
ABSTRACT
CpG islands (CGI) are critical genomic regulatory elements that support transcriptional initiation and are associated with the promoters of most human genes. CGI are distinguished from the bulk genome by their high CpG density, lack of DNA methylation, and euchromatic features. While CGI are canonically known as strong promoters, thousands of ‘orphan’ CGI lie far from any known transcript, leaving their function an open question. We undertook a comprehensive analysis of the epigenetic state of orphan CGI across over 100 cell types. Here we show that most orphan CGI display the chromatin features of active enhancers (H3K4me1, H3K27Ac) in at least one cell type. Relative to classical enhancers, these enhancer CGI (ECGI) are stronger, as gauged by chromatin state and in functional assays, are more broadly expressed, and are more highly conserved. Likewise, ECGI engage in more genomic contacts and are enriched for transcription factor binding relative to classical enhancers. In human cancers, these epigenetic differences between ECGI vs. classical enhancers manifest in distinct alterations in DNA methylation. Thus, ECGI define a class of highly active enhancers, strengthened by the broad transcriptional activity, CpG density, hypomethylation, and chromatin features they share with promoter CGI. In addition to indicating a role for thousands of orphan CGI, these findings suggests that enhancer activity may be an intrinsic function of CGI in general and provides new insights into the evolution of enhancers and their epigenetic regulation during development and tumorigenesis.
Introduction
Vertebrate genomes are heavily methylated, CpG-poor, predominantly heterochromatic terrains disrupted by CpG islands (CGI), essential CpG dense regulatory elements. CGI are defined by a lack of DNA methylation, transcriptional competence, and heightened euchromatic features,Citation1 such as enrichment of H3K4me3 and H3K9/27Ac. These regions are found at the promoters of nearly two-thirds of human protein-coding genes. As such, CGI have been canonically studied for their role in permitting transcriptional initiation. CGI-associated promoters demonstrate broader expression patterns across tissues and tend to be stronger than CpG-poor promoters.Citation2 These properties are contingent on remaining unmethylated, as hypermethylation of CGI leads to the recruitment of repressive complexes containing histone deacetylases and chromatin remodelersCitation3 resulting in the silencing of the associated gene.Citation4–6 This is a cardinal method by which cancer cells inactivate tumor-suppressor genes.Citation7–9
CpG sites are targeted for DNA methylation during developmentCitation10 unless actively protected by methylation of histone H3 lysine 4 (H3K4), a mark found principally at promoters and enhancers and associated with transcriptional initiation.Citation11 Methylated cytosine is mutagenic due to a propensity to undergo deamination to thymine, leading to the paucity of CpG sites throughout the genomeCitation12 and marking CpG-rich regions as being of potential functional importance. Yet, half of CGI do not in fact overlap known transcription start sites (TSS) and thousands lie far from any known transcript. While many of these ‘orphan’ CGI are likely promoters of unannotated transcripts, their prevalence suggests there may be roles for CGI other than as strict promoters.
Enhancers are regulatory elements that act at a distance to promote gene transcription independent of position or orientation. Enhancers are similar to CGI in that their levels of DNA methylation are inversely correlated with their activity.Citation13,14 We also recently demonstrated that following treatment with decitabine, a chemotherapeutic demethylating agent, strong enhancers and promoter CGI are more resilient to de novo methylation than weak enhancers.Citation15 Indeed, recent work has demonstrated that enhancers and promoters possess a unified chromatin architecture, characterized by the presence of H3K4me, H3K9/27Ac, DNAse hypersensitivity, evidence of paired bidirectional transcriptional initiation, and transcription factor binding. Measures of transcript stability provide the most reliable method to distinguish the two, with promoters giving rise to one or two stable transcripts, and enhancers strictly unstable (eRNA) transcripts.Citation16
The striking similarity of enhancers and promoters suggests that CGI, especially those without evidence of a nearby gene, may act as enhancers.Citation17,18 Here, we undertake a comprehensive analysis of orphan CGI chromatin topology across over one hundred cell types. We demonstrate that most orphan CGI appear to be active enhancers in at least one cell type. These enhancer CGI (ECGI) are much more powerful than classical enhancers in their ability to drive transcription by a variety of measures, manifesting open chromatin across a broader variety of cell types, with heightened genomic contacts, and stronger enrichment for transcription factor binding. These features contribute to the evolutionary conservation of CpG density relative to other enhancers and to distinct susceptibilities to alterations in DNA methylation in cancer.
Results
Orphan CpG islands exhibit features of active enhancers
Given the established role for CGI in promoting transcriptional initiation, we first sought to characterize the relationship between CGI across the genome with known transcripts, focusing on UCSC CGICitation19 (n = 27,718) and the GENCODE (V25) transcript databaseCitation20,21 (). Strikingly, only 45% (n = 12,548) of CGI contain an annotated TSS for a protein-coding gene. In contrast, 32% (n = 8899) are found within a protein-coding gene, and 3% (n = 920) are within 2 kb of, but do not directly overlap a gene (perigenic). An additional 6% of CGI overlap or are within 2 kb of noncoding (ncRNA) transcripts [n = 1131 long noncoding (ncRNA), n = 837 other ncRNA, see Methods], and 2.5% are found near or overlapping pseudogenes. We term the 10% of remaining CGI (n = 2693) ‘orphan’ CGI because they cannot be annotated to any known transcript. This distribution suggests that many or most CGI in the genome may not be acting strictly as promoters.
Figure 1. Most orphan CpG islands exhibit an enhancer chromatin state. (A) CpG Islands were categorized hierarchically by distance to the nearest Gencode annotated gene. Shown is the number of CGI that overlap the TSS of a protein-coding gene (TSS), those that overlap a protein coding gene but not the TSS (intragenic), those that lie in proximity (+/−2 kb) of a protein coding gene (perigenic), or those that are within 2 kb of, or overlap, a long non-coding RNA (lncRNA), other non-coding RNAs (ncRNA), or a pseudogene. CGI more than 2 kb from any of these gene classes were considered orphan CGI (see Methods). (B) Enhancers were defined either as the overlap of H3K4me1/H3K27Ac peaks or by an HMM chromatin state as annotated in over 100 cell lines (see Methods). The histograms represent the number of cell lines in which orphan CGIs overlap an enhancer by each definition. (C) Distribution of orphan CGI meeting one or both enhancer definitions in at least one cell line. (D) Stable or Unstable transcript pairs as defined in K562 cellsCitation15 were intersected with promoter CGI, the TSS (+/−500 bp) of other coding genes that not within 2.5 kb of a CGI, ECGI, and classical enhancers active in K562 cells. Shown is the fraction of each set of genomic regions that overlap stable, unstable or mixed transcript pairs. (E) Distribution of the average DNA methylation in WGBS data from normal breast tissue (TCGA) across promoter CGI (those overlapping a coding TSS), ECGI active in HMEC cells, and classical enhancers active in HMEC cells (those orphan CGI or other regions meeting both the H3K27Ac/H3K4me1 peak and HMM enhancer definition). (F) Density of the G+C content (%GC) and CpG content (Observed/Expected) among promoter CGI, vs. ECGI and classical enhancers active in HMEC cells. (G-N) Analysis of H3K27Ac or H3K4me1/2/3 at promoter CGI, HMEC ECGI, and HMEC classical enhancers. (G-J) Distribution of the density (reads/kb) for the indicated chromatin mark among genomic loci in each class. Line indicates median, boxes are the first and third quartiles and whiskers represent the highest and lowest values within 1.5 times the inter-quartile range. (K-N) relative tag densities for the indicated chromatin mark was determined in 10 bp bins for +/−2.5 kb from the center of each genomic feature class as determined from ChIP-seq data from HMEC cells (ENCODE). (O) Distribution of the ratio of H3K4me3 to H3K4me1 tag densities across genomic loci in each class.
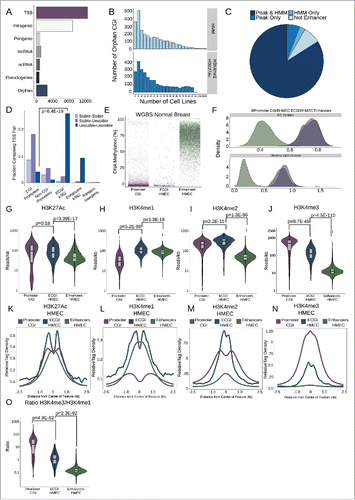
Given that enhancers and promoters are characterized by a similar epigenetic environment, we hypothesized that orphan CGI may be enhancers. To test this, we defined putative enhancers using two established methods: regions containing overlapping peaks of H3K4me1 and H3K27AcCitation22,23 across 120 cell types (22 ENCODE,Citation24 98 Roadmap Epigenomics Project,Citation25 see Methods), as well as regions annotated as enhancers in chromatin state maps based on Hidden Markov Models (chromHMM)Citation26 in 136 cell types (9 ENCODE, 127 Roadmap). Using these definitions, fully 92% of orphan CGI overlapped an enhancer in one or more cell types (2241 overlapped enhancers by both definitions, 197 peak only, 33 HMM only; ).
