishihara M, Fujikawa D, Ishida T, Watanabe T, Yamagishi M. Epigenetic heterogeneity in HIV-1 latency establishment. Sci Rep 2015; 5:7701; PMID:25572573; https://doi.org/10.1038/srep07701
- Fulcher AJ, Jans DA. The HIV-1 Tat transactivator protein: a therapeutic target? IUBMB Life 2003; 55:669-80; PMID:14769003; https://doi.org/10.1080/15216540310001643440
- Mizutani T, Ishizaka A, Tomizawa M, Okazaki T, Yamamichi N, Kawana-Tachikawa A, Iwamoto A, Iba H. Loss of the Brm-type SWI/SNF chromatin remodeling complex is a strong barrier to the Tat-independent transcriptional elongation of human immunodeficiency virus type 1 transcripts. J Virol 2009; 83:11569-80; PMID:19726504; https://doi.org/10.1128/JVI.00742-09
- Sivakumaran H, van der Horst A, Fulcher AJ, Apolloni A, Lin MH, Jans DA, Harrich D. Arginine methylation increases the stability of human immunodeficiency virus type 1 Tat. J Virol 2009; 83:11694-703; PMID:19726520; https://doi.org/10.1128/JVI.00499-09
- Xie B, Invernizzi CF, Richard S, Wainberg MA. Arginine methylation of the human immunodeficiency virus type 1 Tat protein by PRMT6 negatively affects Tat Interactions with both cyclin T1 and the Tat transactivation region. J Virol 2007; 81:4226-34; PMID:17267505; https://doi.org/10.1128/JVI.01888-06
- Sivakumaran H, Lin MH, Apolloni A, Cutillas V, Jin H, Li D, Wei T, Harrich D. Overexpression of PRMT6 does not suppress HIV-1 Tat transactivation in cells naturally lacking PRMT6. Virol J 2013; 10:207; PMID:23800116; https://doi.org/10.1186/1743-422X-10-207
- Church ME, Estrada M, Leutenegger CM, Dela Cruz FN, Pesavento PA, Woolard KD. BRD4 is associated with Raccoon Polyomavirus (RacPyV) genome and mediates viral gene transcription and maintenance of a stem cell state in neuroglial tumor cells. J Gen Virol 2016; 97:2939-48; PMID:27600312; https://doi.org/10.1099/jgv.0.000594
- Wollebo HS, Bellizzi A, Cossari DH, Salkind J, Safak M, White MK. The Brd4 acetyllysine-binding protein is involved in activation of polyomavirus JC. J Neurovirol 2016; 22(5):615-25; PMID:27007123; https://doi.org/10.1007/s13365-016-0435-6
- Zhou H, Schmidt SC, Jiang S, Willox B, Bernhardt K, Liang J, Johannsen EC, Kharchenko P, Gewurz BE, Kieff E, et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microb 2015; 17:205-16; PMID:25639793; https://doi.org/10.1016/j.chom.2014.12.013
- Palermo RD, Webb HM, West MJ. RNA polymerase II stalling promotes nucleosome occlusion and pTEFb recruitment to drive immortalization by Epstein-Barr virus. PLoS Pathogens 2011; 7:e1002334; PMID:22046134; https://doi.org/10.1371/journal.ppat.1002334
- Chang SW, Liu WC, Liao KY, Tsao YP, Hsu PH, Chen SL. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PloS One 2014; 9:e110882; PMID:25340539; https://doi.org/10.1371/journal.pone.0110882
- Helfer CM, Yan J, You J. The cellular bromodomain protein Brd4 has multiple functions in E2-mediated papillomavirus transcription activation. Viruses 2014; 6:3228-49; PMID:25140737; https://doi.org/10.3390/v6083228
- Wang X, Helfer CM, Pancholi N, Bradner JEM, You J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol 2013; 87:3871-84; PMID:23365439; https://doi.org/10.1128/JVI.03068-12
- Jung GS, Jeon JH, Choi YK, Jang SY, Park SY, Kim SW, Byun JK, Kim MK, Lee S, Shin EC, et al. Pyruvate dehydrogenase kinase regulates hepatitis C virus replication. Sci Rep 2016; 6:30846; PMID:27471054; https://doi.org/10.1038/srep30846
- Wang X, Li J, Schowalter RM, Jiao J, Buck CB, You J. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathogens 2012; 8:e1003021; PMID:23144621; https://doi.org/10.1371/journal.ppat.1003021
