
ABSTRACT
Both genetic and lifestyle factors contribute to the risk of non-alcoholic steatohepatitis (NASH). Additionally, epigenetic modifications may also play a key role in the pathogenesis of NASH. We therefore investigated liver DNA methylation, as a marker for epigenetic alterations, in individuals with simple steatosis and NASH, and further tested if these alterations were associated with clinical phenotypes. Liver biopsies obtained from 95 obese individuals (age: 49.5 ± 7.7 years, BMI: 43 ± 5.7 kg/m2, type 2 diabetes [T2D]: 35) as a wedge biopsy during a Roux-en-Y gastric bypass operation were investigated. Thirty-four individuals had a normal liver phenotype, 35 had simple steatosis, and 26 had NASH. Genome-wide DNA methylation pattern was analyzed using the Infinium HumanMethylation450 BeadChip. mRNA expression was analyzed from 42 individuals using the HumanHT-12 Expression BeadChip. We identified 1,292 CpG sites representing 677 unique genes differentially methylated in liver of individuals with NASH (q < 0.001), independently of T2D, age, sex, and BMI. Focusing on the top-ranking 30 and another 37 CpG sites mapped to genes enriched in pathways of metabolism (q = 0.0036) and cancer (q = 0.0001) all together, 59 NASH-associated CpG sites correlated with fasting insulin levels independently of age, fasting glucose, or T2D. From these, we identified 30 correlations between DNA methylation and mRNA expression, for example LDHB (r = −0.45, P = 0.003). We demonstrated that NASH, more than simple steatosis, associates with differential DNA methylation in the human liver. These epigenetic alterations in NASH are linked with insulin metabolism.
Introduction
Non-alcoholic steatohepatitis (NASH), known as the severe form of the most common liver disease worldwide, non-alcoholic fatty liver disease (NAFLD), is a significant risk factor for cirrhosis and hepatic carcinoma.Citation1,2 Importantly, simple steatosis, a less advanced form of NAFLD, may progress to NASH in a considerable proportion of the cases.Citation1,3 NAFLD is often considered as part of the metabolic syndrome due to its close relation with metabolic factors, including obesity and insulin resistance.Citation4 However, the pathogenesis of NAFLD and NASH and the related metabolic disorders are yet to be fully elucidated.
Both genetic and lifestyle factors contribute to the risk of NASH.Citation1,5-7 Therefore, epigenetic modifications, either inherited or induced by lifestyles,Citation8,9 may play a key role in the pathogenesis of the disease. To some extent, the epigenetic mechanisms resulting in aberrant histone modifications, differences in DNA methylation, and dysregulation of microRNAs are the ones that have been related to NAFLD and its transition to more advanced stages.Citation10 So far, the most studied epigenetic mechanisms related to NAFLD in humans are the microRNA serum/plasma profile and DNA methylation.Citation10,11
In human liver, very few studies have demonstrated the importance of DNA methylation in NAFLD,Citation12-15 from which only 2 have applied a genome-wide (GW) approach.Citation13,14 However, none of these GW studies have carefully explored the relation between differential methylation and clinical parameters related to NASH.
In the present study, we investigated liver DNA methylation, as a marker for epigenetic alterations, in obese individuals with either normal liver, simple steatosis, or NASH. To identify epigenetic alterations in liver we used a GW approach where DNA methylation of ∼455,000 sites covering 99% RefSeq genes was analyzed. We further tested if epigenetic changes in the liver were associated with clinical phenotypes that are related to NAFLD.
Results
NASH but not steatosis associates with altered liver DNA methylation
The clinical characteristics of the participants in this study are shown in . While there were no significant differences in age, BMI, and serum cholesterol, individuals with normal liver had lower fasting glucose, insulin levels, and T2D cases compared with individuals with NASH, and lower triglyceride levels than individuals with either simple steatosis or NASH ().
Table 1. Clinical characteristics and liver histology of individuals included in the study.
We first examined if the average DNA methylation levels of different genomic regions in human liver would associate with NASH, either based on their relation to the nearest gene and functional genome distribution () or in relation to the CpG content (). We found no association between NASH or steatosis and differences in average DNA methylation for these genomic regions.
Figure 1. Global DNA methylation in human liver from normal, steatosis and NASH individuals is shown for (A) each gene region and (B) CpG island regions. Global DNA methylation is calculated as average DNA methylation based on all CpG sites in each annotated region on the Infinium HumanMethylation450 BeadChip. TSS, proximal promoter, defined as 200 or 1,500 bp upstream of the transcription start site. Shore, flanking region of CpG islands (0–2,000 bp); Shelf, regions flanking island shores (2,000–4,000 bp from the CpG island). N, northern; S, southern. Pie chart describing the number of sites that exhibit increased or decreased DNA methylation in NASH at (C) q < 0.05 and (D) q < 0.001 and ≥ 5% point difference. (E) CpG sites displaying the most significant increased or decreased DNA methylation (q < 0.001 and at least 5% point change) in NASH.