Given the prevalence with which orphan CGI can confidently be called enhancers, we investigated whether other classes of CGI (containing a TSS or otherwise transcript-associated) could be classified as enhancers (Fig. S1A). We find that promoter CGI are especially likely to contain H3K4me1/H3K27Ac peaks (in ∼60% of cell lines examined; Fig. S1A), although each class, especially perigenic CGI, were likely to contain enhancer peaks in at least a quarter of cell lines. We found no evidence of enhancer activity for 8% (n = 227) of orphan CGI, which we term ‘remnant’ CGI. Overall, orphan CGI overlapped an HMM-defined enhancer in a median of 15 cell types or by H3K4me1/H3K27Ac peak overlap in 20 cell types. We term those orphan CGI that overlap regions that satisfy both enhancer criteria in at least one cell type (n = 2241) enhancer CGI or ECGI.
To study ECGI in greater detail, we focused on those that overlapped both an H3K4me1/H3K27Ac peak and an HMM-defined enhancer in one of 3 cell lines: H1 human embryonic stem cells (H1ESC, n = 180), human mammary epithelial cells (HMEC, n = 205), and K562 (n = 169), a human myelogenous leukemia line. We chose these lines for their phenotypic diversity (stem cell, normal differentiated cell, and cancer cell) and because each is well-studied with an abundance of publicly-available epigenomic data sets. For comparison, we defined non-CGI classical enhancers in each cell line by the same criteria (H3K4me1/H3K27Ac peaks that overlap HMM enhancers, n = 11863 HMEC, n = 12138 H1ESC, n = 7610 K562), as well as CGI overlapping the TSS of protein-coding genes, apparent canonical promoter CGI (n = 12548).
Distinguishing promoters from enhancers is not straightforward; however, Core et al.Citation16 recently characterized initiation regions in mammalian genomes by comparing the TSS of stable transcripts detected by CAGE, which captures 5′ 7-methylguanylate capped, steady-state transcripts like mRNAs or lncRNAs, vs. those detected by GroCap, which detects the TSS of all nascent transcripts, including unstable ones like eRNAs or upstream antisense RNAs (uaRNAs). They found that enhancers could be defined by unstable transcript pairs, and promoters by a stable transcript paired with either an unstable uaRNA or another stable transcript. Focusing on ECGI and non-CGI classical enhancers active in K562 cells (), we found that ECGI, like classical enhancers, exhibit strong enrichment for unstable-unstable pairs, in stark contrast to promoter CGI and other non-CGI promoters (Gencode TSS >2.5 kb from a CGI), which tend to be enriched in stable-stable or stable-unstable pairs (for unstable-unstable pairs in ECGI vs. promoter CGI: Odds Ratio (OR) = 6.36, P = 6.4E-19, Fisher's Exact). Indeed, no other class of transcript-associated CGI exhibited the preponderance of putative eRNA (unstable) pairs displayed by orphan CGI (Fig. S1B,C). Intragenic, perigenic, and ncRNA CGI were equally likely to contain unstable as stable transcripts, which likely mark alternative promoters, a documented role of CGI distinct from enhancer function,Citation27 whereas promoter CGI were enriched in stable pairs as expected (Fig. S1B,C). Notably, CGI associated with known pseudogenes were unlikely to exhibit detected transcript pairs at all, or to overlap enhancer chromatin peaks, consistent with their transcriptional inactivity. Together, the prevalence of enhancer chromatin features and unstable transcripts suggest that many CGI lying near or within known extant transcripts may also exhibit enhancer activity. These data are also robust evidence that ECGI are not simply unannotated promoters, but are in fact enhancers.
DNA hypomethylation is a key feature of promoter CGI, but is also linked to the activity of enhancers.Citation13,14 To assess DNA methylation patterns at ECGI, we used whole-genome bisulfite sequencing (WGBS) data from normal breast tissue (TCGA)Citation28 to compare the average levels of DNA methylation at promoter CGI, ECGI, and enhancers, as defined in HMEC (). We find that, like promoter CGI, active ECGI display minimal DNA methylation (<10%), while classical enhancers exhibit much more variable methylation, typically 50–80%. Because DNA hypomethylation is intrinsically linked to CpG density, we quantified the GC content and CpG density of ECGI (). While ECGI and promoter CGI have similar GC content (median 70% G+C for both, P = 0.45), ECGI are slightly less CpG dense (median 0.82 observed/expected for ECGI vs. median 0.86 for promoter CGI, P = 4.28E-9). However, both CGI classes exhibit far higher GC content and observed/expected CpG density than do classical enhancers.
Given the ways DNA methylation is known to affect histone modifications and other epigenetic features, we next examined the chromatin state of ECGI relative to promoter CGI and other enhancers. Focusing initially on the endogenous chromatin state of features defined in HMEC cells, we found that ECGI tend to have overall higher levels of H3K27Ac than either classical enhancers or promoter CGI (), suggestive of highly active chromatin. Levels of H3K4 methylation have been used to distinguish enhancers from promoters, with enhancers defined as having high levels of H3K4me1, and promoters H3K4me3. However, neither is exclusive as the strongest enhancers exhibit H3K4me3 and it is the ratio of H3K4me3 to H3K4me1 that has been tightly correlated with transcriptional intensity.Citation16 Consistent with this idea, ECGI, like classical enhancers, exhibit substantial H3K4me1 (), which is absent in promoter CGI. Interestingly, ECGI uniquely possess abundant H3K4me2 ( IM), a less-studied mark usually linked to the transition between H3K4me1/3.Citation29,30 ECGI also display modest levels of H3K4me3 () and an intermediate ratio of H3K4me3/H3K4me1 (), much greater than that of classical enhancers, but lower than that of canonical promoter CGI. Together, these data suggest that ECGI display a chromatin state similar to that of the most active enhancers, and distinct from that of promoter CGI.
ECGI are stronger than classical enhancers
The finding that ECGI exhibit a higher H3K4me3/H3K4me1 ratio and less DNA methylation than classical enhancers suggests that they may be more active than classical enhancers. To investigate this relationship, we ascertained the levels of other features often used to gauge enhancer activity: GroSeq tag density, a measure of nascent transcription or eRNA production,Citation31,32 enrichment of H3K27Ac, H3K9Ac, and DNase hypersensitivity, a measure of open chromatin (). We find that ECGI display much stronger enrichment for each of these features of activity than do classical enhancers [ECGI vs. classical enhancers: GroSeq, P = 6.1E-22; H3K27Ac, P = 3.48E-16; H3K9Ac, P = 2.48E-54; DNase hypersensitivity, P = 1.72E-7; Mann-Whitney U].
Figure 2. ECGI are stronger than CpG-poor enhancers (A-D) Nascent transcription and chromatin features associated with open/ active chromatin at ECGI and classical enhancers. Shown is the distribution of tag densities (reads/kb) of nascent transcription (GroSeq, MCF7 cells), active chromatin features (H3K27Ac, H3K9Ac ChIP-seq), or open chromatin (DNase-seq; HMEC cells) across genomic loci in each enhancer class. (E) Promoter CGI, ECGI, and classical enhancers active in the indicated cell type were overlapped with genomic regions called as super enhancers as defined by the super enhancer archive (SEA). Shown is the fraction of genomic loci in each class overlapping any super enhancer in SEA. (F) Distribution of enhancer activity scores, defined by the ratio of GFP reporter mRNA to DNA copy number in lentiMPRA (see Methods).
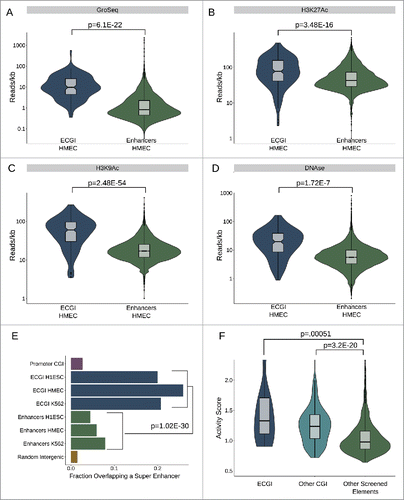
Super enhancers represent a powerful subset of enhancers that exhibit the greatest genomic enrichment of features critical to enhancer function: typically defined by H3K27Ac levels or the degree of binding of transcriptional co-regulators like Mediator and BRD4, among others .33 Utilizing the super enhancer archive (SEA) database,Citation34 we found that ∼20% of ECGI are putative super enhancers, compared with less than 10% of classical enhancers and less than 5% of promoter CGI (combined ECGI vs. enhancers, OR = 3.84, P = 1.02E-30; Fishers exact) ().