- Sharma A, Larue RC, Plumb MR, Malani N, Male F, Slaughter A, Kessl JJ, Shkriabai N, Coward E, Aiyer SS, et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc Nat Acad Sci U S A 2013; 110:12036-41; PMID: 23818621; https://doi.org/10.1073/pnas.1307157110
- Gupta SS, Maetzig T, Maertens GN, Sharif A, Rothe M, Weidner-Glunde M, Galla M, Schambach A, Cherepanov P, Schulz TF. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J Virol 2013; 87:12721-36; PMID:24049186; https://doi.org/10.1128/JVI.01942-13
- Thomas M, Sonntag E, Müller R, Schmidt S, Zielke B, Fossen T, Stamminger T. pUL69 of human cytomegalovirus recruits the cellular protein Arginine Methyltransferase 6 via a domain that is crucial for mRNA export and efficient viral replication. J Virol 2015; 89:9601-15; PMID:26178996; https://doi.org/10.1128/JVI.01399-15
- Friedrich S, Schmidt T, Schierhorn A, Lilie H, Szczepankiewicz G, Bergs S, Liebert UG, Golbik RP, Behrens SE. Arginine methylation enhances the RNA chaperone activity of the West Nile virus host factor AUF1 p45. Rna 2016; 22:1574-91; PMID:27520967; https://doi.org/10.1261/rna.055269.115
- Han T, Wan Y, Wang J, Zhao P, Yuan Y, Wang L, She Y, Broering R, Lu M, Ye L, et al. Set7 facilitates hepatitis C virus replication via enzymatic activity-dependent attenuation of the IFN-related pathway. J Immunol 2015; 194:2757-68; PMID:25681344; https://doi.org/10.4049/jimmunol.1400583
- Wang Y, Zhong H, Xie X, Chen CY, Huang D, Shen L, Zhang H, Chen ZW, Zeng G. Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+ T-cell immune responses in tuberculosis infection. Proc Natl Acad Sci U S A 2015; 112:E3883-92; PMID:26150504; https://doi.org/10.1073/pnas.1501662112
- Ciccarelli M, Vastolo V, Albano L, Lecce M, Cabaro S, Liotti A, Longo M, Oriente F, Russo GL, Macchia PE, et al. Glucose-induced expression of the homeotic transcription factor Prep1 is associated with histone post-translational modifications in skeletal muscle. Diabetologia 2016; 59:176-86; PMID:26453063; https://doi.org/10.1007/s00125-015-3774-6
- Goru SK, Kadakol A, Pandey A, Malek V, Sharma N, Gaikwad AB. Histone H2AK119 and H2BK120 mono-ubiquitination modulate SET7/9 and SUV39H1 in type 1 diabetes-induced renal fibrosis. Biochem J 2016; 473:3937-49; PMID:27582499; https://doi.org/10.1042/BCJ20160595
- Yuan H, Reddy MA, Deshpande S, Jia Y, Park JT, Lanting LL, Jin W, Kato M, Xu ZG, Das S. Epigenetic histone modifications involved in profibrotic gene regulation by 12/15-Lipoxygenase and its oxidized lipid products in diabetic nephropathy. Antioxid Redox Signal 2016; 24:361-75; PMID:26492974; https://doi.org/10.1089/ars.2015.6372
- Paneni F, Costantino S, Battista R, Castello L, Capretti G, Chiandotto S, Scavone G, Villano A, Pitocco D, Lanza G, et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Gen 2015; 8:150-8; PMID:25472959; https://doi.org/10.1161/CIRCGENETICS.114.000671
- Maganti AV, Maier B, Tersey SA, Sampley ML, Mosley AL, Özcan S, Pachaiyappan B, Woster PM, Hunter CS, Stein R, et al. Transcriptional activity of the islet beta cell factor Pdx1 is augmented by lysine methylation catalyzed by the methyltransferase Set7/9. J Biol Chem 2015; 290:9812-22; PMID:25713082; https://doi.org/10.1074/jbc.M114.616219
- Son MJ, Kim WK, Oh KJ, Park A, Lee da S, Han BS, Lee SC, Bae KH. Methyltransferase and demethylase profiling studies during brown adipocyte differentiation. BMB Rep 2016; 49:388-93; PMID:27157542; https://doi.org/10.5483/BMBRep.2016.49.7.062