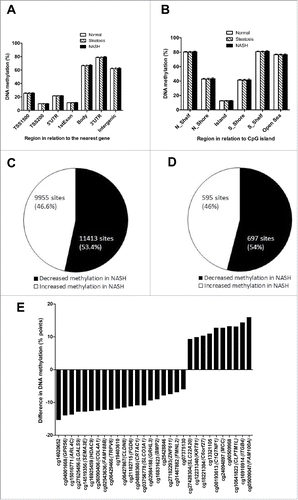
As all calculated variance inflation factors of the covariates (T2D, gender, BMI, and age) included in the linear regression models were close to 1 (1.06–1.41), the problem with multicollinearity among these variables is very limited. Subsequently, based on the linear regression model and after correction for multiple testing, we found only 1 CpG site (cg17468553 in both 5′UTR and 1st Exon of ALKBH5, encoding alkB homolog 5, RNA demethylase) differently methylated in individuals with steatosis (q = 0.0016), which was directly associated with this phenotype. Conversely, we identified 21,368 CpG sites, representing 7788 unique genes, with significant differences in DNA methylation in the liver between individuals with NASH and those with normal liver or simple steatosis at q < 0.05 (Table S1), independently of T2D, gender, BMI, and age.
Approximately half of the significant CpG sites (11,413 sites; 53.4%) displayed decreased DNA methylation in individuals with NASH and the other half (9,955 sites; 46.6%) displayed increased DNA methylation (). In addition, to gain further biologic relevance we filtered out DNA methylation results requiring absolute difference in methylation of ≥ 5% points (β-value) in individuals with NASH and further considered a q < 0.001. Here, we identified 1,292 CpG sites representing 677 unique genes. These CpG sites are presented in Table S2. Again, nearly half of the significant (q < 0.001) CpG sites (697 sites; 54%) displayed decreased DNA methylation associated with NASH and the other half (595 sites; 46%), increased DNA methylation (). The top ranking 30 CpG methylated sites associated with NASH are presented in .
Global methylation in liver estimated by LINE-1 methylation levels is altered in NASH liver
We next analyzed global DNA methylation in a sub-sample of participants (36 individuals: 12 with normal liver, 12 with simple steatosis, and 12 with NASH) by measuring LINE-1 methylation levels as a surrogate marker for global DNA methylation.Citation16,17 We found that NASH, but not steatosis, was associated with lower methylation of LINE-1 in liver (P = 0.03 for NASH and P = 0.52 for steatosis). Models adjusted for T2D, however, attenuated the association with NASH (P = 0.08). Overall, individuals with NASH had lower LINE-1 methylation levels than individuals with simple steatosis or normal liver (75.9 ± 1.68% vs. 78.2 ± 2.37%; P = 0.03).
In addition, we also looked at the expression levels of DNMT1, a maintenance methyl transferase and the enzyme responsible for adding or removing methyl groups to the genome. DNMT1 expression was measured in the HumanHT-12 Expression BeadChip array (Illumina) and remained after pre-processing and filtering of the data.Citation18 The liver DNMT1 expression was ∼5% higher in individuals with NASH compared with individuals with simple steatosis or normal liver (7.1 ± 0.5 vs. 6.7 ± 0.5; P = 0.007 in models adjusted for BMI, age, sex, and T2D).
Methylation of top ranking CpG sites associated with NASH and liver histology
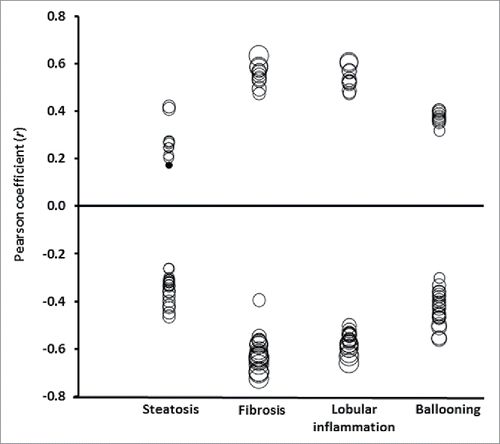
We first explored correlations of the histological features characterizing NAFLD and DNA methylation with the top ranking CpG sites associated with NASH. These correlations are illustrated in (see also Table S3). In general, the correlations between these NASH-associated CpG sites were stronger and more significant with fibrosis stage or lobular inflammation than with steatosis grade and ballooning ().
Figure 2. Pearson correlation coefficients (r; axis Y) between liver histology (axis X) and the 30 top ranking DNA methylated sites inversely or directly associated with NASH. Size of the circles indicates statistical significance (P-value). Filled circles: P > 0.05; empty circles: P < 0.05 ranging from P = 0.048 to P = 1.2 × 10−16
Gene-mapped CpG sites related to NASH are enriched in cancer and metabolic KEGG pathways
We next performed a KEGG pathway analysis using WebGelstat tool (http://bioinfo.vanderbilt.edu/webgestalt/) to identify biologic pathways with enrichment of genes mapped to CpG sites associated with differential DNA methylation in NASH (q < 0.001 and ≥5% difference in methylation points). Interestingly, for inverse associations of DNA methylation and NASH, 2 KEGG pathways, one involved in cancer (11 genes mapped to 17 CpG sites; q = 0.0001) and another in metabolism (18 genes mapped to 22 CpG sites) were significantly enriched (q = 0.0036) (Table S4). From those, 2 CpG sites overlapped with the top ranking sites associated with NASH (Table S2). For direct associations of DNA methylation and NASH, no KEGG pathways were significantly enriched.
