
ABSTRACT
The DNA methylome is re-patterned during discrete phases of vertebrate development. In zebrafish, there are 2 waves of global DNA demethylation and re-methylation: the first occurs before gastrulation when the parental methylome is changed to the zygotic pattern and the second occurs after formation of the embryonic body axis, during organ specification. The occupancy of the histone variant H2A.Z and regions of DNA methylation are generally anti-correlated, and it has been proposed that H2A.Z restricts the boundaries of highly methylated regions. While many studies have described the dynamics of methylome changes during early zebrafish development, the factors involved in establishing the DNA methylation landscape in zebrafish embryos have not been identified. We test the hypothesis that the zebrafish ortholog of H2A.Z (H2afv) restricts DNA methylation during development. We find that, in control embryos, bulk genome methylation decreases after gastrulation, with a nadir at the bud stage, and peaks during mid-somitogenesis; by 24 hours post -fertilization, total DNA methylation levels return to those detected in gastrula. Early zebrafish embryos depleted of H2afv have significantly more bulk DNA methylation during somitogenesis, suggesting that H2afv limits methylation during this stage of development. H2afv deficient embryos are small, with multisystemic abnormalities. Genetic interaction experiments demonstrate that these phenotypes are suppressed by depletion of DNA methyltransferase 1 (Dnmt1). This work demonstrates that H2afv is essential for global DNA methylation reprogramming during early vertebrate development and that embryonic development requires crosstalk between H2afv and Dnmt1.
Introduction
How the epigenome of gametes becomes reprogrammed in zygotes to allow response to signals that regulate cell potency and fate is a central and unanswered question in epigenetics and developmental biology. Epigenome reprogramming in zygotes requires a stripping of the epigenetic marks in gametes that were acquired either during differentiation or in response to environmental cues and a reapplication or redistribution of these marks into a pattern that creates a genome that is competent to support zygotic development. Modulation of epigenetic reprogramming may lead to the retention of acquired epigenetic signatures, which, if sufficiently stable, could be transmitted from one generation to the next.
DNA methylation on cytosines is the primary epigenetic modification of DNA. In most somatic tissue, 5-methylcytosine (5mC) is distributed in blocks of DNA containing CpG dinucleotides, and these methylated blocks of DNA are typically heterochromatic. Other regions, including the CpG islands that characterize most promoters, are protected from methylation.Citation1,Citation2 While most of the methylome is static across cell types of the same species and across developmental time,Citation3-6 different cells have locus-specific differences in methylation levels, and this pattern is essential for embryonic development and developing cell identity.Citation7-9 In most vertebrates, the methylation pattern in the maternal and paternal gametes is erased following fertilizationCitation10-13 and, subsequently, the zygotic methylation pattern is set and re-patterned as cell fate is established.Citation12 However, there are exceptions, as some non-imprinted regions of the genome, such as intracisternal A-particles, can escape demethylation during early development.Citation14 This suggests that mechanisms exist by which regions of the methylome could be impervious to zygotic reprogramming, providing a potential means by which intergenerational epigenetic inheritance might occur.
Recent evidence suggests that DNA methylation is just one of many epigenetic factors that may transmit parental hereditary information, others include histone variants and modifications,Citation15 microRNAs,Citation16 tRNA-derived small RNAs,Citation17,Citation18 and others. As the epigenome is complex and dynamic, it is certain that epigenetic readers, writers, and erasures interact during zygote development so that the epigenome may be shaped by multiple mechanisms. In support of this, studies in somatic tissue and stem cells have shown that crosstalk between histone modifications, histone variants, and DNA methylation contribute to DNA methylation patterning.Citation19 In Arabidopsis thaliana, the location of small RNAs (smRNAs) Citation20 and histone variants Citation21-23 has a direct relationship with DNA methylation homology; however, the conservation of these patterns has not been extensively investigated. Thus, despite intensive study on the mechanisms that control DNA methylome erasure and re-patterning in developing embryos,Citation9 these processes remain poorly understood.