The above data suggest that ECGI represent a subset of enhancers distinct from classical enhancers in terms of strength. Thus, we next sought to ascertain the functional enhancer activity of ECGI. Inoue et al.Citation35 recently screened over 2000 putative enhancer elements by cloning them into a GFP enhancer-reporter vector capable of integrating into the genome in an assay known as lentiviral massively parallel reporter assay, in which the strength of such elements is determined by comparing the GFP reporter mRNA levels with the DNA copy number in transfected cells. We compared the activity of ECGI, other CGI, and other putative enhancers tested in the assay, and found that both ECGI (n = 13) and CGI in general (n = 171), exhibited much stronger ability to enhance transcription than other elements (n = 2055) [; ECGI vs. non-CGI elements, P = 0.00051; other CGI vs. non-CGI elements, P = 3.26E-20; ECGI vs. other CGI, P = 0.2]. We also examined two additional functional enhancer screens conducted in mouse cells, FIREWAChCitation36 and CapStarr-seqCitation37 (See Supplemental Data). In both assays, we found that mouse CGI in general, but especially those conserved as human ECGI, exhibit stronger enhancer activity than classical enhancers (Fig. S2). However, human ECGI that have lost their CpG density in the mouse showed a much reduced ability to enhance transcription. These results indicate that while CGI in general can exhibit potent enhancer activity, ECGI in particular are functionally stronger than classical enhancers, dependent upon the conservation of their CpG density.
ECGI are more broadly active than typical enhancers
We next addressed the degree of cell type specificity exhibited by ECGI vs. classical enhancers by comparing the fraction of tested cell lines in which each feature also exhibited marks of active enhancers (H3K4me1/H3K27Ac peaks). We found that the ECGI in each cell line (H1ESC, HMEC, K562) were active in a median of 50–75% of cell lines examined, compared with a median of just 25–30% of typical enhancers (, combined ECGI vs. enhancers P = 6.39E-116; Mann-Whitney U).
Figure 3. ECGI are more widely active than CpG-poor enhancers A) Distribution of the percent of analyzed cell lines (n = 120) in which the loci in each class exhibit enhancer activity (overlap an annotated H3K4me1/H3K27Ac peak in that cell type). B-G) Distribution of the ChIP-seq or DNase-seq tag density (reads/kb) for the indicated chromatin feature as measured in HMEC cells (ENCODE) among ECGI or classical enhancers active in the indicated cell type.
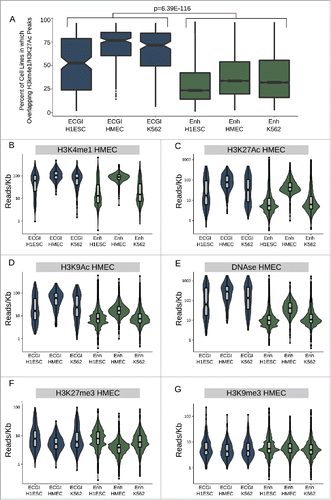
Moreover, ECGI and classical enhancers defined in HMEC exhibit the highest levels of each feature of enhancer activity: H3K4me1, H3K27Ac, H3K9Ac, and DNase hypersensitivity in HMEC cells, as expected (). Yet, H1ESC and K562-derived ECGI also exhibit significant enrichment for each of these features in HMEC cells, consistent with constitutive activity for a substantial proportion. In contrast, classical enhancers from these lines tend to lose these features and appear inactive in HMEC cells, consistent with more cell-type restricted activity. Consistent with this idea, both H1ESC and K562-defined ECGI as well as classical enhancers were enriched in H3K27me3 in HMEC relative to the cell type of origin (), demonstrating Polycomb-mediated repression. However, in contrast to classical enhancers, ECGI were unlikely to be marked by H3K9me3 (), consistent with their DNA hypomethylation. We found similar trends toward cell type restricted activity when comparing the chromatin state of ECGI vs. classical enhancers in H1ESC and K562 cells (Fig. S3). These data indicate that ECGI are more likely than classical enhancers to be active across multiple cell types. When they do undergo silencing, ECGI and classical enhancers both exhibit H3K27me3, but only classical enhancers appear prone to H3K9me3-mediated repression.
ECGI are hubs of genomic contacts
The ability of enhancers to form physical loops with gene promoters is a critical feature of their activity.Citation38 We find that ECGI exhibit greater enrichment than classical enhancers for proteins involved in enhancer/promoter contacts including CTCF,Citation39 Cohesin,Citation40 and BRD4Citation41,42 () (combined P-value for ECGI in all 3 lines vs. classical enhancers in all 3 lines CTCF P = 1.44E-12, Cohesin P = 4.45E-18, BRD4 P = 7.11E-90]. Using K562 Hi-C data, which detects all physical contacts in an unbiased manner,Citation43 we find that ECGI, like promoter CGI, exhibit a much greater contact frequency than classical enhancers do (), regardless of the cell line in which they are active. However, ECGI defined in K562 exhibited the strongest enrichment, indicating modest cell-type specificity (combined ECGI vs. enhancers P = 5.52E-129, K562 ECGI vs. other ECGI P = 2.21E-5; Mann-Whitney U). Similar results were observed in CTCF and RNA Polymerase II ChiAPet data (ENCODE), which exposes only contacts between fragments containing these factors and thus more likely to be of functional relevanceCitation44 [combined CTCF OR = 5.12, P = 1.96E-51, Pol II OR = 4.48, P = 1.55E-56; Fisher's exact; ].
Figure 4. ECGI are hubs for genomic contacts (A) Distribution of the average CTCF (from H1ESC), cohesin/Rad21 (from MCF7 cells), or BRD4 (from MM.1S) ChIP-seq tag densities (reads/kb) among promoter CGI vs. ECGI or classical enhancers active in the indicated cell type. Genomic loci defined as in . (B) Annotated intrachromosomal contacts as defined by Rao et al.Citation42 were extracted from K562 cell Hi-C data, overlapped with the genomic loci in each class, and the average strength of all contacts per locus was determined. Shown is the distribution of the mean intrachromosomal contact strength among promoter CGI vs. ECGI or classical enhancers active in the indicated cell type. (C) The fraction of loci in each genomic class that overlap an annotated contact as determined by ChIA-Pet of CTCF in K562 cells or of RNA polymerase (POLR2A) in MCF7 cells (ENCODE). (D) Median distance from TAD boundaries among genomic loci in each class as called in GM12878 cell Hi-C data.Citation42
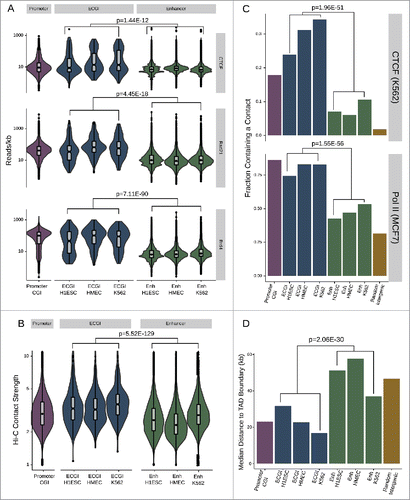
Topologically associated domains (TADs) are large genomic regions within which physical interactions are likely to occur. TADs are vital to gene regulation and nuclear organization, often serving to partition active from inactive chromatin.45 Because CTCF and Cohesin are important to the formation of TADs, we examined the distance to TAD boundaries for ECGI and classical enhancers previously determined in high-resolution Hi-C studies in GM12878 cells.Citation43 We find that ECGI, like promoter CGI, on average lay much closer to TAD boundaries than do classical enhancers (combined ECGI vs. enhancers P = 2.06E-30; Mann Whitney U; ), suggesting a role for ECGI in global nuclear organization, or a requirement for tight regulation of their chromatin state.
