
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
DNA methylation (DNAm) is an important epigenetic process involved in the regulation of gene expression. While many studies have identified thousands of loci associated with age, few have differentiated between linear and non-linear DNAm trends with age. Non-linear trends could indicate early- or late-life gene regulatory processes. Using data from the Illumina 450K array on 336 human peripheral blood samples, we identified 21 CpG sites that associated with age (P<1.03E-7) and exhibited changing rates of DNAm change with age (P<1.94E-6). For 2 of these CpG sites (cg07955995 and cg22285878), DNAm increased with age at an increasing rate, indicating that differential DNAm was greatest among elderly individuals. We observed significant replication for both CpG sites (P<5.0E-8) in a second set of peripheral blood samples. In 8 of 9 additional data sets comprising samples of monocytes, T cell subtypes, and brain tissue, we observed a pattern directionally consistent with DNAm increasing with age at an increasing rate, which was nominally significant in the 3 largest data sets (4.3E-15<P<0.039). cg07955995 and cg22285878 reside in the promoter region of KLF14, which encodes a protein involved in immune cell differentiation via the repression of FOXP3. These findings may suggest a possible role for cg07955995 and cg22285878 in immunosenescence.
Introduction
Gene expression is regulated via highly coordinated epigenetic changes, of which the most studied is DNA methylation (DNAm). DNAm is the binding of a methyl group (−CH3) to DNA, which, in mammals, occurs most commonly at a cytosine nucleotide that resides 5′ to a guanine nucleotide, referred to as a CpG site.Citation1 Clusters of CpG-rich regions, known as CpG islands (CGIs), are often found in gene promoters and are typically hypomethylated.Citation2 The position of the CpG site within the transcription unit has a substantial influence on how DNAm impacts expression of the downstream gene,Citation3 and evidence suggests that DNAm in gene promoters can result in gene silencing.Citation4
While patterns of DNAm across the genome vary according to tissue type and environmental exposure, many studies have shown that age explains a substantial portion of the variation in human DNAm.Citation5-13 DNAm has been observed to decrease genome-wide with age,Citation14 although at a regional level the relationship between DNAm and age is much more nuanced. For example, CGIs are associated with an increasing rate of DNAm with age, whereas non-CGIs are associated with a decreasing rate of DNAm with age.Citation7 Furthermore, postnatal DNA is hypomethylated and undergoes a rapid increase in DNAm in early life before stabilizing in adulthood, followed by a gradual decrease later in life.Citation15
While linear trends can provide a useful summary of age-related DNAm patterns, deviations from linearity can provide insight into the relationship between DNAm and senescence. Heretofore, studies investigating non-linear patterns in age-related DNAm have uncovered patterns of rapid DNAm changes in early life, followed by stabilization in later life. For example, differences in the rates of DNAm have been observed between pediatric and adult subjects,Citation13 with many CpG sites showing a decreasing rate of change with age. Such patterns may reflect early-life developmental processes. Perhaps more pertinent to the study of senescence and age-related diseases is an investigation of the reverse pattern: stable DNAm levels in early life followed by rapid methylation/demethylation in late life. To our knowledge, this work is the first to investigate the reverse pattern. Here, we analyze genome-wide DNAm data in a set of peripheral blood samples with the goal of identifying loci for which DNAm increases or decreases at an increasing rate with age. We follow up this analysis in 10 additional data sets comprising distinct tissue types including peripheral blood, dorsolateral prefrontal cortex (dlPFC) tissue, neurons isolated from PFC tissue, glial cells isolated from PFC tissue, and purified blood cell types isolated from peripheral blood, including monocytes, CD4+ T cells, and CD8+ T cells.
Results
Using DNAm data obtained from peripheral blood samples from 336 individuals aged 15–78 from the Grady Trauma Project (GTP, ), we performed separate linear regressions for each of 483,399 CpG sites to model DNAm as a function of age and other covariates, including estimated cell type proportions (Methods). A total of 25,723 CpG sites were significantly associated with age (P<1.03E-7). Subsequently, we tested for non-linear trends in these 25,723 CpG sites by fitting quadrilinear models that included a term for age2. For the subsequent analysis, 21 CpG sites had a Bonferroni-significant age2 term (P<1.94E-6=0.05/25,723). Among these 21 CpG sites (Table S1 and Figure S1), we observed 3 general patterns:
Table 1. Reference information regarding each of the 11 data sets analyzed.
Pattern 1
Two of the 21 CpG sites (cg07955995 and cg22285878 in KLF14) exhibited a low level of DNAm (≤ 0.04) and a low rate of change (slope or ΔDNAm proportion per year = −8.7E-4 and −5.7E-4) at the minimum age of the GTP age range (agemin= 15.9 years) that positively accelerated as age increased (slope at age 70 = 3.1E-3 and 1.7E-3; first row of and ).
Table 2. Statistics corresponding to the age2 term of the regression model fitted separately to each of the 11 data sets for the CpG site, cg07955995 (Equation Equation1(Equation 1)
(Equation 1) ). The regression model includes covariates. Columns 1–3 include abbreviations of the data set, cell/tissue type, and the sample size. Columns 4–6 include the estimated rate of change in DNAm proportion per year at 50, 60, and 70 years of age. Columns 7–10 include the coefficient, standard error, T-statistic, and P-value on the age2 term.
Table 3. Statistics corresponding to the age2 term of the regression model fitted separately to each of the 11 data sets for the CpG site, cg22285878 (Equation Equation1(Equation 1)
(Equation 1) ).
Pattern 2
One CpG site (cg14293223 in GFI1B) exhibited high DNAm at agemin and a small slope that became increasingly negative with age (Table S1, Figure S1). It appeared that the oldest individual, who also had the lowest level of DNAm, was driving the quadrilinear effect between DNAm and age: when the oldest individual was removed from the model, both the age term and the age2 term were no longer significant.
Pattern 3
The remaining 18 CpG sites that were modeled exhibited a positive relationship between DNAm and age at agemin, whose slope decreased with age, becoming negative among older individuals (Table S1, Figure S1).
Patterns 1 and 2 are of particular interest because they indicate stable DNAm in early life, and a more rapidly changing rate of DNAm late in life. To follow up on the 2 CpG sites (cg07955995 and cg22285878) exhibiting Pattern 1 and the one CpG site (cg14293223) exhibiting Pattern 2 in the GTP, we attempted replication in a second set of samples derived from peripheral whole blood from the Take Off Pounds Sensibly (TOPS) Family Study of Epigenetics, whose subjects were members of the TOPS Club. We observed strong replication at cg07955995 (P = 4.3E-15) and cg22285878 (P = 5.0E-8), with extremely similar effect sizes in TOPS and GTP ( in ). We did not find a significant quadrilinear relationship between DNAm and age at the CpG site exhibiting Pattern 2 (cg14111928; P = 0.16).