Association of DNA methylation with clinical phenotypes
To explore the relationship between NASH-related differential DNA methylation and clinical phenotypes, we first focused on the top ranking 30 CpG sites associated with NASH. Additionally, we also tested for correlations between clinical phenotype and methylation of the additional 37 CpG sites negatively associated with NASH that mapped the genes enriched in the KEGG pathways in cancer and metabolism.
As illustrated in and , age was correlated with DNA methylation of CpG sites associated with NASH, but overall, the strongest correlations were found with serum fasting insulin followed by plasma fasting glucose (see also Table S5). Without distinction, methylation of each of the CpG sites that correlated negatively or positively with fasting insulin were, respectively, inversely or directly associated with NASH ( and ).
Figure 3. Pearson correlation coefficients (r; axis Y) between clinical phenotype (axis X) and (A) the 30 top ranking DNA methylated sites inversely or directly associated with NASH or (B) CpG methylated sites that mapped to the genes in the KEGG enriched pathways negatively associated with NASH. Size of the circles indicates statistical significance (P-value). Filled circles: P > 0.05; empty circles: P < 0.05 in panel A ranging from P = 0.049 to P = 7.1 × 10−6 and in panel B ranging from P = 0.049 to P = 4.0 × 10−6, where a larger circle indicates a higher statistical significance. C: cholesterol; TG: triglycerides; Glucose: fasting glucose; Insulin: fasting insulin.
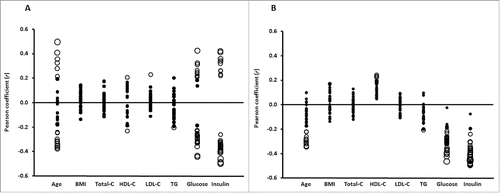
Taken together, a total of 59 out of 67 NASH-associated methylated sites correlated with fasting insulin levels, and remained significant also after adjustments for age, fasting glucose, or T2D (Table S5).
Correlation between DNA methylation and mRNA expression
To further support the findings on the relationship between DNA methylation and fasting insulin levels, we correlated DNA methylation of the 59 CpG sites associated with fasting insulin levels with the expression for transcripts located in the genomic region around these CpGs (within the cis distance 500 kb upstream and 100 kb downstream of the gene).
We identified 30 correlations (9 negative and 21 positive) between DNA methylation and mRNA expression at P < 0.05 (). Among the strongest (−0.40 < r > 0.40) correlations were the ones between methylation of cg08836954 and mRNA expression of both RNF167 (encoding the ring finger protein 167) (r = −0.46) and CANX (encoding calnexin) (r = 0.44) (). Other examples are DNA methylation of the cg04949489 in the LDHB gene (encoding the enzyme lactate dehydrogenase B) and of cg06429466, which were respectively negatively correlated with LDHB (r = −0.45) and ZYX (encoding zyxin) mRNA expression levels (r = −0.43) (). We also found positive correlations between DNA methylation of cg00193613, cg21172319, and cg22220467, respectively, with the mRNA expression of LGALS3BP (encoding lectin, galactoside-binding, soluble 3 binding protein), NAT2 (encoding N-Acetyltransferase 2), and CEACAM1 (encoding carcinoembryonic antigen-related cell adhesion molecule 1) ().
Discussion
In the present study, we demonstrated that NASH was associated with differential DNA methylation compared with normal liver and simple steatosis. More important, we showed that the majority of the top-ranking CpG sites associated with NASH correlated with fasting insulin, independently of the presence of T2D, and that, at least to some extent, this relationship is mediated by the expression of genes involved in insulin signaling.
Our first main finding was that global liver methylation based on the GW methylation array was not associated either with simple steatosis or NASH. Nevertheless, when we assessed liver global DNA methylation by LINE-1 methylation levels, NASH was associated with hypomethylation when compared with simple steatosis or normal liver. Therefore, it is possible that LINE-1 methylation levels vary independently of genomic CpG islands.Citation19,20
On the other hand, Murphy et al. have earlier demonstrated that individuals with advanced NAFLD had livers generally more hypomethylated than those with mild NAFLD.Citation14 Although the platform used for measuring DNA methylation was the same in both studies, the approach for analyzing the differences between liver phenotypes in this regard was not necessarily similar to ours, therefore limiting straight comparison of the results between these studies. Still, other differences may have accounted for differences concerning global DNA methylation and GW methylation results. In our study, participants had either normal liver, simple steatosis (presence of steatosis grade <5%, no presence of fibrosis, ballooning, or inflammation), or NASH; while in the study by Murphy et al. the difference in liver histology between the study groups (mild and advanced NAFLD) was based on the degree of fibrosis and activity score. Moreover, in both mild and advanced NAFLD groups various subjects also presented lobular or portal inflammation and ballooning. Consequently, the clinical characteristics of the participants in Murphy et al. did not differ between the study groups to the same extent they did in our study. The study design was also different considering the fact that we had a reference group (normal liver) for comparisons of the results derived from the GW array methylation, while in Murphy et al. the reference group was studied only for validation of the array results that consisted in the measurement of only 3 CpG sites. In our study, the main analyses were controlled not only for age and sex, but also for BMI and T2D.