- LeBlanc SE, Wu Q, Lamba P, Sif S, Imbalzano AN. Promoter-enhancer looping at the PPARgamma2 locus during adipogenic differentiation requires the Prmt5 methyltransferase. Nucl Acid Res 2016; 44:5133-47; PMID:26935580; https://doi.org/10.1093/nar/gkw129
- Yi SA, Um SH, Lee J, Yoo JH, Bang SY, Park EK, Lee MG, Nam KH, Jeon YJ, Park JW, et al. S6K1 phosphorylation of H2B mediates EZH2 trimethylation of H3: A determinant of early adipogenesis. Mol Cell 2016; 62:443-52; PMID:27151441; https://doi.org/10.1016/j.molcel.2016.03.011
- Claycombe KJ, Vomhof-DeKrey EE, Garcia R, Johnson WT, Uthus E, Roemmich JN. Decreased beige adipocyte number and mitochondrial respiration coincide with increased histone methyl transferase (G9a) and reduced FGF21 gene expression in Sprague-Dawley rats fed prenatal low protein and postnatal high-fat diets. J Nutrit Biochem 2016; 31:113-21; PMID:27133430; https://doi.org/10.1016/j.jnutbio.2016.01.008
- Kim DI, Park MJ, Choi JH, Kim IS, Han HJ, Yoon KC, Park SW, Lee MY, Oh KS, Park SH. PRMT1 and PRMT4 regulate oxidative stress-induced retinal pigment epithelial cell damage in SIRT1-dependent and SIRT1-independent manners. Oxidat Med Cell Longev 2015; 617919; PMID:26583059; https://doi.org/10.1155/2015/617919
- Wang F, Liu H, Blanton WP, Belkina A, Lebrasseur NK, Denis GV. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem J 2010; 425:71-83; PMID:19883376; https://doi.org/10.1042/BJ20090928
- Wang F, Deeney JT, Denis GV. Brd2 gene disruption causes “metabolically healthy” obesity: epigenetic and chromatin-based mechanisms that uncouple obesity from type 2 diabetes. Vitam Horm 2013; 91:49-75; PMID:23374712; https://doi.org/10.1016/B978-0-12-407766-9.00003-1
- Liu X, Chen Z, Xu C, Leng X, Cao H, Ouyang G, Xiao W. Repression of hypoxia-inducible factor alpha signaling by Set7-mediated methylation. Nucl Acid Res 2015; 43:5081-98; PMID:25897119; https://doi.org/10.1093/nar/gkv379
- Rodriguez H, Rafehi H, Bhave M, El-Osta A. Metabolism and chromatin dynamics in health and disease. Mol Aspect Med 2016; PMID:27697603; https://doi.org/10.1016/j.mam.2016.09.004
- Barsyte-Lovejoy D, Li F, Oudhoff MJ, Tatlock JH, Dong A, Zeng H, Wu H, Freeman SA, Schapira M, Senisterra GA, et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc Natl Acad Sci U S A 2014; 111:12853-8; PMID:25136132; https://doi.org/10.1073/pnas.1407358111
- Han HS, Choi D, Choi S, Koo SH. Roles of protein arginine methyltransferases in the control of glucose metabolism. Endocrinol Metabol 2014; 29:435-40; PMID:25559572; https://doi.org/10.3803/EnM.2014.29.4.435
- Deeney JT, Belkina AC, Shirihai OS, Corkey BE, Denis GV. BET Bromodomain Proteins Brd2, Brd3 and Brd4 selectively regulate metabolic pathways in the Pancreatic beta-Cell. PloS One 2016; 11:e0151329; PMID:27008626; https://doi.org/10.1371/journal.pone.0151329
- Krivega I, Dale RK, Dean A. Role of LDB1 in the transition from chromatin looping to transcription activation. Gen Dev 2014; 28:1278-90; PMID:24874989; https://doi.org/10.1101/gad.239749.114
- Sunshine HR, Hofrichter J, Eaton WA. Requirement for therapeutic inhibition of sickle haemoglobin gelation. Nature 1978; 275:238-40; PMID:692700; https://doi.org/10.1038/275238a0
- Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, Yao H, Trowbridge JJ, Mandel G, Orkin SH. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A 2013; 110:6518-23; PMID:23576758; https://doi.org/10.1073/pnas.1303976110