To investigate whether the quadrilinear patterns observed for cg07955995 and cg22285878 were specific to whole blood, we fit a similar model in 9 additional data sets that were derived from whole blood, subtypes of whole blood cells (monocytes, CD8+ T cells, and CD4+ T cells), and brain [tissue samples from dorsolateral prefrontal cortex (dlPFC) and samples of glial cells and neurons, both isolated from the prefrontal cortex tissue] (). All data sets exhibited increasing DNAm with age at cg07955995 and cg22285878 (Table S2 and S3). In the quadrilinear model, the increasing rate of increase in DNAm with age that was observed in the GTP and TOPS analyses was observed in 7 of 9 data sets for cg07955995 and 8 of 9 data sets for cg22285878 ( and , ). The age2 terms were significant in MESA-M (samples of isolated monocytes), GOLDN (CD4+ T-cells), and CTX (dlPFC) for both CpG sites (4.3E-15<P<0.039; and ). The coefficient on age2 had a positive sign with comparable effect sizes for nearly all data sets, with the exceptions of EGC-PBL at cg07955995 and NICHD-G at both cg07955995 and cg22285878. These exceptions could potentially be attributable to a lack of older individuals in NICHD-G, and a lack of individuals of ages 23–72 years in the EGC study, given the difficulty of identifying non-linear patterns using just 2 extreme endpoints. However, the pattern is consistent overall and the comparable effect sizes suggest that the main difference between data sets with a significant vs. non-significant age2 term is statistical power due to differences in both age range and sample size.
Figure 1. Scatterplots of DNAm (y-axis) vs. age (x-axis) with fitted quadratic regression lines for each of the 11 data sets, holding covariates constant at their mean values.
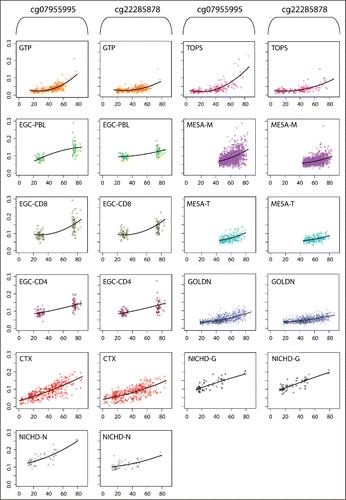
These results show a consistent pattern in age-related DNAm at these 2 CpG sites across white blood cell subtypes. In particular, MESA-M (samples of isolated monocytes) and GOLDN (samples of isolated CD4+ T cells) exhibited evidence of an age-quadratic relationship with DNAm. However, it is well-known that peripheral blood is a heterogeneous tissue, and cell type proportions may vary across samples.Citation16 Analyses of peripheral blood were adjusted for cell type proportion (see Methods), but to further investigate the possible role of cell type in the observed relationships, we examined correlations between age and estimated cell type proportions in the GTP. Of the 6 cell types considered, only CD8+ T cells showed a small but significant correlation with age (r=−0.188, P=0.005; Figure S2). Notably, the 2 oldest individuals (who are also the 2 individuals with the highest DNAm) had similar proportions of CD8+ T cells compared with the remaining individuals (Figure S2). For MESA-M, we investigated whether age associated with the cell type contamination proportions estimated by Reynolds et al. (Figure S3).Citation56 We found small but significant correlations with age with proportions of B cells (r = 0.0584, P=0.0428), natural killer cells (r = 0.1034, P = 0.0003), and T cells (r=−0.0703, P=0.0148). We also plotted cell type contamination proportion against DNAm at both cg07955995 and cg22285878 (Figures S4 and S5). At cg07955995, we observed a small but significant correlation between DNAm and estimated B cell contamination (r=−0.0906, P = 0.0017). At cg22285878, we observed small but significant correlations with estimated proportions of B cells (r=−0.0689, P = 0.0169) and natural killer cells (r = 0.0645, P=0.0254).
Because an increased variation in DNAm among older subjects appeared to be driving the quadrilinear relationship between DNAm and age in GTP, TOPS, MESA-M, and GOLDN, we investigated whether the variance in DNAm differed significantly between younger and older individuals in each data set. The EGC data set consisted solely of 2 age groups: a young age group (≤ 34 years) and an old age group (≥ 73 years). For comparability, we split the GTP, TOPS, CTX, and GOLDN data sets into young (≤ 34 years) and old ≥ 73 years) age groups. MESA-M and MESA-T have age ranges of 45–79 and 44–83, so we could not split these data sets in the same age groups; similarly, NICHD-G and NICHD-N only included 2 individuals ≥ 73 years. For these data sets, we instead used the median values (58 for MESA-M, 60 for MESA-T, and 23 for both NICHD-G and NICHD-N) to split the data sets into younger (≤median) and older (>median) age groups. We then computed the variance of DNAm at cg07955995 and cg22285878 separately for the 2 age groups. and plot the deviation of these variances from the line for all data sets. When comparing all data sets GTP and TOPS had the highest variances among the old age groups at cg07955995. GTP also had the highest variance among the old age group at cg22285878, followed by CTX. When comparing EGC-CD4, EGC-CD8, and EGC-PBL (derived from the same set of subjects) EGC-CD8 exhibited the highest variance in the old age group at cg07955995 while EGC-CD4 exhibited the lowest variance. At cg22285878, EGC-CD4 exhibited the highest variance of the 3 in the old age group whereas PBL exhibited the lowest variance. Across all data sets, variance was significantly greater in the old age groups compared with the young age groups ( and ; ), suggesting that the quadrilinear pattern is in part driven by increased variability in DNAm across older subjects.
Figure 2. Plot of the variance of methylation across subjects at cg07955995 (left) and cg22285878 (right) for the older age group (73 years or older) against the younger age group for each data set. MESA-M, MESA-T, NICHD-G, and NICHD-N appear with different symbols because the age groups were calculated differently than the other data sets. For these 4 data sets, the median ages (in years) were used (58 for MESA-M, 60 for MESA-T, and 23 for both NICHD-G and NICHD-N) to create young groups (≤median) and old groups (>median) because MESA-M and MESA-T had no individuals less than 34 years of age and NICHD-G and NICHD-N only had 2 individuals older than 73 years.
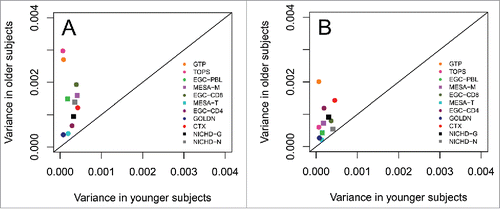
Table 4. Variances of the young age group, variances of the old age group, and P-values corresponding to the F-statistic calculated as the ratio of variances of the young and old age groups for each of the 11 data sets.