Secondly, we observed more than 20,000 CpG sites differently methylated in NASH. After adopting more stringent criteria of statistical significance, still a large number of methylated sites were significantly associated with NASH. Among those, there was differential methylation at genes involved in cholesterol transport and inflammation (ARL4C, encoding ADP-ribosylation factor-like 4C),Citation21,22 glucose metabolism and epigenetics (HDAC9, encoding histone deacetylase 9),Citation23 liver inflammation and coronary artery disease (COL4A1, encoding collagen, type IV, α 1),Citation24,25 and liver fibrogenesis (SEMA3E, encoding class 1 semaphorin and ITGB4, encoding integrin β 4 subunit, which is a receptor for laminins).Citation26,27 These observations may reflect, at the epigenetic level, the presence of hepatic inflammation in NASH and the liver fibrosis contributing to the progression of the disease,Citation1 in addition to disturbances in cholesterol transport that may participate in NAFLD progressionCitation28 and the increased risk of cardiovascular disease.Citation29 On the other hand, we found only one CpG site differently methylated in steatosis, mapped to ALKBH5, which encodes an enzyme that demethylates RNA. More specifically, this enzyme demethylates N(6)-methyladenosine (m6A), in which inhibition of methylation may disrupt the circadian clock, thereby increasing susceptibility to obesity, diabetes, and cancer.Citation30,31 Supporting these findings, we found that the top-ranking CpG sites associated with NASH were more strongly correlated with liver fibrosis stage and lobular inflammation than with liver steatosis grade in the individuals who participated in this study.
Interestingly, pathway analyses revealed that significant hypomethylation in NASH represents genes enriched in pathways in cancer and metabolism. Pathways in cancer possibly represent the association of NASH with higher risk of developing cancer, e.g., hepatocellular carcinoma.Citation2 Metabolic pathways representing genes within, e.g., glycosphingolipid biosynthesis (ST3GAL4, B3GNT3) and ketolisys (ACSS2), potentially reflecting the suggested benefit of glycophospholipid and decreased ketone body metabolism in NASH.Citation32,33
Importantly, we found that methylation of 59 out of 67 selected CpG sites (either from the top-ranked 30 or pathway analysis) correlated with fasting insulin independently of age, presence of T2D and fasting plasma glucose. Fasting insulin levels can be used to estimate insulin resistance,Citation34 which has been considered the primary factor underlying hepatic steatosis.Citation1 Accordingly, methylation of PPARGC1A promoter has been associated with differential liver DNA methylation in NAFLD and with fasting insulin levels.Citation15 In our study, most of the CpGs mapped to PPARGC1A that were nominally directly associated with NASH also correlated with fasting insulin levels (data not shown).
The correlations of DNA methylation with fasting insulin are most logically explained by the fact that changes in DNA methylation may lead to changes in transcription of the nearby genes regulating insulin action. At the molecular level, our findings indicate that the disruption of insulin signaling pathway in NAFLD could be a result of altered epigenetic regulation. For example, we were able to replicate the findings from the previous GW study by Ahrens et al. that has shown NAFLD-specific differences at DNA methylation levels of IGF1, a gene coding key enzymes of in insulin/insulin-like signaling.Citation13 This hypothesis is also partially supported by our findings that 30 out of the 67 of the differentially methylated CpG sites demonstrated a correlation of methylation on this site both with fasting insulin (n = 51) and transcription of a nearby gene. For example, the correlations between these NASH-associated CpG sites with the mRNA expression of genes such as LDHB and SQSTM1. The LDHB encodes an enzyme that drives the conversion of lactate to pyruvate. In higher amount, pyruvate could lead to impairment of liver mitochondrial function, which has been related to oxidative stress and insulin resistance.Citation35,36 SQSTM1 encodes a multifunctional protein that besides binding ubiquitin and regulating activation of the nuclear factor kappa-B (NF-kB) signaling pathway, has also effects related to impairment of autophagic flux,Citation37 thereby contributing to hepatic insulin sensitivity.Citation38–40 Moreover, correlations of NASH-associated CpG sites with mRNA expression of CEACAM1, SH3BP4 (encoding transferrin receptor-trafficking protein), and NAT2 also support a role for our findings relating differential DNA methylation in NASH with impaired insulin signaling/action.Citation41–47
The mechanisms that could alter DNA methylation in the insulin-resistant liver remain to be elucidated. However, these differences in methylation may be due to altered levels of enzymes responsible for adding or removing methyl groups to the genome, such as DNMTs. Supporting this hypothesis, liver DNMT1 expression in our study is in fact higher in individuals with NASH compared with individuals with simple steatosis or normal liver. Therefore, differential methylation in the insulin-resistant liver could also be a result of pro-inflammatory cytokine signaling inducing DNMT1 expression and activity.Citation48 Furthermore, we cannot rule out the possibility that insulin resistance or higher insulin levels would be in fact modifying DNA methylation in liver. One can hypothesize that due to insulin resistance, increased release into the circulation of free-fatty acids from the adipose tissue is delivered to the liver,Citation1 which could potentially mediate the effect of insulin resistance on DNA methylation levels.Citation49–52
Due to the cross-sectional nature of this study, it is not possible to determine causality in our study. However, it is unethical to have serial liver biopsies including both histological analysis and epigenetics, in individuals with clearly defined liver phenotypes. We were also not able to differentiate the liver cell type composition between hepatocytes and inflammatory cells, which has been also the case in previous GW studies.Citation13,14 Thus, some changes in individuals with NASH could be related to changes in cell type composition. Nevertheless, we observed similar strength in the relationship between top-ranking NASH-associated CpG sites with both liver fibrosis and lobular inflammation. Moreover, we used robust statistical analyses in our GW analyses, also adjusting for confounding factors such as the presence of T2D, and studied unique liver samples from a well age- and BMI-matched study groups with normal liver, simple steatosis and NASH. We acknowledge all individuals in this study were obese and therefore the results cannot be generalized to lean individuals with NASH.