ECGI are enriched in GC and CpG-rich transcription factor binding sites
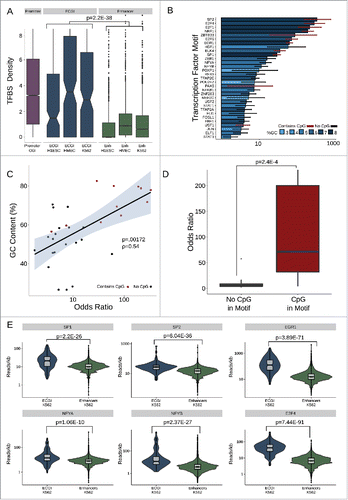
Enhancers are enriched in transcription factor (TF) binding sites and TF binding is correlated with their activity and chromatin state,Citation33,46 leading us to investigate the identity and degree of TF binding at ECGI. The JASPAR database contains approximately 1.1 million annotated binding sites for ∼130 TFs based on binding motif sequence.Citation47 We find that ECGI, like promoter CGI, contain ∼2–4 potential TF binding sites per kilobase, while most classical enhancers had one or no sites (combined P = 2.16E-38; Mann-Whitney U; ). Overall, binding sites for 33 TFs were significantly enriched (OR > 1; P < 0.05) in ECGI relative to classical enhancers (). Strikingly, we find that the top 7 most enriched TF motifs (SP2, E2F4, E2F1, NRF1, ZBTB33, E2F6, and EGR1) had GC contents greater than 50% and contained a CpG site, leading us to quantify the association between these intrinsic features of CGI and TF motif density. Consistent with the high GC content and CpG density of CGI in general, especially relative to that of classical enhancers (), we find that there is a direct correlation between the GC content of a motif and its relative enrichment in ECGI vs. classical enhancers (Pearson correlation ρ = 0.54; P = 0.00172; ). Furthermore, even within a GC-rich context, motifs containing a CpG were much more likely to be enriched in ECGI vs. classical enhancers (median CpG-motif OR = 62.5 vs. median non-CpG-motif OR 5.2, P = 2.5E-4; Mann-Whitney U; ). To determine whether this enrichment of GC/CpG-rich motifs corresponded to the degree of actual TF binding to chromatin in cells, we used ENCODE ChIP-Seq data performed in K562 cells to compare binding of 6 of the top-scoring TFs at K562-specific ECGI and classical enhancers. Binding for each of these factors was strongly enriched at ECGI relative to classical enhancers [SP1, P = 2.18E-26; SP2, P = 6.04E-35; EGR1, P = 3.89E-71; NFYA, P = 1.06E-10; NFYB, P = 2.37E-27; E2F4, P = 7.44E-91; Mann-Whitney U; ], demonstrating that not only are ECGI highly enriched in TF motifs, they are much more likely to be bound by these TF proteins in chromatin relative to classical enhancers.
Figure 5. ECGI are enriched in transcription factor binding (A) Distribution of the density (per kb) of transcription factor binding sites (Jaspar database) among promoter CGI vs. ECGI or classical enhancers active in the indicated cell type. (B) Relative enrichment of each transcription factor binding motif in the cumulative pool of ECGI called in HMEC, K562, and H1ESCs relative to that in all classical enhancers from the same cell lines. Shown is the odds ratio (OR) of enrichment of each motif among ECGI vs. that of classical enhancers, +/−95% confidence interval for each motif for which the ratio was >1 (P<0.05, Fisher's exact). The GC content of each motif is indicated by the blue shaded bar color, and the error bar color indicates whether the motif contains a CpG (red, with CpG; black, without CpG). (C) Relationship between GC content of enriched motifs vs. OR of enrichment. Shown is the linear regression of the relationship, with shadows representing the 95% confidence interval. (D) Distribution of Odds Ratios of enrichment for those enriched motifs that contain or do not contain a CpG site. (E) Distribution of the ChIP-seq tag densities (reads/kb) for representative transcription factors whose motifs are enriched in ECGI among genomic loci classified as ECGI or classical enhancers active in K562 cells.
ECGI are conserved as CpG islands
The concentration of CpG-rich TF motifs in ECGI suggests that their CpG density may be fundamental to their ability to act as enhancers. We thus took two approaches to determine the conservation of ECGI across mammals and vertebrates: examining the frequency with which ECGI also met the UCSC criteria for CGI in other animals, and the phyloPCitation48 conservation scores for individual residues. This analysis revealed that ECGI are typically conserved as CGI throughout placental mammals (), although not to the same extent as promoter CGI, and are rarely conserved as CGI in non-mammalian vertebrates (). Notably, the mouse has significantly fewer CGI than most mammals (n = 16026 vs. human n = 28691 based on UCSC criteria), and they appear to have preferentially lost ECGI, rather than canonical promoter CGI (). CpG dinucleotides in ECGI were likewise selectively conserved both among placental mammals (combined P = 1.46E-65; Mann-Whitney U; ) and among vertebrates (combined P = 3.18E-27; Mann-Whitney U; ) relative to those in classical enhancers, while non-CpG residues exhibited similar mammalian conservation rates in ECGI and classical enhancers (combined P = 0.101) and were even slightly less conserved across vertebrates than were those in classical enhancers (combined P = 2.44E-5). Promoters are known to be more conserved than enhancers in general,Citation49 and promoter CGI have even greater retention of CpG dinucleotides than ECGI do across mammals and vertebrates (), consistent with their higher likelihood of meeting CGI criteria in other animals. This may reflect the biologic role of ECGI as enhancers that serve as adaptable accessories to genes, rather than as promoters critical to the integrity of specific genes.
Figure 6. ECGI CpG density is highly conserved in mammals Genomic coordinates annotated as CGI from each species indicated (UCSC) were lifted over to the human genome (hg19). Shown is the fraction of human CGI in each class that overlapped a CGI in the indicated mammals (A) or other vertebrates (B). Distribution of the average placental mammal (C) or vertebrate (D) phyloP score for CpG dinucleotides (top) or other residues (bottom) among human promoter CGI, ECGI, or classical enhancers active in the indicated cell type. E) Distribution of average ChIP-Seq tag densities (read/kb) for the indicated chromatin feature (CTCF, H3K27me3, H3K9me3) among ECGI, classical enhancers and Remnant CGI as defined in HMEC cells. F) Density of the mean DNA methylation level for ECGI or Remnant CGI as determined from H1ESC whole genome bisulfite sequencing.
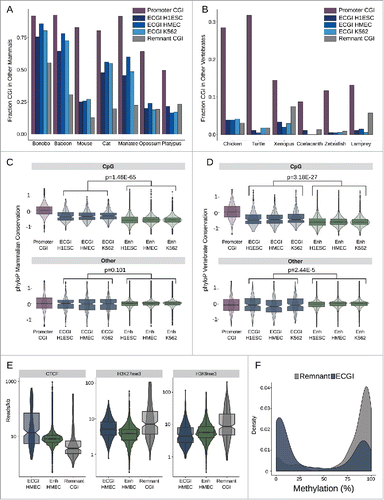
We noted that relative to ECGI, remnant CGI (orphan CGI with no evidence of enhancer activity in any of the analyzed cell lines) are less often conserved as CGI in placental mammals, prompting us to examine their chromatin state (). We find that Remnants are strikingly depleted in CTCF binding, and instead exhibit heterochromatic H3K9me3 and H3K27me3 when compared with ECGI and classical enhancers active in HMEC. Consistent with their relative absence of CTCF binding,Citation50 remnant CGI are almost completely DNA methylated in embryonic stem cells (ENCODE), whereas most ECGI are protected from methylation (). This distinction in conservation between ECGI and remnants disappears in the more distantly related marsupials and monotremes, as well as in non-mammalian vertebrates (), suggesting that ECGI may have diverged functionally early in the evolution of placental mammals, initiating the decay of remnant CGI CpG density coinciding with the loss of selective pressure to remain unmethylated.
Active ECGI are resistant, and inactive ECGI are prone, to methylation changes in cancer
The selective conservation of CpG sites within ECGI relative to other enhancers suggests that, as seen at promoter CGI, there has been a selection against CpG methylation in these regions in the germline across evolutionary time, and that their hypomethylated status is critical to their function. To investigate possible mechanisms underlying the persistent hypomethylation of ECGI, we first examined the levels of 5-hydroxymethylation (5hmC), a known intermediate in passive and active DNA demethylation,Citation51,52 using a 5hmC Capture-Seq data set from IMR90 cellsCitation53 (). Strikingly, we find that ECGI from each cell line examined were uniquely marked by high levels of 5hmC, compared with both promoter CGI and classical enhancers from the same cell line, suggesting active turnover of DNA methylation. Promoter CGI are also suggested to be protected from DNA methylation by R-loops, RNA-DNA hybrids that form co-transcriptionally preferentially at G-skewed and GC-rich regions.Citation2,54,55 Interestingly, ECGI exhibit substantial enrichment for R-loops, approaching that of promoter CGI (). In contrast, classical enhancers exhibit almost no detectable R-loop formation, consistent with the differences in DNA methylation and nascent transcription between these groups (see and ).