Although many of the studies described above had limited numbers of older individuals, this pattern was also observed in an additional blood-based data set (peripheral blood mononuclear cells) that included 122 nonagenarians and 21 younger controls aged 19–30 from the Vitality 90+ Study (Gene Expression Omnibus accession number GSE58888);Citation17 this data set was not analyzed with the others because individual ages were unavailable. Figure S6 shows that for both CpG sites, the nonagenarians in the study had greater mean DNAm levels (P<6E-13) and variance of DNAm (P=8.7e-6 for cg07955995 and 0.0014 for cg22285878) than the younger controls, supporting the results observed in the other data sets.
To test the robustness of the quadrilinear model and ensure we did not miss other interesting non-linear patterns, we also performed a secondary analysis in which we fit spline regressions for the 25,723 age-associated CpG sites in GTP (see Methods for details). Results were similar to those of the quadrilinear model: 20 CpG sites were significant for the spline-based model, whereas 21 CpG sites were significant for the quadrilinear model. Among these, 15 were significant for both models (Figure S7).
As technical validation of the array-based results for cg07955995 and cg22285878, we used the Sequenom EpiTyper technology to interrogate a section of the promoter region that included cg07955995 and cg22285878 for the 2 oldest and 2 of the youngest GTP individuals in the GTP data set (subject ages = 21, 21, 75, and 78). Standard curve analysis based on commercially available methylation standards (Figure S8) suggested that the assay underestimates DNAm at these 2 sites. For cg07955995, DNAm levels from the targeted assay of young vs. old subjects were consistent with the original array-based results, though the difference between older and younger subjects was somewhat compressed (Figure S9). For cg22285878, the EpiTyper DNAm levels show little difference between the older and younger subjects; this may be due to a combination of the smaller differences observed in GTP and other data sets for cg22285878 and the underestimation of DNAm indicated in Figure S8. Notably, increased DNAm for the older subjects was observed at many of the 22 CpG sites in the region, displaying more striking differences at some of the 20 flanking CpG sites than at cg07955995 and cg22285878 (Figure S10).
The 2 CpG sites (cg07955995 and cg22285878) exhibiting Pattern 1 are located 14 base pairs apart in the promoter region of the gene KLF14, and show high pairwise correlation in all data sets (0.70 < r < 0.87; Figure S11). KLF14 is an imprinted, maternally expressed gene with a hypomethylated CpG island.Citation18 Previous work has found associations between DNAm at KLF14 and age in pancreatic islets, adipose tissue, and whole blood.Citation19-22 In addition, KLF14 has been suggested to be involved in the regulation of inflammation.Citation23 To investigate a possible relationship between DNAm and inflammation, we compared interleukin 6 (IL-6) and C-reactive protein (CRP) across GTP subjects (Figure S12 and S13). The 2 oldest individuals, who also exhibited the highest β-values at both cg07955995 and cg22285878, did not have plasma levels of inflammatory markers that differed from the rest of the GTP subjects. There was also no association between levels of either inflammatory marker and DNAm levels at cg07955995 or cg22285878 (0.4972 ≤ P ≤ 0.9780). We also investigated CRP and IL-6 in GOLDN, along with 3 additional inflammatory markers (TNFα, MCP1, and sIL2Rα). No significant associations were found between DNAm and these 5 markers at either CpG site.
Discussion
Many studies have investigated the relationship between DNAm and human age. While significantly different rates of DNAm have been observed between pediatric and adult populations,Citation13 we are unaware of studies investigating changing rates of DNAm with age in the same cohort. Such investigations are important to unravel the complex relationship between DNAm and senescence. In particular, CpG sites demonstrating an increasing rate of DNAm change with age may be involved in processes relevant to aging. To understand the importance of these results, we discuss the tissue and cell type specific patterns we observe, and we outline a possible pathway whereby suppression of KLF14 via DNAm at cg07955995 and cg22285878 may play a role in immunosenescence. In light of previous findings linking KLF14 to metabolic outcomes, we discuss whether our results are consistent with aging-related onset of metabolic disorders, immunosenescence, or both. Finally, we discuss limitations of our study and implications for future research.
While an increased rate of change in DNAm with age suggests relevance to aging-specific processes, it remains unclear what biologic phenomenon might be responsible for such age-related DNAm. Many studies have found not only tissue-specific but also cell type-specific DNAm patterns.Citation24,25 Furthermore, such tissue- and cell type-specific DNAm patterns could be involved in cell lineage differentiation.Citation16 We investigated age-related DNAm patterns among data sets of different tissue and cell types to explore the possibility that the changing rate of DNAm that we observed was specific to a tissue, a cell type, or group of cell types. We found that CpG sites cg07955995 and cg22285878 had DNAm that increased at an increasing rate with age across nearly all of the 11 data sets we investigated. While our investigation is not exhaustive, the patterns so far suggest that our findings generalize across several sub-types of whole blood. It remains to be seen whether this pattern will generalize to other tissues. In PFC tissue (CTX, NICHD-N), the 2 CpG sites exhibited a pattern consistent with the analyses in blood (an increasing rate of increase and larger variance in older subjects). While this pattern was significant in dlPFC tissue (CTX), it was insignificant in PFC neurons (NICHD-N), and was not observed in PFC glial cells (NICHD-G). Future studies should investigate these patterns in additional tissues as additional data sets become available.
The analyses of peripheral whole-blood data (GTP and TOPS) revealed a significant quadrilinear relationship between β-values and age that was consistent with an increasing rate of DNAm change with age. In subsequent analyses, we investigated whether this finding replicated in blood cell subtypes ( and ). Isolated monocytes (MESA-M) also showed a significant quadrilinear relationship between β and age at both cg07955995 and cg22285878. In isolated CD8+ T cells (EGC-CD8), the relationship was not significant, but the coefficient for the age2 term was comparable to those observed in monocytes, suggesting that the age range (no individuals between the ages of 23 and 72 years) and small sample sizes (n = 100) hindered our ability to detect a quadrilinear association. However, the older subjects exhibited significantly greater variance than the younger subjects, consistent with what was observed for other cell subtypes. In isolated CD4+ T cells (EGC-CD4, MESA-T, and GOLDN), we observed quadrilinear associations the same direction with one exception (EGC-CD4 for cg22285878) but the associations were significant in GOLDN only. Like EGC-CD8, EGC-CD4 had a small sample size (n = 99) and was limited to individuals at the endpoints of the age distribution; MESA-T also had a small sample size (n = 214). Effect sizes were comparable across the 3 CD4+ T cell data sets: the coefficients on the quadratic terms were actually larger in MESA-T than in GOLDN for both CpG sites, and the coefficients for EGC-CD4 and GOLDN were similar in magnitude for cg07955995.