We conclude that while NASH is associated with differential DNA methylation, simple steatosis had only minor effects on methylation in human liver. The finding that the level of methylation in the majority of the top-ranking CpG sites related to NASH correlated with fasting insulin indicates that these epigenetic alterations associate with changes in insulin action. All together, these results also support the hypothesis that genetic and lifestyle factors contributing to NASH may interact at the level of DNA methylation.
Patients and methods
Study participants and analyses of clinical and metabolic parameters
Participants were selected from an ongoing study recruiting all subjects undergoing bariatric surgery Kuopio Obesity Bariatric Study (KOBS).Citation18,33,53 Ninety-five individuals who were accepted for the Roux-en-Y gastric bypass (RYGB) operation, were selected for this study from the KOBS cohort to obtain balance study groups with normal liver steatosis, simple steatosis and NASH (see below histological analysis).
Liver histology
Liver biopsies were obtained using Trucut needles (Radiplast AB, Uppsala, Sweden) or as a biopsy during elective gastric bypass operations as described previously.Citation33,53 Briefly, overall histological assessment of liver biopsy samples was performed by one pathologist according to the standard criteria.Citation54,55 Histological diagnosis was divided into 3 categories: 1) Normal liver without any steatosis, inflammation, ballooning or fibrosis; 2) simple steatosis (steatosis >5%) without evidence of hepatocellular ballooning, inflammation or fibrosis; and 3) NASH. Of the 95 individuals, 35 had a normal liver phenotype, 34 had simple steatosis, and 26 had NASH (). Type 2 diabetes (T2D) was defined according to WHO's criteria of diabetes. The study was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants and the study protocol was approved by the Ethics Committee of the Northern Savo Hospital District (54/2005, 104/2008, and 27/2010).
GW analysis of DNA methylation in human liver
After DNA extraction from human liver biopsies DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany), nucleic acid concentration and purity were determined, and DNA methylation was analyzed in liver from all 95 individuals using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA), as described previously.Citation18,56 The raw methylation data in β-values were converted to M-values {M = log2[β/(1-β)]}, which were then used for data normalization, to correct for probe design bias and batch effects as well as statistical tests, as described previously.Citation18 To easier interpret the results, M-values were reconverted to β-values, which were used when describing the data and creating figures. A total of 455,526 CpG sites were studied for DNA methylation differences associated with liver phenotypes.
LINE-1 DNA methylation
For the measurement of LINE-1 DNA methylation, 200 ng of DNA from a subset of the original cohort of 95 individuals (36 individuals: 12 with normal liver, 12 with simple steatosis, and 12 with NASH) was bisulfite converted (EpiTect Bisulfite Kit, Qiagen, Hilden, Germany). Next, 2 ul of DNA was used for PCR together with the PyroMark Q24 CpG LINE-1 assay and the PyroMark PCR kit according to the manufacturers' recommendation (Qiagen). Pyrosequencing was performed on the PyroMark Q96ID (Qiagen) using all PCR product (40 ul), with the recommended dispensation order (GCTCGTGTAGTCAGTCG). This assay quantifies methylation levels of 3 CpG sites of LINE-1, in positions 318 to 331. We used the averaged value of these 3 CpG sites to estimate global DNA methylation.
GW analysis of gene expression in human liver
RNA expression was analyzed in liver samples from a subset of individuals included in this study (42 individuals: 14 with normal liver, 13 with simple steatosis, and 15 with NASH) using the HumanHT-12 Expression BeadChip (Illumina), which covers 28,688 coding transcripts, according to the manufacturer's recommendations, as described previously and validated.Citation18
Statistical analyses
Since we have earlier published results related to differential DNA methylation and T2D,Citation18 we now carefully adjusted our analyses for T2D. To identify differences in DNA methylation and mRNA expression in the liver associated with NASH and steatosis, a linear regression model was used including T2D, gender, BMI, and age as covariates and DNA methylation or mRNA expression as the dependent variable. Variance inflation factors, which provide information about potential multicollinearity of studied phenotypes, were calculated. To account for multiple testing in the GW analysis, we applied false discovery rate (FDR).