Figure 7. Active ECGI are resilient to aberrant methylation in cancer (A) Distribution in tag density (reads/kb) of 5-hydroxymethylcytosine (5hmC) among promoter CGI vs. ECGI or classical enhancers active in the indicated cell type (5hmC Capture-seq data; IMR90 cells). (B) Mean normalized DRIP-seq (primary fibroblasts) tag densities in 10 bp bins for +/−2.5 kb from the center of promoter CGI vs. ECGI and classical enhancers active in HMEC. (C, D) Density of the average methylation level (β) per feature as determined by 450K Methylation Array in normal breast tissue (n = 97) or primary breast tumors (n = 781, TCGA). ECGI active in HMEC vs. inactive in HMEC are those CGI that overlap H3K27Ac/H3K4me1 peak and HMM enhancer definitions in HMEC vs. those called active in at least one other cell line assayed by the same criteria but absent from HMEC. Classical enhancers active vs. inactive in HMEC were similarly defined but do not overlap a CGI. (E) Volcano plot showing the change in the average DNA methylation (β value) per genomic feature among normal or breast tumor samples. Blue features are those ECGI/Enhancers significantly hypomethylated (FDR<0.01 and change in β < −0.2), and red features are those significantly hypermethylated (FDR<0.01 and change in β > 0.2). Active and inactive ECGI vs. classical enhancers are as defined in panels C and D. (F) Relationship between the average DNA methylation (β value) for each ECGI/ Enhancer between normal and breast tumor samples. Lines represent an average change in β of 0.2. Active and inactive ECGI vs. classical enhancers are as defined in panels C and D.
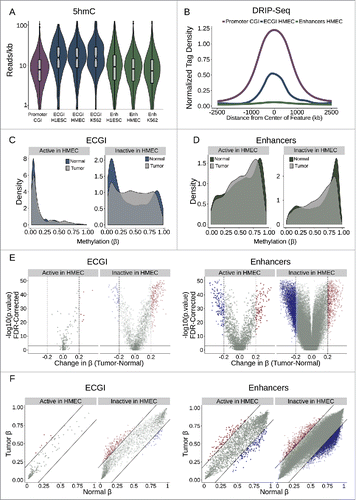
The aberrant hypermethylation of typically unmethylated promoter CGI has been linked to tumor suppressor gene silencing.Citation56 Likewise, alterations in enhancer methylation state have been implicated in tumorigenesis, cancer progression, and metastasis.Citation13,57 To investigate ECGI methylation in human cancers, we used 450K Infinium Methylation array data from 97 normal breast and 781 breast tumor samples (TCGA consortium) to compare the average methylation state of ECGI and classical enhancers active in HMEC vs. those only active in other cell lines. We defined significant methylation changes as those with an FDR <0.01 and an absolute change in β > 0.2.
ECGI active in HMEC were unmethylated in normal tissue and were resistant to methylation changes in primary breast tumors (3.4% hypermethylated, none hypomethylated). While most ECGI inactive in HMEC were also unmethylated, a substantial proportion are methylated in normal breast tissue (26.5% with average β > 0.7). In cancer, these inactive ECGI were prone to hypermethylation, while few undergo hypomethylation (10.3% hypermethylated, 2.1% hypomethylated). These data suggest that persistent ECGI activity is necessary to repel aberrant DNA methylation ( C-F). In contrast, active classical HMEC enhancers exhibit a variable methylation pattern in normal breast tissue (median β = 0.6, ), and are more prone to methylation changes than active ECGI (4.4% hypermethylated, 6.1% hypomethylated in active classical enhancers vs. 3.4% hypermethylated and 0% hypomethylated in active ECGI). As expected, classical enhancers inactive in HMEC tended to be more methylated in normal breast tissue than active ones (median β = 0.72), but unlike inactive ECGI, inactive classical enhancers were more prone to hypomethylation (1.1% hypermethylated, 5.9% hypomethylated) in breast cancers. Thus, ECGI are highly resistant to methylation while active, but a subset of silent ECGI may gain methylation either normally during cell-type specification or aberrantly during carcinogenesis. In contrast, classical enhancers appear less resistant to DNA methylation than ECGI, even if active, which can lead to aberrant hypo- or hyper-methylation in tumors. Together, these data show that the pervasive disparities between classical and ECGI in chromatin state, architecture, and conservation manifest as sweeping differences in DNA methylation dynamics during development and carcinogenesis.
Discussion
Research has largely focused on the canonical role of CGI as strong promoters, even though many ‘orphan’ CGI in the human genome exist far from any known transcript. At promoter CGI, the lack of DNA methylation ensures the preservation of CpG density, the binding of GC and CpG-binding transcription factors, and recruitment of active chromatin modifiers, ultimately creating a permissive environment for transcriptional initiation.Citation1 Indeed, several groups have also documented transcriptional initiation at orphan CGI, typically ascribing it to promoter function.Citation27,58 However, Mendizabal et al.Citation18 recently noted that many CGI resemble enhancers in terms of chromatin state. Indeed, transcriptional initiation is also a key feature of enhancers, and recent work has shown that promoters and enhancers share a common epigenetic architecture, with the strongest enhancers resembling weak promoters in terms of their chromatin state.Citation16 Here, we demonstrate the extension of this promoter-enhancer relationship, establishing that most orphan CGI are in fact putative enhancers, or ECGI.
ECGI resemble classical enhancers in many ways, but possess the elevated GC content, hypomethylation, and CpG density that empower promoter CGI. Like at promoter CGI, these features license recruitment of TFs and chromatin modifiers, but result in enhancer-like H3K4me1 and unstable transcripts, rather than promoter-like H3K4me3 and stable transcripts. Just as promoter CGI are stronger and more euchromatic than other promoters, ECGI display higher eRNA production, histone acetylation, H3K4me3/me1 ratios, and functional ability to drive gene expression than do classical enhancers. The greater strength of ECGI is likely a direct reflection of their importance, and ECGI are also enriched in highly active ‘super’ enhancers that have been assigned pivotal roles in development, pluripotency, and oncogenesis by driving the expression of genes essential to lineage and proliferation regulation.Citation33,59,60
Indeed, enhancer activity may be a feature of CGI in general, not just orphans. Critically, we find that many transcript-associated CGI also frequently overlap putative enhancers and exhibit strictly unstable transcripts. Additionally, in screens of enhancer activity even gene-associated CGI were more capable than other elements of enhancing expression. This suggests a role as enhancers for thousands of additional genic CGI that lack a TSS. Embedded enhancer activity also may be a mechanism by which canonical CGI promoters achieve higher and broader gene expression across cell types than other promoters.Citation1
Nuclear architecture, the formation and location of DNA:DNA contacts, is central to gene regulation, enforcing chromatin boundaries and enabling enhancers to act on promoters.Citation61 ECGI display far more of these contacts that classical enhancers do, and they exhibit far greater enrichment of factors like CTCF, cohesin, and Brd4 that orchestrate these loops, suggesting they engage in more or tighter contacts. CTCF, which regulates recruitment of cohesin, likely prefers ECGI to classical enhancers because it is unable to bind methylated DNA.Citation50,62 As enhancers must contact promoters to act, these loops are likely essential to ECGI activity, and recent work has linked aberrant nuclear disorganization to carcinogenesis.Citation63 Indeed, aberrant methylation of the PTSG2 promoter CGI in cancer cells abolishes CTCF/cohesin binding, silences the gene, and disrupts architecture across the locus.Citation64
CTCF is just one of many methylation-sensitive transcription factors that enhancers rely on for their activity.Citation52 As a whole, TF binding motifs have higher G+C content than the genome average,Citation1 and are generally more common in both promoter CGI and ECGI than in classical enhancers. In chromatin, TFs with GC-rich and CpG-containing motifs accumulate to much greater levels at ECGI than at classical enhancers, and several these play critical roles in carcinogenesis. SP1 overexpression occurs in many cancers and is linked to poor survival,Citation65 ELK4 translocations can drive prostate cancer,Citation66 and EGR1 regulates the survival of endocrine-resistant breast cancer cells,Citation67 for example. Enrichment of these factors suggests that ECGI may play integral roles in carcinogenesis by mediating the ability of TFs to activate their target genes, or by acting as deep sinks to titrate TF pools, an effect which may be exacerbated by cancer-related methylation changes, perturbing TF binding and activity elsewhere.Citation68
The CpG density and hypomethylation that permit TF binding are linked evolutionarily by the mutagenicity of methylated cytosine. The conservation of promoter CGI stems from the specific conservation of CpG sites, rather than other nucleotides, suggesting that there is selective pressure to maintain CpG density and that this is the critical factor in their activity.Citation69 ECGI exhibit the same phenomena, but to a lesser extent than promoter CGI. This likely reflects the diminished H3K4me state of ECGI relative to promoter CGI, and the fact that enhancers evolve more rapidly than promoters.Citation49 Indeed, we find evidence for previously active ECGI in remnant CGI: those CGI that are not transcript-associated and did not exhibit evidence of enhancer activity in any cell line. Unlike ECGI, remnants are heavily methylated and heterochromatic, similar to the promoter CGI associated with pseudogenes, leading to their loss over time. Although some remnants may represent the promoters of lost transcripts, many are likely decommissioned ECGI, given their distance from detectable pseudogenes. This finding highlights that a persistent function, and selection to remain unmethylated, is necessary to maintain CpG density, and suggests that ECGI have had important roles in mammalian evolution.