Given the patterns observed in T cell subtypes, it is notable that cg07955995 and cg22285878 reside in the promoter region of a gene called KLF14, which is involved in immune cell differentiation, particularly the conversion of CD4+CD25− naïve T cells to CD4+CD25+ regulatory T cells. KLF14 belongs to a family of 17 proteins known as Kruppel-like factor (KLF) proteins, which are involved in immune cell differentiation. KLF proteins exert their effect via the binding to gene promoters and enhancers.Citation23 A recent study found that KLF14 represses FOXP3 via epigenetic regulation at the Treg-specific demethylated region (TSDR) of FOXP3 in vitro and in vivo using a mouse model.Citation23 Previous work has shown that FOXP3 induces and is required for the conversion of CD4+CD25− naïve T cells to CD4+CD25+ regulatory T cells via the joint stimulation of TCR and TGF-β.Citation26-28 A study of CD25-deficient mice found that the overexpression of proinflammatory cytokines, including IL-2 and IFN-γ, in response to bacterial superantigen stimulation, particularly staphylococcal enterotoxin B, was curbed by injection of CD4+CD25+, suggesting that CD4+CD25+ plays a role in regulating antigen-induced inflammatory responses.Citation29 Moreover, evidence suggests a relationship between immunosenescence (the general deterioration of the immune system over the course of the lifespan) and CD4+CD25+ regulatory T cell levels and function.Citation30,31 In consideration of the aforementioned studies and our findings, we outline one possible biologic pathway whereby late-life DNAm at cg07955995 and cg22285878 in CD4+ T cells could be a response to immunosenescence (Figure S14): (1) Age-related DNAm in the promoter region of KLF14 downregulates expression of KLF14; (2) In the absence of normal levels of KLF14, which otherwise inhibits FOXP3, FOXP3 expression is upregulated; (3) Higher levels of FOXP3 induces a greater number of naïve CD4+25− T cells to convert to CD4+25+ T cells (sometimes referred to as Treg cells); and (4) CD4+25+ T cells regulate the heightened inflammation observed in immunosenescent individuals. To investigate the conjectured pathway outlined above, we assessed correlation between DNAm at cg07955995 and cg22285878 and levels of inflammatory markers in GTP and GOLDN individuals. However, there was no association between inflammatory markers and DNAm at cg07955995 and cg22285878 in either GTP (Figures S12 and S13) or GOLDN, and the 2 oldest individuals in the GTP data set, who also exhibited the highest β-values at both cg07955995 and cg22285878, were well within the normal range for plasma levels of IL-6 and CRP. Nevertheless, Pontoux et al. investigated levels of IL-2 and IFN-γ in response to injection of CD4+CD25+.Citation29 Thus, it is possible that we were unable to find any indication of immunosenescence because this particular pathway affects inflammatory markers that we did not investigate. Future studies should collect additional inflammatory markers, including IL-2 and IFN-γ, on a larger subset of elderly individuals to further investigate this putative pathway and its relationship to immunosenescence.
While we suggest that DNAm at cg22285878 and cg07955995 could regulate KLF14 expression and possibly contribute to an immunosenescent phenotype in older adults, we did not investigate expression of KLF14 because it did not show sufficient expression to pass standard quality control procedures in the available blood-based expression data sets (GTP, MESA-M, MESA-T, and a small subset of GOLDN). Because KLF14 did not show expression in these data sets, it is unlikely KLF14 is expressed over long periods of time. Data from the Common Fund (CF) Genotype-Tissue Expression Project (GTEx) indicates that KLF14 expression in whole blood exhibits a very low mean gene expression level with a large upward skew relative to other tissue types (Figure S15), which is what one would expect if a gene is transiently expressed in a tissue.Citation32 Many genes have been found to undergo transient stints of expression.Citation33-35 In fact, one study found transient expression of cell surface proteins in monocytes in response to inflammatory cytokines.Citation36 If KLF14 regulates T cell conversion via intermittent expression followed by long periods of inactivity, then it is unlikely that we would be able to detect KLF14 gene expression in our samples. If this is the case, it is possible that KLF14 is activated in the blood to decrease excessive levels of CD4+CD25+ regulatory T cells. If heightened DNAm at cg22285878 and cg07955995 in immunosenescent individuals regulates KLF14 expression by blocking or partially blocking brief and intermittent periods of KLF14 expression in some CD4+ T cells, then we would expect Treg cells to accumulate in the blood of immunosenescent individuals. Interestingly, previous research has found that Treg cells accumulate with age.Citation30,31 Another possible explanation for lack of detectable levels of KLF14 gene expression is a technical failure of the Illumina probe, which has been suggested in a previous study.Citation44
While aging affects all aspects of the immune system, T cells are the most severely affected.Citation37 While many factors may contribute to an immunosenescent phenotype, one such factor, namely, cytomegalovirus (CMV), infects an estimated 50–80% of North Americans by age 40 and is most commonly symptomatic among immunodeficient individuals, such as the elderly.Citation38 CMV has been observed to induce CD8+ T cell proliferation,Citation39 which has been hypothesized to play a role in age-related DNAm change.Citation40 Moreover, a substantial portion of peripheral blood CD4+ T cell and CD8+ T cell responses has been observed in CMV-seropositive individuals comprising approximately 10% of T cell memory compartments.Citation41 Our investigation found a significant quadrilinear effect between DNAm and age at cg07955995 and cg22285878 in peripheral whole-blood (GTP and TOPS), isolated CD4+ T cells, and, to a lesser (but still significant) extent, in isolated monocytes (MESA-M). These findings suggest that CD4+ T cells could be partially driving the quadrilinear effect, which is consistent with the putative pathway outlined above. In GTP (at cg07955995 and cg22285878) and TOPS (at cg07955995), the oldest individuals had the highest DNAm levels, and in several of the remaining data sets (MESA-M, EGC-PBL, EGC-CD8, EGC-CD4; ), we observed the highest DNAm levels among a subset of the older individuals, which could indicate a possible immunosenescent phenotype in these individuals.
In addition to the possible role for KLF14 in immune cell differentiation and inflammation, previous studies have linked it to metabolic outcomes. Genetic variants in the promoter region of KLF14 associate robustly with risk for type II diabetes (T2D) and high-density lipoprotein cholesterol.Citation42,43 It has also been reported that DNAm of cg22285878 in pancreatic islets associates both with aging and with measures of insulin secretion independent of age.Citation19 GTEx data indicate that KLF14 is expressed in the adrenal gland as well as in both visceral and subcutaneous adipose tissue (Figure S15), and it has been reported that KLF14 acts as a master regulator in adipose tissue, regulating a set of genes correlated with metabolic traits.Citation44 A growing body of literature suggests that inflammation is involved in insulin resistance and chronic metabolic disorders, such as T2D,Citation45-49 so it is possible that the roles of KLF14 in inflammation and in chronic metabolic disorders represent 2 sides of the same coin. The high levels of DNAm at cg07955995 and cg22285878 that we observed among elderly individuals may reflect immunosenescence, onset of a chronic metabolic disorder, or both.