- Bradner JE, Mak R, Tanguturi SK, Mazitschek R, Haggarty SJ, Ross K, Chang CY, Bosco J, West N, Morse E, et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci U S A 2010; 107:12617-22; PMID:20616024; https://doi.org/10.1073/pnas.1006774107
- Rank G, Cerruti L, Simpson RJ, Moritz RL, Jane SM, Zhao Q. Identification of a PRMT5-dependent repressor complex linked to silencing of human fetal globin gene expression. Blood 2010; 116:1585-92; PMID:20495075; https://doi.org/10.1182/blood-2009-10-251116
- Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nature Medicine 2013; 19:291-4; PMID:23416702; https://doi.org/10.1038/nm.3101
- Krivega I, Byrnes C, de Vasconcellos JF, Lee YT, Kaushal M, Dean A, Miller JL. Inhibition of G9a methyltransferase stimulates fetal hemoglobin production by facilitating LCR/gamma-globin looping. Blood 2015; 126:665-72; PMID:25979948; https://doi.org/10.1182/blood-2015-02-629972
- Renneville A, Van Galen P, Canver MC, McConkey M, Krill-Burger JM, Dorfman DM, Holson EB, Bernstein BE, Orkin SH, Bauer DE, et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood 2015; 126:1930-9; PMID:26320100; https://doi.org/10.1182/blood-2015-06-649087
- Kadoch C, Copeland RA, Keilhack H. PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry 2016; 55:1600-14; PMID:26836503; https://doi.org/10.1021/acs.biochem.5b01191
- Fedorov O, Castex J, Tallant C, Owen DR, Martin S, Aldeghi M, Monteiro O, Filippakopoulos P, Picaud S, Trzupek JD, et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci Adv 2015; 1:e1500723; PMID:26702435; https://doi.org/10.1126/sciadv.1500723
- Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res 2015; 75:3865-78; PMID:26139243; https://doi.org/10.1158/0008-5472.CAN-14-3798
- Gerstenberger BS, Trzupek JD, Tallant C, Fedorov O, Filippakopoulos P, Brennan PE, Fedele V, Martin S, Picaud S, Rogers C, et al. Identification of a Chemical Probe for Family VIII Bromodomains through Optimization of a Fragment Hit. J Med Chem 2016; 59:4800-11; PMID:27115555; https://doi.org/10.1021/acs.jmedchem.6b00012
- Gonzales-Cope M, Sidoli S, Bhanu NV, Won KJ, Garcia BA. Histone H4 acetylation and the epigenetic reader Brd4 are critical regulators of pluripotency in embryonic stem cells. BMC Genomics 2016; 17:95; PMID:26847871; https://doi.org/10.1186/s12864-016-2414-y
- Hofstetter C, Kampka JM, Huppertz S, Weber H, Schlosser A, Müller AM, Becker M. Inhibition of KDM6 activity during murine ESC differentiation induces DNA damage. J Cell Sc 2016; 129:788-803; PMID:26759175; https://doi.org/10.1242/jcs.175174
- Kamikawa YF, Donohoe ME. Histone demethylation maintains Prdm14 and Tsix expression and represses xIst in embryonic stem cells. Plos One 2015; 10:e0125626; PMID:25993097; https://doi.org/10.1371/journal.pone.0125626
- Kang SC, Kim SK, Chai JC, Kim SH, Won KJ, Lee YS, Jung KH, Chai YG. Transcriptomic Profiling and H3K27me3 distribution reveal both Demethylase-Dependent and independent regulation of developmental gene transcription in cell differentiation. Plos One 2015; 10:e0135276; PMID:26263556; https://doi.org/10.1371/journal.pone.0135276
- Korb E, Herre M, Zucker-Scharff I, Darnell RB, Allis CD. BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat Neurosci 2015; 18:1464-73; PMID:26301327; https://doi.org/10.1038/nn.4095
- Xu Y, Cao W, Zhou M, Li C, Luo Y, Wang H, Zhao R, Jiang S, Yang J, Liu Y, et al. Inactivation of BRD7 results in impaired cognitive behavior and reduced synaptic plasticity of the medial prefrontal cortex. Behav Brain Res 2015; 286:1-10; PMID:25721744; https://doi.org/10.1016/j.bbr.2015.02.031