Pearson correlations were used to relate clinical phenotype with the top ranking differentiated CpG methylated sites. As post-hoc analyses, partial correlations were further applied to control for confounding factors (e.g., age and T2D). We also correlated these top ranking CpG sites with the mRNA expression of their nearby gene(s) (within the cis distance 500 kb upstream and 100 kb downstream of the gene). For these analyses, a P < 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1294305_supplementary_data.zip
Download Zip (3.6 MB)Acknowledgments
We thank Päivi Turunen, Tiina Sistonen and Matti Laitinen for their technical assistance in the KOBS study. We thank SCIBLU (Swegene Center for Integrative Biology at Lund University) Genomics Facility for help with DNA methylation and mRNA expression analyses.
Funding
Academy of Finland (Contract no. 120979; 138006; 131593), the Finnish Diabetes Research Foundation, the North-Savo Finnish Cultural Foundation and the Kuopio University Hospital EVO and VTR funding, the Swedish Research Council, Region Skåne (ALF), Knut and Alice Wallenberg Foundation, Novo Nordisk Foundation, EFSD/Lilly Fellowship, Söderberg Foundation, The Swedish Diabetes Foundation, Påhlsson Foundation, EXODIAB and a Linné grant.
Related Research Data
References
- Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci 2016; 10:1969-1987; PMID:26894897; http://dx.doi.org/10.1007/s00018-016-2161-x
- Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010; 51:1820-1832; PMID:20432259; http://dx.doi.org/10.1002/hep.23594
- McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J Hepatol 2015; 62:1148-1155; PMID:25477264; http://dx.doi.org/10.1016/j.jhep.2014.11.034
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016; 64:73-84; PMID:26707365; http://dx.doi.org/10.1002/hep.28431
- Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, Nguyen P, Hernandez C, Richards L, Salotti J, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015; 149:1784-1793; PMID:26299412; http://dx.doi.org/10.1053/j.gastro.2015.08.011
- Watanabe S, Hashimoto E, Ikejima K, Uto H, Ono M, Sumida Y, Seike M, Takei Y, Takehara T, Tokushige K, et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. J Gastroenterol 2015; 50:364-377; PMID:25708290; http://dx.doi.org/10.1007/s00535-015-1050-7
- Zelber-Sagi S, Lotan R, Shlomai A, Webb M, Harrari G, Buch A, Nitzan Kaluski D, Halpern Z, Oren R. Predictors for incidence and remission of NAFLD in the general population during a seven-year prospective follow-up. J Hepatol 2012; 56:1145-1151; PMID:22245895; http://dx.doi.org/10.1016/j.jhep.2011.12.011
- de Mello VD, Pulkkinen L, Lalli M, Kolehmainen M, Pihlajamaki J, Uusitupa M. DNA methylation in obesity and type 2 diabetes. Ann Med 2014; 46:103-113; PMID:24779963; http://dx.doi.org/10.3109/07853890.2013.857259
- Gillberg L, Perfilyev A, Brons C, Thomasen M, Grunnet LG, Volkov P, Rosqvist F, Iggman D, Dahlman I, Riserus U, et al. Adipose tissue transcriptomics and epigenomics in low birthweight men and controls: Role of high-fat overfeeding. Diabetologia 2016; 59:799-812; PMID:26750116; http://dx.doi.org/10.1007/s00125-015-3852-9
- Gallego-Duran R, Romero-Gomez M. Epigenetic mechanisms in non-alcoholic fatty liver disease: An emerging field. World J Hepatol 2015; 7:2497-2502; PMID:26523202; http://dx.doi.org/10.4254/wjh.v7.i24.2497
- Lee J, Kim Y, Friso S, Choi SW. Epigenetics in non-alcoholic fatty liver disease. Mol Aspects Med 2016; Mol Aspects Med. 2017 54:78-88; PMID:27889327; http://dx.doi.org/10.1016/j.mam.2016.11.008
- Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, Ness T, Masson S, Mathers JC, French J, White S, et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics 2015; 7:110, 25-015-0056-6. eCollection 2015; PMID:26473022; http://dx.doi.org/10.1186/s13148-015-0056-6
- Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab 2013; 18:296-302; PMID:23931760; http://dx.doi.org/10.1016/j.cmet.2013.07.004
- Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, Garrett ME, Ashley-Koch A, Suzuki A, Tillmann HL, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013; 145:1076-1087; PMID:23916847; http://dx.doi.org/10.1053/j.gastro.2013.07.047
- Sookoian S, Rosselli MS, Gemma C, Burgueno AL, Fernandez Gianotti T, Castano GO, Pirola CJ. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010; 52:1992-2000; PMID:20890895; http://dx.doi.org/10.1002/hep.23927
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32:e38; PMID:14973332; http://dx.doi.org/10.1093/nar/gnh032