To further our understanding of the role of age-related DNAm at cg07955995 and cg22285878 and its possible relationship with metabolic disorders and immunosenescence, future research should focus on cell type specific data with large sample size and a wide age range. Moreover, our study suggests a possible role of T cell subtypes, specifically, Treg cells (CD4+CD25+ T cells), which are difficult to isolated from other CD4+CD25+ T cells. As we overcome challenges involving the isolation of cell subtypes, particularly T cell subtypes, future studies should investigate DNAm of T cell subtypes to further investigate the biologic pathway conjectured here. In addition, research should pay particular attention to DNAm data on T2D, obese, and immunosenescent individuals, including those who are CMV-seropositive.
Several limitations of this study are worth noting. First, all 11 data sets are cross-sectional, so it is not possible to infer that an increase in age yields an increase in DNAm, merely that there is an association. Ideally, longitudinal data could better assess the relationship in question. Second, the primary analysis was conducted on a data set with a small sample size (n = 336) and few older individuals, as were some of the follow-up analyses (NICHD-N and NICHD-G). These data sets may have lacked the statistical power to detect all CpG sites that exhibit a non-linear trend. A related concern is that the small number of older individuals in these samples could potentially exert undue influence as outliers, which could lead to false positive results. However, the consistency of our initial result in 9 of 11 data sets derived from diverse tissues and the corroboration of observed patterns in a larger data set of nonagenarians suggest that the patterns observed for these 2 CpG sites reflect replicable and generalizable biologic differences and are robust rather than outlier-driven. Third, it is possible that unobserved environmental factors, such as pollutants, accumulate in the body over time and help drive the age-related relationship at these 2 CpG sites. Finally, we were unable to detect KLF14 expression in the available data sets, which limited the possible follow-up analyses. While it is possible that KLF14 is generally not expressed in blood or its subtypes, it is also possible that it is expressed only transiently, since it is unlikely that transient expression of KLF14 would be detected in a small cross-sectional sample. Future studies in other tissues, such as adipose tissue, will be necessary to assess the possible role of these CpG sites in KLF14 expression, though it is notable that a large genome-wide study of methylation and expression in adipose tissue did not report significant associations (FDR<0.01) involving cg07955995, cg22285878, or KLF14.Citation50
Overall, our study is the first to utilize methylome-wide association studies across data sets derived from diverse cell and tissue types to investigate a quadratic relationship between DNAm and age. We found 2 CpG sites that exhibit stable DNAm early in life followed by a rapid increase in DNAm in late life in peripheral whole blood, monocytes, and isolated CD4+ T cells. These CpG sites reside in the promoter region of KLF14, which has recently been shown to be involved in CD4+ T cell differentiation via the suppression of FOXP3. These findings highlight the importance of DNAm as a means to further our understanding of aging, immunology, and biologic pathways.
Methods
Samples
A total of 11 data sets were analyzed (). The primary discovery analysis was performed in data from the Grady Trauma project, and the remaining data sets were analyzed to replicate or follow up findings of the primary analysis.
Grady Trauma Project (GTP)
The primary analysis was performed using a subset of 336 African-American individuals ranging in age from 16 to 78 from data collected as part of the GTP, a study investigating the effects of genetic and environmental factors on individuals' response to stressful life events. Participants were recruited from waiting rooms at Grady Memorial Hospital in Atlanta, GA between 2005 and 2008. The Institutional Review Boards of Emory University School of Medicine and Grady Memorial Hospital approved all procedures of the Grady Trauma Project.Citation51-53
Take Off Pounds Sensibly family study (TOPS)
The TOPS Family Study of Epigenetics included methylation data collected from peripheral blood of 192 individuals. Individual ages ranged from 6 to 85 and each individual belonged to 1 of 7 extended families of Northern European descent. To be included in the study, each nuclear family was required to have 2 obese siblings and at least one parent or sibling who was never obese. The NCBI Gene Expression Omnibus accession number corresponding to the TOPS Family Study is GSE60132.Citation54
Multi-Ethnic Study of Atherosclerosis (MESA)
The Multi-Ethnic Study of Atherosclerosis (MESA) collected methylation data for samples of CD4+ T cells (MESA-CD4; 214 subjects) and monocytes (MESA-M; 1,202 subjects) isolated from peripheral blood. The age range was 45–79 for MESA-CD4 subjects and 44–83 for MESA-M subjects. The MESA study was conducted to collect population-based information on the prevalence and progression of subclinical cardiovascular disease. Subjects were recruited from 6 sites: Baltimore City and Baltimore County, Maryland; Chicago, Illinois; Forsyth County, North Carolina; Los Angeles County, California; New York, New York; and St. Paul, Minnesota. The NCBI Gene Expression Omnibus accession number corresponding to MESA-CD4 and MESA-M are GSE56581 and GSE56046, respectively.Citation55,56
Estonian genome center investigation of age-related epigenetics and immune system function in PBL, CD4+, and CD8+ T cells (EGC-PBL, EGC-CD4, EGC-CD8)
Peripheral blood leukocytes (EGC-PBL) were collected, in addition to CD4+ T cells (EGC-CD4) and CD8+ T cells (EGC-CD8), which were isolated from peripheral blood from healthy donors of the Estonian Genome Center of the University of Tartu. A total of 101 subjects were in the study, and were divided into a young age group and an old age group whose ranges were 22–34 and 73–84, respectively. After quality control, a total of 296 samples were collected from the 101 subjects: 99 CD4+ T cell samples, 100 CD8+ T cell samples, and 97 PBL samples. The NCBI Gene Expression Omnibus accession number corresponding to the Estonian Genome Center Investigation of Age-related epigenetics and immune system function in PBL, CD4+, and CD8+ T cells is GSE59065.Citation40
Schizophrenia-related DNAm and gene expression (CTX)
Samples of dlPFC tissue were surgically removed from post-mortem brain samples donated to the NIMH Brain Tissue Collection at the National Institute of Health in Bethesda, Maryland. Our investigation focused on 346 samples from the control group with an age range of 2–85. The NCBI Gene Expression Omnibus accession number corresponding to the Schizophrenia-related DNAm and gene expression data are GSE74193.Citation57
Genetics Of Lipid Lowering Drugs and Diet Network (GOLDN)
The GOLDN study recruited families that had at least 2 siblings from the National Heart, Lung, and Blood Institute Family Heart Study from Minneapolis and Salt Lake City. The study collected methylation data from CD4+ T cells isolated from peripheral blood samples. After quality control, 991 individuals remained in the analysis. All individuals self-identified as European American and provided written informed consent, and the study protocol was approved by Institutional Review Boards at the University of Minnesota, University of Utah, Tufts University/New England Medical Center, and University of Alabama at Birmingham.