The conservation of CpG sites and hypomethylation suggest ECGI have mechanisms to repel DNA methylation. The TET enzymes, which catalyze the oxidation of 5-methyl cytosine residues, have been implicated in maintaining enhancer activity by preserving DNA hypomethylation.Citation70 Consistent with this idea, 5hmC is found at especially high levels in super enhancers,Citation71 and DNA methylation preferentially accumulates in enhancer regions in cancers that experience loss of TET2 function.Citation72 We find that 5hmC is also heavily enriched at ECGI, compared with either promoter CGI or other enhancers. H3K4me3 and R-loops inhibit DNA methyltransferase recruitment at promoter CGI, but ECGI may lack full protection because of their lower H3K4me states and R-loop formation. Thus, ECGI may experience higher rates of DNA methylation, eliciting a greater need to remove it. Alternatively, hydroxymethylation may serve a functional role at ECGI independent of its role as an intermediate in 5mC turnover, by either buffering the binding of 5hmC-sensitive TFsCitation73,74 or by specifically recruiting 5hmC readers, several candidates of which have recently been identified.75
It is well established that hypermethylation of promoter CGI often silences tumor suppressor genes during cancer progression, and that hypomethylation of intragenic CGI can unleash certain oncogenes like hTERT.Citation76 More recently, methylation changes at classical enhancers during carcinogenesis have been linked to altered activity and changes in gene expression.Citation57,77 In fact, it has been suggested that enhancers exhibit more DNA methylation changes in cancer than other genomic compartments, and that these changes can modulate the expression of known oncogenes like KIT and ESR1 at a distance.Citation77 Here, we show that ECGI inactive in HMEC cells (but active in another cell type) are especially prone to methylation changes, with more than 10% exhibiting significant hypermethylation in primary breast tumors. Bae et al.Citation17 recently suggested broad hypermethylation of enhancer-like CGI that lack TSS in cancer. However, they did not distinguish intragenic and other transcript-associated CGI in their analysis, which we find often contain stable transcripts (Fig. S1B) making many likely promoters. Indeed, while we do find that more than half of ECGI active in HMEC cells have lost H3K27Ac (and presumably their activity) in MCF7 breast cancer cells, few of these become hypermethylated in primary breast tumors. Furthermore, we also find that dozens of ECGI inactive in HMEC acquire aberrant H3K27Ac in MCF7 cells (Fig. S4). These findings suggest that, rather than indiscriminant hypermethylation, there is frequent decommissioning of active ECGI (often independent of methylation changes) as well as cryptic activation of inactive ECGI during oncogenesis. Given their massive activity and role in genome organization, even modest changes in the unique chromatin state of ECGI may impact cancer progression and survival by directly or indirectly perturbing gene expression.
Thus, we have identified that ECGI represent a novel class of enhancers, more powerful than classical enhancers by every measure and prone to aberrant DNA methylation changes in cancer. These findings point to a common evolutionary origin of CpG-rich promoters and enhancers, with enhancer CGI representing a subset of enhancers that possess many of the features of promoter CGI likely due to similar evolutionary pressures to maintain activity. This helps resolve the longstanding mystery of orphan CGI function and illuminates new aspects of enhancer and CpG island biology critical to understanding the chromatin dynamics that drive development and carcinogenesis.
Methods
CpG island and enhancer annotation
CpG Islands, as defined by UCSC (hg19), were annotated based on their relationship with GenCode (V25, hg19) transcripts, hierarchically as those: 1) containing a protein-coding TSS; 2) contained within a protein-coding gene; 3) within 2 kb of a protein-coding gene (perigenic); or within 2 kb or overlapping 4) a lncRNA; 5) other ncRNA (miRNA, rRNA, scRNA, snRNA, snoRNA, ribozyme, sRNA, antisense RNA, or scaRNA); 6) pseudogenes (including protein-coding or any other). Other CGI were considered to be ‘orphan’ CGI.
For enhancer definitions, cell lines examined as part of the ENCODE and Roadmap Epigenomics project that had available H3K4me1 and H3K27Ac ChIP-Seq called peaks or an HMM chromatin state map were used. ENCODE lines with peak definitions were: A549, astrocytes, CD14+, skin fibroblasts, DND41, GM12878, H1ESC, HCT116, HeLa, HepG2, HMEC, HUVEC, K562, keratinocytes, lung fibroblasts, myotubes, osteoblasts, Panc1, and skeletal myoblasts; and with HMM definitions: GM12878, H1ESC, HepG2, HMEC, HSMM, HUVEC, K562, NHEK, and NHLF.
Roadmap cells with the following EIDs were used for peaks and HMM definition: E003, E004, E005, E006, E007, E008, E011, E012, E013, E014, E015, E016, E017, E019, E020, E021, E022, E026, E029, E032, E034, E037, E038, E039, E040, E041, E042, E043, E044, E045, E046, E047, E048, E049, E050, E055, E056, E058, E059, E061, E062, E063, E065, E066, E067, E068, E069, E071, E072, E073, E074, E075, E076, E078, E079, E080, E084, E085, E087, E089, E090, E091, E092, E093, E094, E095, E096, E097, E098, E099, E100, E101, E102, E103, E104, E105, E106, E108, E109, E111, E112, E113, E114, E115, E116, E117, E118, E119, E120, E121, E122, E123, E124, E125, E126, E127, E128, E129, or just for HMM: E001, E002, E009, E010, E018, E023, E024, E025, E027, E028, E030, E031, E033, E035, E036, E051, E052, E053, E054, E057, E070, E077, E081, E082, E083, E086, E088, E107, E110. For ENCODE HMMs, categories 4–7 (Strong and Weak Enhancers) were considered, and for Roadmap HMMs, categories 6, 7, and 12 were used (Genic Enhancers, Enhancers, Bivalent Enhancers). For peak definitions, enhancers were defined as the overlap of H3K4me1 and H3K27ac peaks within each cell line. Orphan CGI that overlapped enhancers by both peak and HMM definition were used for ECGI analysis in the text. Given the lower resolution of HMM, where enhancers are used as a comparison, they represent regions of overlapping H3K4me1 and H3K27Ac peaks, that at least partially overlap an HMM enhancer.
As a negative control for the transcription stability analysis, 5 thousand random intergenic regions (at least 2 kb from any Gencode transcript) of 500 bp (for similarity to ECGI and enhancers) were chosen. Non-CGI promoters were defined as +/− 500 bp from any protein-coding transcript in Gencode without a CGI within 2 kb of the TSS.
For CGI from other animals, CGI tables were downloaded from UCSC, and lifted to hg19 using chains available from UCSC. Genomes used are as follows: bonobo (Pan paniscus, PanPan1), baboon (Papio anubis, PapAnu2), mouse (Mus musculus, mm9), cat (Felis catus, FelCat5), manatee (Trichechus manatus, TriMan1), opossum (Monodelphis domestica, MonDom5), platypus (Ornithorhynchus anatinus, OrnAna1), chicken (Gallus gallus, GalGal4), alligator (Alligator mississippiensis, AllMis1), turtle (Chrysemys picta, ChrPic1), western clawed frog (Xenopus tropicalis, XenTro3), coelacanth (Latimeria chalumnae, LatCha1), zebrafish (Danio rerio, DanRer10), and lamprey (Petromyzon marinus, PetMar2).
ChIP-Seq and chromatin analyses
ENCODE ChIP-Seq data sets were downloaded as mapped BAM files. Tag densities for genomic features were determined using R/Bioconductor packages GenomicRanges, GenomicAlignments,Citation78 and Rtracklayer,Citation79 and visualized using ggplot2.Citation80 The Mann-Whitney U test is used for significance tests of differences in tag density. Accession numbers are as follows: HMEC H3K4me1 GSM733705, HMEC H3K4me2 GSM733654, HMEC H3K4me3 GSM733712, HMEC H3K27Ac GSM733660, HMEC H3K9Ac GSM733713, HMEC DNase ENCSR000ENV, K562 H3K4me1 GSM733692, K562 H3K27Ac GSM733656, K562 H3K9Ac GSM733778, K562 DNase ENCFF000SVI, K562 H3K27me GSM733658, K562 H3K9me3 GSM733776, H1Esc H3K4me1 GSM733782, H1Esc H3K27Ac GSM733718, H1Esc H3K9Ac GSM733773, H1Esc DNase ENCFF658MCK, H1Esc H3K27me GSM733748, H1Esc H3K9me3 ENCFF769VJB. For data sets for which mapped files were unavailable (MCF7 GroSeq GSE27463), reads were mapped to hg19 using Bowtie2Citation81 with default settings. For DRIP-Seq data (GSE57353), tag densities for the DRIP-Seq library (normal control fibroblasts) were normalized to that of the input library.
DNA methylation analysis
Processed (Tier 3) data for WGBS and Illumina Infinium 450K methylation array data were downloaded from TCGA (ID numbers in Table S1). DNA methylation levels at each feature were calculated by averaging the β values (450K) or percent methylation (WGBS) for each CpG site or probe present in each genomic locus using the GenomicRanges R package.Citation78 The significance of hyper- and hypo-methylation was determined by an FDR-corrected Wilcoxon rank sum test, with a 0.2 change in β used as an additional cutoff for significance.