- Anwar SL, Krech T, Hasemeier B, Schipper E, Schweitzer N, Vogel A, Kreipe H, Lehmann U. Loss of DNA methylation at imprinted loci is a frequent event in hepatocellular carcinoma and identifies patients with shortened survival. Clin Epigenetics 2015; 7:110- 015-0145-6. eCollection 2015; PMID:26473022; http://dx.doi.org/10.1186/s13148-015-0145-6
- Nilsson E, Matte A, Perfilyev A, de Mello VD, Kakela P, Pihlajamaki J, Ling C. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J Clin Endocrinol Metab 2015; 100:E1491-501; PMID:26418287; http://dx.doi.org/10.1210/jc.2015-3204
- Ehrlich M, Jiang G, Fiala E, Dome JS, Yu MC, Long TI, Youn B, Sohn OS, Widschwendter M, Tomlinson GE, et al. Hypomethylation and hypermethylation of DNA in wilms tumors. Oncogene 2002; 21:6694-6702; PMID:12242669; http://dx.doi.org/10.1038/sj.onc.1205890
- Iacopetta B, Grieu F, Phillips M, Ruszkiewicz A, Moore J, Minamoto T, Kawakami K. Methylation levels of LINE-1 repeats and CpG island loci are inversely related in normal colonic mucosa. Cancer Sci 2007; 98:1454-1460; PMID:17640302; http://dx.doi.org/10.1111/j.1349-7006.2007.00548.x
- Engel T, Lueken A, Bode G, Hobohm U, Lorkowski S, Schlueter B, Rust S, Cullen P, Pech M, Assmann G, et al. ADP-ribosylation factor (ARF)-like 7 (ARL7) is induced by cholesterol loading and participates in apolipoprotein AI-dependent cholesterol export. FEBS Lett 2004; 566:241-246; PMID:15147902; http://dx.doi.org/10.1016/j.febslet.2004.04.048
- Hong C, Walczak R, Dhamko H, Bradley MN, Marathe C, Boyadjian R, Salazar JV, Tontonoz P. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: Identification of ARL7 as a direct target. J Lipid Res 2011; 52:531-539; PMID:21187453; http://dx.doi.org/10.1194/jlr.M010686
- Chen J, Wang N, Dong M, Guo M, Zhao Y, Zhuo Z, Zhang C, Chi X, Pan Y, Jiang J, et al. The metabolic regulator histone deacetylase 9 contributes to glucose homeostasis abnormality induced by hepatitis C virus infection. Diabetes 2015; 64:4088-4098; PMID:26420860; http://dx.doi.org/10.2337/db15-0197
- Staten NR, Welsh EA, Sidik K, McDonald SA, Dufield DR, Maqsodi B, Ma Y, McMaster GK, Mathews RW, Arch RH, et al. Multiplex transcriptional analysis of paraffin-embedded liver needle biopsy from patients with liver fibrosis. Fibrogenesis Tissue Repair 2012; 5:21- 1536-5-21; PMID:23270325; http://dx.doi.org/10.1186/1755-1536-5-21
- CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013; 45:25-33; PMID:23202125; http://dx.doi.org/10.1111/j.1478-3231.2006.01312.x
- Lydatakis H, Hager IP, Kostadelou E, Mpousmpoulas S, Pappas S, Diamantis I. Non-invasive markers to predict the liver fibrosis in non-alcoholic fatty liver disease. Liver Int 2006; 26:864-871; PMID:16911470; http://dx.doi.org/10.1111/j.1478-3231.2006.01312.x
- Yagai T, Miyajima A, Tanaka M. Semaphorin 3E secreted by damaged hepatocytes regulates the sinusoidal regeneration and liver fibrosis during liver regeneration. Am J Pathol 2014; 184:2250-2259; PMID:24930441; http://dx.doi.org/10.1016/j.ajpath.2014.04.018
- Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta 2015; 1852:1765-1778; PMID:26027904; http://dx.doi.org/10.1016/j.bbadis.2015.05.015
- Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol 2015; 62:S47-64; PMID:25920090; http://dx.doi.org/10.1016/j.jhep.2014.12.012
- Zelinski EL, Deibel SH, McDonald RJ. The trouble with circadian clock dysfunction: Multiple deleterious effects on the brain and body. Neurosci Biobehav Rev 2014; 40:80-101; PMID:24468109; http://dx.doi.org/10.1016/j.neubiorev.2014.01.007
- Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG. N6-methyl-adenosine (m6A) in RNA: An old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics 2013; 11:8-17; PMID:23453015; http://dx.doi.org/10.1016/j.gpb.2012.12.002
- Zigmond E, Tayer-Shifman O, Lalazar G, Ben Ya'acov A, Weksler-Zangen S, Shasha D, Sklair-Levy M, Zolotarov L, Shalev Z, Kalman R, et al. Beta-glycosphingolipids ameliorated non-alcoholic steatohepatitis in the psammomys obesus model. J Inflamm Res 2014; 7:151-158; PMID:25336983; https://doi.org/10.2147/JIR.S50508
- Männisto VT, Simonen M, Hyysalo J, Soininen P, Kangas AJ, Kaminska D, Matte AK, Venesmaa S, Käkelä P, Karja V, et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int 2015; 35:1853-1861; PMID:25533197; http://dx.doi.org/10.1111/liv.12769
- Laakso M. How good a marker is insulin level for insulin resistance? Am J Epidemiol 1993; 137:959-965; PMID:8317453; http://dx.doi.org/10.1093/oxfordjournals.aje.a116768