National Institute of Child Health and Human Development (NICHD) brain bank of developmental disorders – cell heterogeneity study (NICHD-G, NICHD-N)
The NICHD study included post-mortem samples from subjects diagnosed with major depressive disorder (MDD) and matched controls. The study collected methylation data from glial cells (n = 50) isolated from prefrontal cortex tissue, in addition to methylation data collected from neurons (n = 50) isolated from prefrontal cortex tissue. The NCBI Gene Expression Omnibus accession number corresponding to NICHD is GSE4182658.
Analysis
For each of the studies described above, DNA was collected from tissue samples and was bisulfite-treated for cytosine to thymine conversion and hybridized to the Illumina Infinium HumanMethylation450 (450K) BeadChip and processed according to the instructions of the manufacturer. For GTP and GOLDN, the methylated signal (M) and unmethylated signal (U) as well as detection P-values were obtained from GenomeStudio after processing. For all other studies, data were downloaded from the NCBI Gene Expression Omnibus (GEO).
For all data sets except GOLDN, the cpg.qc function within the R package CpGassocCitation59 was used to perform quality control. Quality control included the removal of probes with missing data for >5% of samples, removal of samples with >5% missing probe sites, and the removal of samples with a detection P-value > 0.001. Methylated and unmethylated signals were then quantile normalized for each data set separately. A β-value was then computed from the methylated signal (M) and unmethylated signal (U) as follows: . In GOLDN, quality control was performed in R to exclude samples with >1.5% missing data, β-values with a detection P-value >0.01, CpG sites where probe sequences mapped multiple loci or to a location that did not match the annotation file, and CpG sites where >10% of samples had inadequate intensity. In data sets derived from whole-blood samples (GTP and TOPS), cell type proportions were estimated using the Houseman method as implemented in the R package minfi,Citation60,61 which utilizes Illumina 450K methylation data from 6 isolated cell types as reference data.Citation16
For our initial discovery analysis in GTP, the R function cpg.assoc within the CpGassoc packageCitation59 was used to perform a separate linear regression for each CpG site where β was regressed on age, age2, sex, and estimated cell type proportions, and an indicator for row on chip as fixed effects. Chip ID was included as a random effect to absorb potential batch effects. The quadratic term for age was included to allow identification of CpG sites demonstrating an increasing or decreasing rate of change with age. The model used for each of the 11 data sets appears below:(Equation 1)
(Equation 1)
: proportion of cells methylated at a CpG site of the ith individuals
: intercept
: slope coefficient corresponding to the jth independent variable
: number of covariates in the model. Covariates differ between data sets (Table S4).
: between-chip error of the ith individual
: within-chip error of the ith individual
CpG sites were considered to have a significantly increasing rate of methylation change if had the same sign as
and the P-value corresponding to the t-statistic on
was smaller than the Bonferroni-adjusted level of significance (α = 1.03E-07).
In addition to the quadrilinear model described in the preceding paragraph, we also performed a spline analysis. We chose 19 knots corresponding to 19 quantiles across the age-distribution and fit a model using the same fixed effects and random effects in the above paragraph, with the exclusion of the age-quadratic term and the inclusion of an age-knot interaction term. A separate model was fit for each of the 19 knots across each of the 25,723 CpG sites found to be linearly associated with age in the primary analysis. To compare with our quadrilinear results, we plotted the t-statistics corresponding to the knot that maximized the absolute value of the t-statistic on the age-knot interaction term among the 19 knots across each of 25,723 CpG sites against the t-statistics on the age-quadratic term from the quadrilinear model (Figure S7). The minimum P-value was selected among the 19 P-values corresponding to the age-knot interaction term for each of the 25,723 CpG sites in the spline model, and tested against a Bonferroni threshold of 9.5E-8 to determine significance while adjusting for both the 19 knots considered and the 25,723 CpG sites tested.
For CpG sites demonstrating an increasing rate of change with age in GTP, follow-up analyses were performed on the remaining data sets. Only CpG sites with a significant quadratic term were included in the subsequent analyses, which were performed using the R function lme if random effects were included and lm if no random effects were included (with the exception of GOLDN, where the model was fit using SOLARCitation62 to account for familial relationships between subjects). The response variable β was regressed on age and age2 using the model shown in Equation Equation1(Equation 1)
(Equation 1) . Chip ID was included as a random effect for GTP, EGC-PBL, MESA-M, EGC-CD8, MESA-T, EGC-CD4, NICHD-G, NICHD-N. Details on fixed and random effect covariates for all data sets are shown in Table S4. The MESA-T and MESA-M models included estimated measures of contamination by other cell types, namely, proportions of B-cells, monocytes, natural killer cells, and neutrophils, which were provided with GEO data sets GSE56581 and GSE5604656. Likewise, the CTX model included estimated cell type proportions (embryonic stem cells, neural progenitor cells, mature adult neurons, non-neuronal cells, and ES-derived dopamine neurons), which were provided with the GEO data set (GSE74193).Citation57
Based on Equation Equation1(Equation 1)
(Equation 1) , we derived the instantaneous slope between DNAm and age at ages 50, 60, and 70 y using the following computations ( and ):
For comparisons of variance in DNAm between older and younger subjects, GTP, TOPS, CTX, EGC-PBL, EGC-CD4, and EGC-CD8 were each partitioned into an older (≥ 73 years) and a younger group (≤ 34 years). Because the age ranges of MESA-M and MESA-T were 45–79 and 44–83 years, respectively, the younger group in these data sets comprised individuals ≤ the median age (58 for MESA-M and 60 for MESA-T) and the older age group comprised individuals > the median age. We used the median (23 for both NICHD-G and NICHD-N) to separate NICHD-N and NICHD-G into young and old age groups in a similar fashion as MESA because NICHD-G and NICHD-N both only had 2 individuals aged ≥ 73 years. The variances of DNAm at cg07955995 and cg22285878 were computed for the young age group and the old age group and an F statistic was computed as the ratio of these variances ().
As technical validation to address the possibility that artifacts were driving our key results, we performed targeted analysis of samples from the 2 oldest and 2 of the youngest individuals in the GTP cohort. We designed an assay for the promoter region of KLF14 that included 20 additional CpG sites flanking cg07955995 and cg22285878. We used commercially available standards to validate the assay across the range of possible methylation values (0–100%) and generated standard curves of observed vs. expected DNAm at key CpG sites (Figure S8). We then used Sequenom EpiTyper technology to measure DNAm at 22 CpG sites for the 2 older vs. the 2 younger subjects.