Transcription factor binding sites
JASPARCitation47 database putative TFBS bed files for hg19 (∼1.1 million binding sites for ∼130 TFs) were downloaded. Motif GC content was determined by averaging the combined G+C likelihood of each residue in the motif. Motifs were considered to have a CpG site if they contained a site with at least a 50% likelihood of containing a C followed by a site with at least a 50% likelihood of containing a G. Odds ratios are the prevalence of a motif occurring within total kb contained by ECGI vs. the prevalence in classical enhancers (see above).
Enhancer screens
For lentiMPRA, a file containing each screened region and its score was downloaded from the supplement of the article.Citation35 In lentiMPRA, putative enhancer regions are cloned into a lentiviral GFP reporter vector that then integrates into the genome. Targeted RNA and DNA sequencing are performed, and the enhancer activity is determined by the ratio of RNA to DNA copy number. We present only the integrase-competent lentiMPRA analysis, but obtained similar results with the integrase-deficient library, although Inoue et al. note that the integrase-competent method is much more robust in detecting enhancers.
For mouse assays, FIREWAChCitation36 and CapStarr-Seq,Citation37 files containing of regions in the input vs. captured libraries (and enhancer scores and categories for CapStarr-Seq) were downloaded from the supplementary materials of each publication. Both assays clone genomic regions into GFP enhancer-reporter vectors with subsequent transfection into cells, but FIREWACh clones putative enhancers upstream of the GFP promoter, while CapStarr-Seq clones the putative enhancers downstream of the reporter, enabling expression of the element in a GFP fusion transcript. FIREWACh relies on the isolation of GFP+ cells, identifying any element able to drive strong GFP reporter transcription, but without further quantification of strength. CapStarr-Seq utilizes targeted RNA sequencing to determine relative enrichment of each screened region (RNA copy number), relative to DNA copy number in the input library (cloned plasmids before transfection) to assign each element an enhancer score [fold change (FC) in input DNA copy number vs. RNA copy number in transfected cells]. Based on this FC, the authors assigned each element to a category: Inactive (FC < 1.5), Weak (FC 1.5–3), or Strong (FC > 3).
Hi-C and ChIA-PET
For Hi-C, intrachromosomal combined contact matrices with scores for each annotated interaction in K562 cells were downloaded from GEO, as were TAD boundary locations in GM12878 (GSE63525). For ChIA-PET, bed files of annotated contacts were downloaded from ENCODE (CTCF in K562 cells ENCSR000CAC, POLR2A in MCF7 cells ENCSR000CAA).
Conservation
PhylopCitation48 conservation score files (bigWig) for mammalian and vertebrate genomes were downloaded from UCSC. For each feature, the average score for CpG residues and non-CpG residues was calculated independently.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1297910_Supplementary_Figures_and_legend.pdf
Download PDF (676 KB)Acknowledgments
We would like to acknowledge Benjamin Barwick for thoughtful editing of the manuscript. We would also like to acknowledge the patients, donors, and scientists of the TCGA, ENCODE, and Roadmap Epigenome projects, without which this project would not be possible.
Funding
This work was supported by NIH grants R01-CA077337 (to PMV) and NIH NRSA pre-doctoral fellowship F31-CA186676 (to JSKB).
Related Research Data
References
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25(10):1010-22; PMID:21576262; https://doi.org/10.1101/gad.2037511
- Kellner WA, Bell JS, Vertino PM. GC skew defines distinct RNA polymerase pause sites in CpG island promoters. Genome Res 2015; 25(11):1600-9; PMID:26275623; https://doi.org/10.1101/gr.189068.114
- Zhang Y, Ng H-H, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 1999; 13(15):1924-35; PMID:21576262; https://doi.org/10.1101/gad.13.15.1924
- Baylln SB, Herman JG, Graff JR, Vertino PM, Issa J-P. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 1997; 72:141-96; PMID:9338076
- Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res 2000; 60(22):6236-42; PMID:11103776
- Issa J, Vertino PM, Boehm CD, Newsham IF, Baylin SB. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Pro Natl Acad Sci U S A 1996; 93(21):11757-62; PMID:8876210; https://doi.org/10.1073/pnas.93.21.11757
- Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999; 59(4):793-97; PMID:10029064
- Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park J-G. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000; 26(1):16-7; PMID:10973239; https://doi.org/10.1038/79120
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997; 57(5):808-11; PMID:9041175
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99(3):247-57; PMID:10555141; https://doi.org/10.1016/S0092-8674(00)81656-6
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007; 130(1):77-88; PMID:17632057; https://doi.org/10.1016/j.cell.2007.05.042
- Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res 1980; 8(7):1499-504; PMID:6253938; https://doi.org/10.1093/nar/8.7.1499
- Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell 2013; 154(1):11-3; PMID:23827668; https://doi.org/10.1016/j.cell.2013.06.018
- Barwick BG, Scharer CD, Bally AP, Boss JM. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol 2016; 17(10):1216-25; PMID:27500631; https://doi.org/10.1038/ni.3519
- Bell JS, Kagey JD, Barwick BG, Dwivedi B, McCabe MT, Kowalski J, Vertino PM. Factors affecting the persistence of drug-induced reprogramming of the cancer methylome. Epigenetics 2016; 11(4):273-87; PMID:27082926; https://doi.org/10.1080/15592294.2016.1158364
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 2014; 46(12):1311-20; PMID:25383968; https://doi.org/10.1038/ng.3142
- Bae MG, Kim JY, Choi JK. Frequent hypermethylation of orphan CpG islands with enhancer activity in cancer. BMC Medical Genomics 2016; 9(1):38; PMID:27534853; https://doi.org/10.1186/s12920-016-0198-1
- Mendizabal I, Soojin VY. Whole-genome bisulfite sequencing maps from multiple human tissues reveal novel CpG islands associated with tissue-specific regulation. Hum Mol Genet 2015:ddv449; PMID:26512062; https://doi.org/10.1093/hmg/ddv449
- Bock C, Walter J, Paulsen M, Lengauer T. CpG island mapping by epigenome prediction. PLoS Comput Biol 2007; 3(6):e110; PMID:17559301; https://doi.org/10.1371/journal.pcbi.0030110
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 2012; 22(9):1775-89; PMID:22955988; https://doi.org/10.1101/gr.132159.111
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012; 22(9):1760-74; PMID:22955987; https://doi.org/10.1101/gr.135350.111
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Pro Natl Acad Sci U S A 2010; 107(50):21931-6; PMID:21106759; https://doi.org/10.1073/pnas.1016071107
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470(7333):279-83; PMID:21160473; https://doi.org/10.1038/nature09692
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489(7414):57-74; PMID:22955616; https://doi.org/10.1038/nature11247
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518(7539):317-30; PMID:25693563; https://doi.org/10.1038/nature14248
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 2012; 9(3):215-6; PMID:22373907; https://doi.org/10.1038/nmeth.1906
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466(7303):253-7; PMID:20613842; https://doi.org/10.1038/nature09165
- Network CGA. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490(7418):61-70; PMID:23000897; https://doi.org/10.1038/nature11412
- Barrero MJ, Sese B, Kuebler B, Bilic J, Boue S, Martí M, Belmonte JCI. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep 2013; 3(4):1005-11; PMID:23545500; https://doi.org/10.1016/j.celrep.2013.02.029
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan G-C, Lian C. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell 2010; 39(2):222-33; PMID:20670891; https://doi.org/10.1016/j.molcel.2010.07.008
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008; 322(5909):1845-8; PMID:19056941; https://doi.org/10.1126/science.1162228
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, Siepel A. Identification of active transcriptional regulatory elements from GRO-seq data. Nat Methods 2015; 12(5):433-8; PMID:25799441; https://doi.org/10.1038/nmeth.3329
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013; 153(2):307-19; PMID:23582322; https://doi.org/10.1016/j.cell.2013.03.035
- Wei Y, Zhang S, Shang S, Zhang B, Li S, Wang X, Wang F, Su J, Wu Q, Liu H. SEA: a super-enhancer archive. Nucleic Acids Res 2016; 44(D1):D172-9; PMID:26578594; https://doi.org/10.1093/nar/gkv1243
- Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, Shendure J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. bioRxiv 2016:061606; PMID:27831498; https://doi.org/10.1101/gr.212092.116