- Surapaneni KM, Jainu M. Pioglitazone, quercetin and hydroxy citric acid effect on hepatic biomarkers in non alcoholic steatohepatitis. Pharmacognosy Res 2014; 6:153-162; PMID:24761121; http://dx.doi.org/10.4103/0974-8490.129037
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res 2008; 102:401-414; PMID:18309108; http://dx.doi.org/10.1161/CIRCRESAHA.107.165472
- Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillon J, Lo Iacono O, Corazzari M, Fimia GM, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis 2014; 5:e1179; PMID:24743734; http://dx.doi.org/10.1038/cddis.2014.162
- Ke B, Zhao Z, Ye X, Gao Z, Manganiello V, Wu B, Ye J. Inactivation of NF-ΰB p65 (RelA) in liver improves insulin sensitivity and inhibits cAMP/PKA pathway. Diabetes 2015; 64:3355-3362; PMID:26038580; http://dx.doi.org/10.2337/db15-0242
- Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem 2009; 284:31484-31492; PMID:19758991; http://dx.doi.org/10.1074/jbc.M109.033936
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006; 116:1793-1801; PMID:16823477; http://dx.doi.org/10.1172/JCI29069
- Knowles JW, Xie W, Zhang Z, Chennemsetty I, Assimes TL, Paananen J, Hansson O, Pankow J, Goodarzi MO, Carcamo-Orive I, et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J Clin Invest 2015; 125:1739-1751; PMID:25798622; http://dx.doi.org/10.1172/JCI74692
- Al-Share QY, DeAngelis AM, Lester SG, Bowman TA, Ramakrishnan SK, Abdallah SL, Russo L, Patel PR, Kaw MK, Raphael CK, et al. Forced hepatic overexpression of CEACAM1 curtails diet-induced insulin resistance. Diabetes 2015; 64:2780-2790; PMID:25972571; http://dx.doi.org/10.2337/db14-1772
- Alexaki A, Gupta SD, Majumder S, Kono M, Tuymetova G, Harmon JM, Dunn TM, Proia RL. Autophagy regulates sphingolipid levels in the liver. J Lipid Res 2014; 55:2521-2531; PMID:25332431; http://dx.doi.org/10.1194/jlr.M051862
- Kim YM, Stone M, Hwang TH, Kim YG, Dunlevy JR, Griffin TJ, Kim DH. SH3BP4 is a negative regulator of amino acid-rag GTPase-mTORC1 signaling. Mol Cell 2012; 46:833-846; PMID:22575674; http://dx.doi.org/10.1016/j.molcel.2012.04.007
- Zoncu R, Efeyan A, Sabatini DM. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21-35; PMID:21157483; http://dx.doi.org/10.1038/nrm3025
- DeAngelis AM, Heinrich G, Dai T, Bowman TA, Patel PR, Lee SJ, Hong EG, Jung DY, Assmann A, Kulkarni RN, et al. Carcinoembryonic antigen-related cell adhesion molecule 1: A link between insulin and lipid metabolism. Diabetes 2008; 57:2296-2303; PMID:18544705; http://dx.doi.org/10.2337/db08-0379
- Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab 2002; 13:240-245; PMID:12128284; http://dx.doi.org/10.1016/S1043-2760(02)00608-2
- Kim AY, Park YJ, Pan X, Shin KC, Kwak SH, Bassas AF, Sallam RM, Park KS, Alfadda AA, Xu A, et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat Commun 2015; 6:7585; PMID:26139044; http://dx.doi.org/10.1038/ncomms8585
- Olsen AS, Sarras MP, Jr, Leontovich A, Intine RV. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 2012; 61:485-491; PMID:22228713; http://dx.doi.org/10.2337/db11-0588
- Dhliwayo N, Sarras MP, Jr, Luczkowski E, Mason SM, Intine RV. Parp inhibition prevents ten-eleven translocase enzyme activation and hyperglycemia-induced DNA demethylation. Diabetes 2014; 63:3069-3076; PMID:24722243; http://dx.doi.org/10.2337/db13-1916
- Hall E, Volkov P, Dayeh T, Bacos K, Ronn T, Nitert MD, Ling C. Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets. BMC Med 2014; 12:103- 7015-12-103; http://dx.doi.org/10.1186/1741-7015-12-103
- Silva-Martinez GA, Rodriguez-Rios D, Alvarado-Caudillo Y, Vaquero A, Esteller M, Carmona FJ, Moran S, Nielsen FC, Wickstrom-Lindholm M, Wrobel K, et al. Arachidonic and oleic acid exert distinct effects on the DNA methylome. Epigenetics 2016; 11:321-334; PMID:27088456; http://dx.doi.org/10.1080/15592294.2016.1161873
- Männisto VT, Simonen M, Soininen P, Tiainen M, Kangas AJ, Kaminska D, Venesmaa S, Käkelä P, Karja V, Gylling H, et al. Lipoprotein subclass metabolism in nonalcoholic steatohepatitis. J Lipid Res 2014; 55:2676-2684; PMID:25344588; http://dx.doi.org/10.1194/jlr.P054387
- Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am J Gastroenterol 1999; 94:2467-2474; PMID:10484010; http://dx.doi.org/10.1111/j.1572-0241.1999.01377.x
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005; 41:1313-1321; PMID:15915461; http://dx.doi.org/10.1002/hep.20701
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288-295; PMID:21839163; http://dx.doi.org/10.1016/j.ygeno.2011.07.007