Disclosure of potential conflicts of interest
The authors declare no competing financial interests.
Author contributions
N.J. performed all data analyses except those specific to GOLDN, wrote the main manuscript text, and prepared all Figures and Tables. H.W. performed all data analyses in the GOLDN study, and contributed figure panels from these analyses. A.S. contributed to study design and data collection in the GTP and provided a critical read of the manuscript. S.N. and A.S. performed the Sequenom EpiTyper assay for the technical validation of results. D.M.A., D.K.A., and S.A. contributed to study design and data collection in the GOLDN study. S.A. supervised data analyses in the GOLDN study and provided a critical read of the manuscript. K.C. conceived of the study, supervised all data analyses, contributed to study design and data collection in the GTP, and made critical revisions to the manuscript. All authors reviewed and approved the manuscript.
KEPI_A_1314419_s02.docx
Download MS Word (1.2 MB)Acknowledgments
We gratefully acknowledge Drs. Kerry Ressler and Elisabeth Binder for use of data from the Grady Trauma Project. The Grady Trauma Project was supported by the National Institutes of Health Grants MH071537 and MH096764 (Ressler) and MH085806 (A.K.S.). This work was also supported by a NARSAD YI award (A.K.S.) and the Howard Hughes Medical Institute (Ressler). GOLDN was supported by National Institutes of Health grant HL104135 (D.K.A.). We are also very appreciative of Zachary V. Johnson for his help formatting .
Related Research Data
References
- Mendizabal I, Keller TE, Zeng J, Yi SV. Epigenetics and evolution. Integr Comp Biol 2014; 54:31-42; PMID:24838745; https://doi.org/10.1093/icb/icu040
- Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 2013; 14:R102; PMID:24034465; https://doi.org/10.1186/gb-2013-14-9-r102
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33 Suppl:245-54; PMID:12610534; https://doi.org/10.1038/ng1089
- Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; https://doi.org/10.1038/nrg3230
- Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet 2012; 8:e1002629; PMID:22532803; https://doi.org/10.1371/journal.pgen.1002629
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. Jama 2008; 299:2877-83; PMID:18577732; https://doi.org/10.1001/jama.299.24.2877
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:e1000602; PMID:19680444; https://doi.org/10.1371/journal.pgen.1000602
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005; 102:10604-9; PMID:16009939; https://doi.org/10.1073/pnas.0500398102
- Fraga MF, Esteller M. Epigenetics and aging: The targets and the marks. Trends Genet 2007; 23:413-8; PMID:17559965; https://doi.org/10.1016/j.tig.2007.05.008
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013; 14:R115; PMID:24138928; https://doi.org/10.1186/gb-2013-14-10-r115
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 2010; 20:434-9; PMID:20219945; https://doi.org/10.1101/gr.103101.109
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 2010; 20:440-6; PMID:20219944; https://doi.org/10.1101/gr.103606.109
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations. Genome Res 2012; 22:623-32; PMID:22300631; https://doi.org/10.1101/gr.125187.111
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 2009; 130:234-9; PMID:19150625; https://doi.org/10.1016/j.mad.2008.12.003
- Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell 2015; 14:924-32; PMID:25913071; https://doi.org/10.1111/acel.12349
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS One 2012; 7:e41361; PMID:22848472; https://doi.org/10.1371/journal.pone.0041361
- Marttila S, Kananen L, Häyrynen S, Jylhävä J, Nevalainen T, Hervonen A, Jylhä M, Nykter M, Hurme M. Ageing-associated changes in the human DNA methylome: Genomic locations and effects on gene expression. BMC Genomics 2015; 16:179; PMID:25888029; https://doi.org/10.1186/s12864-015-1381-z
- Parker-Katiraee L, Carson AR, Yamada T, Arnaud P, Feil R, Abu-Amero SN, Moore GE, Kaneda M, Perry GH, Stone AC, et al. Identification of the imprinted KLF14 transcription factor undergoing human-specific accelerated evolution. PLoS Genet 2007; 3:e65; PMID:17480121; https://doi.org/10.1371/journal.pgen.0030065
- Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, Gjesing AP, Eiberg H, Tuomi T, Almgren P, et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun 2016; 7:11089; PMID:27029739; https://doi.org/10.1038/ncomms11089
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49:359-67; PMID:23177740; https://doi.org/10.1016/j.molcel.2012.10.016
- Kananen L, Marttila S, Nevalainen T, Jylhava J, Mononen N, Kahonen M, Raitakari OT, Lehtimaki T, Hurme M. Aging-associated DNA methylation changes in middle-aged individuals: The Young Finns study. BMC Genomics 2016; 17:103; PMID:26861258; https://doi.org/10.1186/s12864-016-2421-z
- Rönn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, Jørgensen SW, Brøns C, Jansson P-A, Eriksson K-F, et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet 2015; 24:3792-813; PMID:25861810; https://doi.org/10.1093/hmg/ddv124
- Sarmento OF, Svingen PA, Xiong Y, Xavier RJ, McGovern D, Smyrk TC, Papadakis KA, Urrutia RA, Faubion WA. A novel role for Kruppel-like factor 14 (KLF14) in T-regulatory cell differentiation. Cell Mol Gastroenterol Hepatol 2015; 1:188-202. e4; PMID:25750932; https://doi.org/10.1016/j.jcmgh.2014.12.007
- Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, Koltsina M, Nilsson TK, Vilo J, Salumets A, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol 2014; 15:r54; PMID:24690455; https://doi.org/10.1186/gb-2014-15-4-r54
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013; 153:1134-48; PMID:23664764; https://doi.org/10.1016/j.cell.2013.04.022
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198:1875-86; PMID:14676299; https://doi.org/10.1084/jem.20030152
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330-6; PMID:12612578; https://doi.org/10.1038/ni904
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299:1057-61; PMID:12522256; https://doi.org/10.1126/science.1079490
- Pontoux C, Banz A, Papiernik M. Natural CD4 CD25(+) regulatory T cells control the burst of superantigen-induced cytokine production: The role of IL-10. Int Immunol 2002; 14:233-9; PMID:11809742; https://doi.org/10.1093/intimm/14.2.233
- Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: A mini-review. Gerontology 2014; 60:130-7; PMID:24296590; https://doi.org/10.1159/000355303
- Wang L, Xie Y, Zhu LJ, Chang TT, Mao YQ, Li J. An association between immunosenescence and CD4(+)CD25(+) regulatory T cells: A systematic review. Biomed Environ Sci 2010; 23:327-32; PMID:20934123; https://doi.org/10.1016/S0895-3988(10)60072-4