- Murtha M, Tokcaer-Keskin Z, Tang Z, Strino F, Chen X, Wang Y, Xi X, Basilico C, Brown S, Bonneau R. FIREWACh: high-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat Methods 2014; 11(5):559-65; PMID:24658142; https://doi.org/10.1038/nmeth.2885
- Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LT, Fernandez N, Ballester B, Andrau JC, Spicuglia S. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nature communications 2015; 6; PMID:25872643; https://doi.org/10.1038/ncomms7905
- Nolis IK, McKay DJ, Mantouvalou E, Lomvardas S, Merika M, Thanos D. Transcription factors mediate long-range enhancer–promoter interactions. Pro Natl Acad Sci U S A 2009; 106(48):20222-7; PMID:19923429; https://doi.org/10.1073/pnas.0902454106
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CWH, Ye C, Ping JLH, Mulawadi F. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 2011; 43(7):630-38; PMID:21685913; https://doi.org/10.1038/ng.857
- Ong C-T, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 2011; 12(4):283-93; PMID:21358745; https://doi.org/10.1038/nrg2957
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun H-W, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 2014; 21(12):1047-57; PMID:25383670; https://doi.org/10.1038/nsmb.2912
- Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal AK, Rosenfeld MG. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013; 155(7):1581-95; PMID:24360279; https://doi.org/10.1016/j.cell.2013.10.056
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014; 159(7):1665-80; PMID:25497547; https://doi.org/10.1016/j.cell.2014.11.021
- Zhang J, Poh HM, Peh SQ, Sia YY, Li G, Mulawadi FH, Goh Y, Fullwood MJ, Sung W-K, Ruan X. ChIA-PET analysis of transcriptional chromatin interactions. Methods 2012; 58(3):289-99; PMID:22926262; https://doi.org/10.1016/j.ymeth.2012.08.009
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012; 485(7398):376-80; PMID:22495300; https://doi.org/10.1038/nature11082
- Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, Natoli G. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell 2014; 54(5):844-57; PMID:24813947; https://doi.org/10.1016/j.molcel.2014.04.006
- Mathelier A, Fornes O, Arenillas DJ, Chen C-y, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2015:gkv1176; PMID:26531826; https://doi.org/10.1093/nar/gkv1176
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010; 20(1):110-21; PMID:19858363; https://doi.org/10.1101/gr.097857.109
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ. Enhancer evolution across 20 mammalian species. Cell 2015; 160(3):554-66; PMID:25635462; https://doi.org/10.1016/j.cell.2015.01.006
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000; 405(6785):486-9; PMID:10839547; https://doi.org/10.1038/35013106
- Guo JU, Su Y, Zhong C, Ming G-l, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011; 145(3):423-34; PMID:21496894; https://doi.org/10.1016/j.cell.2011.03.022
- Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 2012:gks155; PMID:22362737; https://doi.org/10.1093/nar/gks155
- Wang T, Wu H, Li Y, Szulwach KE, Lin L, Li X, Chen I-P, Goldlust IS, Chamberlain SJ, Dodd A. Subtelomeric hotspots of aberrant 5-hydroxymethylcytosine-mediated epigenetic modifications during reprogramming to pluripotency. Nat Cell Biol 2013; 15(6):700-11; PMID:23685628; https://doi.org/10.1038/ncb2748
- Ginno PA, Lott PL, Christensen HC, Korf I, Chédin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 2012; 45(6):814-25; PMID:22387027; https://doi.org/10.1016/j.molcel.2012.01.017
- Lim YW, Sanz LA, Xu X, Hartono SR, Chédin F. Genome-wide DNA hypomethylation and RNA: DNA hybrid accumulation in Aicardi–Goutieres syndrome. Elife 2015; 4:e08007; PMID:26182405; https://doi.org/10.7554/eLife.08007
- McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res 2009; 15(12):3927-37; PMID:19509173; https://doi.org/10.1158/1078-0432.CCR-08-2784
- Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol 2013; 14(3):1; PMID:23497655; https://doi.org/10.1186/gb-2013-14-3-r21
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet 2010; 6(9):e1001134; PMID:20885785; https://doi.org/10.1371/journal.pgen.1001134
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell 2013; 155(4):934-47; PMID:24119843; https://doi.org/10.1016/j.cell.2013.09.053
- Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, Young RA. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 2015; 58(2):362-70; PMID:25801169; https://doi.org/10.1016/j.molcel.2015.02.014
- Van Bortle K, Corces VG. The role of chromatin insulators in nuclear architecture and genome function. Cur Opin Genet Dev 2013; 23(2):212-8; PMID:23298659; https://doi.org/10.1016/j.gde.2012.11.003
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell 2009; 137(7):1194-211; PMID:19563753; https://doi.org/10.1016/j.cell.2009.06.001
- Taberlay PC, Achinger-Kawecka J, Lun AT, Buske FA, Sabir K, Gould CM, Zotenko E, Bert SA, Giles KA, Bauer DC. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res 2016; 26(6):719-31; PMID:27053337; https://doi.org/10.1101/gr.201517.115
- Kang J, Song S, Yun J, Jeon M, Kim H, Han S, Kim T. Disruption of CTCF/cohesin-mediated high-order chromatin structures by DNA methylation downregulates PTGS2 expression. Oncogene 2015; 34(45):5677-84; PMID:25703332; https://doi.org/10.1038/onc.2015.17
- Beishline K, Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. FEBS J 2015; 282(2):224-58; PMID:25393971; https://doi.org/10.1111/febs.13148
- Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, de la Taille A, Kuefer R, Tewari AK, Setlur SR, Demichelis F. SLC45A3-ELK4 is a novel and frequent erythroblast transformation–specific fusion transcript in prostate cancer. Cancer Res 2009; 69(7):2734-8; PMID:19293179; https://doi.org/10.1158/0008-5472.CAN-08-4926
- Shajahan-Haq AN, Cheema A, Jin L, Boca S, Gusev Y, Bhuvaneshwar K, Demas D, Raghavan K, Madhavan S, Clarke R. Integration of transcriptomic and metabolomic data reveals a central role for EGR1 in regulating survival and cellular metabolism in endocrine-resistant breast cancer. Cancer Res 2016; 76(14 Supplement):1508; PMID:23918603; https://doi.org/10.1158/1538-7445.AM2016-1508
- Brewster RC, Weinert FM, Garcia HG, Song D, Rydenfelt M, Phillips R. The transcription factor titration effect dictates level of gene expression. Cell 2014; 156(6):1312-23; PMID:24612990; https://doi.org/10.1016/j.cell.2014.02.022
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engström PG, Frith MC. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 2006; 38(6):626-35; PMID:16645617; https://doi.org/10.1038/ng1789
- Hon GC, Song C-X, Du T, Jin F, Selvaraj S, Lee AY, Yen C-a, Ye Z, Mao S-Q, Wang B-A. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell 2014; 56(2):286-97; PMID:25263596; https://doi.org/10.1016/j.molcel.2014.08.026
- Johnson KC, Houseman EA, King JE, von Herrmann KM, Fadul CE, Christensen BC. 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat Commun 2016; 7:13177; PMID:27886174; https://doi.org/10.1038/ncomms13177
- Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, Porse BT, Bernard OA, Christensen J, Helin K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev 2015; 29(9):910-22; PMID:23352388; https://doi.org/10.1101/gad.260174.115
- Liu Y, Zhang X, Blumenthal RM, Cheng X. A common mode of recognition for methylated CpG. Trends Biochem Sci 2013; 38(4):177-83; PMID:23352388; https://doi.org/10.1016/j.tibs.2012.12.005
- Wang D, Hashimoto H, Zhang X, Barwick BG, Lonial S, Boise LH, Vertino PM, Cheng X. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res 2016:gkw1184; PMID:27903915; https://doi.org/10.1093/nar/gkw1184
- Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Münzel M, Wagner M, Müller M, Khan F. Dynamic readers for 5-(hydroxy) methylcytosine and its oxidized derivatives. Cell 2013; 152(5):1146-59; PMID:23434322; https://doi.org/10.1016/j.cell.2013.02.004
- Nagarajan RP, Zhang B, Bell RJ, Johnson BE, Olshen AB, Sundaram V, Li D, Graham AE, Diaz A, Fouse SD. Recurrent epimutations activate gene body promoters in primary glioblastoma. Genome Res 2014; 24(5):761-74; PMID:24709822; https://doi.org/10.1101/gr.164707.113
- Bell RE, Golan T, Malcov H, Amar D, Salamon A, Liron T, Sheinboim D, Gelfman S, Gabet Y, Shamir R. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res:gr. 2016:197194.115; PMID:26907635; https://doi.org/10.1101/gr.197194.115
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol 2013; 9(8):e1003118; PMID:23950696; https://doi.org/10.1371/journal.pcbi.1003118
- Lawrence M, Gentleman R, Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 2009; 25(14):1841-2; PMID:19468054; https://doi.org/10.1093/bioinformatics/btp328
- Wickham H. ggplot2: elegant graphics for data analysis. Springer Science & Business Media, 2009; Book. ISBN 978-0-387-98141-3
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9(4):357-9; PMID:22388286; https://doi.org/10.1038/nmeth.1923