- GTEx. The genotype-tissue expression (GTEx) project. Nat Genet 2013; 45:580-5; PMID:23715323; https://doi.org/10.1038/ng.2653
- Coussens PM, Jeffers A, Colvin C. Rapid and transient activation of gene expression in peripheral blood mononuclear cells from Johne's disease positive cows exposed to Mycobacterium paratuberculosis in vitro. Microb Pathog 2004; 36:93-108; PMID:14687562; https://doi.org/10.1016/j.micpath.2003.09.007
- Galea E, Reis DJ, Xu H, Feinstein DL. Transient expression of calcium-independent nitric oxide synthase in blood vessels during brain development. FASEB J 1995; 9:1632-7; PMID:8529843
- Neumann-Haefelin T, Wiessner C, Vogel P, Back T, Hossmann KA. Differential expression of the immediate early genes c-fos, c-jun, junB, and NGFI-B in the rat brain following transient forebrain ischemia. J Cereb Blood Flow Metab 1994; 14:206-16; PMID:8113317; https://doi.org/10.1038/jcbfm.1994.27
- Farina C, Theil D, Semlinger B, Hohlfeld R, Meinl E. Distinct responses of monocytes to Toll-like receptor ligands and inflammatory cytokines. Int Immunol 2004; 16:799-809; PMID:15096475; https://doi.org/10.1093/intimm/dxh083
- Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol 2004; 5:133-9; PMID:14749784; https://doi.org/10.1038/ni1033
- Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 2010; 20:202-13; PMID:20564615; https://doi.org/10.1002/rmv.655
- Pawelec G, Akbar A, Beverley P, Caruso C, Derhovanessian E, Fülöp T, Griffiths P, Grubeck-Loebenstein B, Hamprecht K, Jahn G, et al. Immunosenescence and Cytomegalovirus: Where do we stand after a decade? Immun Ageing 2010; 7:13; PMID:20822513; https://doi.org/10.1186/1742-4933-7-13
- Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, Saare M, Vilo J, Metspalu A, Milani L, et al. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep 2015; 5:13107; PMID:26286994; https://doi.org/10.1038/srep13107
- Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 2005; 202:673-85; PMID:16147978; https://doi.org/10.1084/jem.20050882
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical, and population relevance of 95 loci for blood lipids. Nature 2010; 466:707-13; PMID:20686565; https://doi.org/10.1038/nature09270
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010; 42:579-89; PMID:20581827; https://doi.org/10.1038/ng.609
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin SY, Richards HB, Soranzo N, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet 2011; 43:561-4; PMID:21572415; https://doi.org/10.1038/ng.833
- Gutierrez DA, Puglisi MJ, Hasty AH. Impact of increased adipose tissue mass on inflammation, insulin resistance, and dyslipidemia. Curr Diab Rep 2009; 9:26-32; PMID:19192421; https://doi.org/10.1007/s11892-009-0006-9
- Lee J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Arch Pharm Res 2013; 36:208-22; PMID:23397293; https://doi.org/10.1007/s12272-013-0023-8
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010; 72:219-46; PMID:20148674; https://doi.org/10.1146/annurev-physiol-021909-135846
- Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS, Lomenick JP, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A 2012; 109:E1143-52; PMID:22493234; https://doi.org/10.1073/pnas.1200498109
- Yang M, Ren Y, Lin Z, Tang C, Jia Y, Lai Y, Zhou T, Wu S, Liu H, Yang G, et al. Kruppel-like factor 14 increases insulin sensitivity through activation of PI3K/Akt signal pathway. Cell Signal 2015; 27:2201-8; PMID:26226221; https://doi.org/10.1016/j.cellsig.2015.07.019
- Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet 2013; 93:876-90; PMID:24183450; https://doi.org/10.1016/j.ajhg.2013.10.004
- Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, Gapen M, Weiss T, Schwartz AC, Cubells JF, Ressler KJ. Trauma exposure and stress-related disorders in inner city primary care patients. Gen Hosp Psychiatry 2009; 31:505-14; PMID:19892208; https://doi.org/10.1016/j.genhosppsych.2009.05.003
- Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, Klengel T, Mehta D, Binder EB, Epstein MP, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol 2014; 38:231-41; PMID:24478250; https://doi.org/10.1002/gepi.21789
- Zannas AS, Arloth J, Carrillo-Roa T, Iurato S, Röh S, Ressler KJ, Nemeroff CB, Smith AK, Bradley B, Heim C, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: Relevance of glucocorticoid signaling. Genome Biol 2015; 16:1-12; PMID:25583448; https://doi.org/10.1186/s13059-015-0828-5
- Ali O, Cerjak D, Kent JW, Jr., James R, Blangero J, Carless MA, Zhang Y. An epigenetic map of age-associated autosomal loci in northern European families at high risk for the metabolic syndrome. Clin Epigenetics 2015; 7:12; PMID:25806089; https://doi.org/10.1186/s13148-015-0048-6
- Ding J, Reynolds LM, Zeller T, Muller C, Lohman K, Nicklas BJ, Kritchevsky SB, Huang Z, de la Fuente A, Soranzo N, et al. Alterations of a cellular cholesterol metabolism network are a molecular feature of obesity-related type 2 diabetes and cardiovascular disease. Diabetes 2015; 64:3464-74; PMID:26153245; https://doi.org/10.2337/db14-1314
- Reynolds LM, Taylor JR, Ding J, Lohman K, Johnson C, Siscovick D, Burke G, Post W, Shea S, Jacobs DR, Jr., et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat Commun 2014; 5:5366; PMID:25404168; https://doi.org/10.1038/ncomms6366
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 2016; 19:40-7; PMID:26619358; https://doi.org/10.1038/nn.4181
- Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 2013; 8:290-302; PMID:23426267; https://doi.org/10.4161/epi.23924
- Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: An R function for analysis of DNA methylation microarray data. Bioinformatics (Oxford, England) 2012; 28:1280-1; PMID:22451269; https://doi.org/10.1093/bioinformatics/bts124
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: A flexible and comprehensive Bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics (Oxford, England) 2014; 30:1363-9; PMID:24478339; https://doi.org/10.1093/bioinformatics/btu049
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13:1-16; PMID:22214541; https://doi.org/10.1186/1471-2105-13-86
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 1998; 62:1198-211; PMID:9545414; https://doi.org/10.1086/301844
- Irvin MR, Zhi D, Joehanes R, Mendelson M, Aslibekyan S, Claas SA, Thibeault KS, Patel N, Day K, Jones LW, et al. Epigenome-wide association study of fasting blood lipids in the genetics of lipid-lowering drugs and diet network study. Circulation 2014; 130:565-72; PMID:24920721; https://doi.org/10.1161/CIRCULATIONAHA.114.009158