One possibility is that epigenome patterning in the embryo could be dictated by stable epigenetic modifications that serve to bookmark regions of the genome during the period when the parental methylome is erased, so that the de novo methylation machinery could use this bookmark as a guide to regions that should become methylated in the zygotic genome. Epigenetic features that are anti-correlated with DNA methylation could serve as a mark for regions to be protected from methylation. Histone variants are good candidates to serve this function, as some of the best-characterized functions of histone variants are to direct the pattern of epigenetic modifications. In particular, the histone variant H2afva and its paralog, H2afvb (known as H2A.Z in mammalian species) is anti-correlated with DNA methylation in plants and animalsCitation24 and may serve as a mark to restrict where DNA methylation is deposited following methylome erasure.
H2A.Z is present at a less than 1:100 ratio with H2A and becomes incorporated into chromatin in place of H2A at specific loci, which are correlated with active transcription; in addition, it is most often observed at the 5′ regulatory regions of genes.Citation25,Citation26 Therefore, it is proposed that H2A.Z repels repressive epigenetic marks to promote transcription. In mice, H2A.Z is expressed during spermatogenesisCitation27 and in the nuclei of oocytes,Citation28 where it is enriched at transcriptionally active promoters. Genome-wide studies in A. thaliana,Citation29,Citation30 and in pufferfishCitation31,Citation32 have revealed that heavily methylated regions are devoid of H2A.Z, suggesting it marks genomic loci designated to remain methylation-free. However, this concept has been challenged, since genetic elimination of H2A.Z in A. thaliana did not lead to DNA hypermethylation.Citation33 Moreover, another study demonstrating that blocking the DNA methyltransferase (Dnmt1) causes H2A.Z to reposition to promoters,Citation34 suggesting that, in this case, loss of DNA methylation can reorganize histone occupancy, indicating that crosstalk between the methylome and H2A.Z is bidirectional.
We recently hypothesized that, in rat sperm, H2A.Z occupancy may be modulated in response to environmental toxins and serve as an epigenetic “memory” for non-Mendelian paternal transmission of developmental signals in the zygote.Citation35 If the paternal genome retains its H2A.Z patterning in the fertilized zygote, then it can influence developmental reprogramming of DNA methylation. However, we lack direct evidence that zygotic H2afv influences DNA methylation and subsequent embryo development.
Zebrafish provide a tractable system to study the mechanisms of DNA methylome patterning in early embryo development. As in mammals, a wave of global DNA demethylation occurs just after fertilization in zebrafish, and the zygotic genome is re-methylated by 3 h post-fertilization (hpf; ∼1000 cells stage), just before zygotic genome activation.Citation12,Citation36,Citation37 Detailed genome-wide maps of the paternal, maternal, and zygotic methylomes have recently been described and extensively analyzed.Citation5,Citation37-42 One of the findings is that the sperm methylome is resistant to re-patterning compared with the maternal genome.Citation36,Citation37 However, the sperm genome does not seem to dictate where methylation occurs in the embryonic genome, as patterning occurs normally in embryos derived from activated eggs, which contain only the maternal genome.Citation37 During a later stage of development, after the embryonic axes have been established and the nervous system and somite formation is underway (i.e., mid-somitogenesis), a second wave of DNA erasure occurs at ∼13 hpf, with the genome becoming re-methylated by 24 hpf.Citation43-45 The functional significance of global demethylation and re-methylation during somitogenesis has not yet been established and there is nothing known about which factors regulate this stage of methylome patterning. Additionally, although there is evolutionary conservation of histone and DNA methylation crosstalk during the post-fertilization stage of vertebrate embryogenesis,Citation46 experimental evidence demonstrating that the histone code impacts the zygotic DNA methylome is lacking.
H2A.Z is highly conserved between mammals and zebrafish and h2afv mutant zebrafish die at the larval stages of development.Citation47 The relatively late onset of the mutant phenotype is attributed to ample maternally provided supply of h2afv mRNA and protein. Interestingly, H2afva overexpression in transgenic zebrafish is well tolerated,Citation48 suggesting that depletion of this histone variant has the most dramatic impact on the chromatin landscape. The effects of modulating H2afv levels on the zygotic methylome have not been examined and there is no published data on the genomic occupancy pattern of either H2afv in zebrafish embryos. We therefore investigated bulk genome methylation combined with knockdown approaches in zebrafish embryos using morpholino, which allowed for depletion of H2afv derived from maternal and zygotic transcripts. This provides an optimal and straightforward approach to examine effects on DNA methylation during the early stages of development supported by maternal h2afva mRNA. We report that depletion of H2afv from early zebrafish embryos results in a global increase in 5mC levels during methylome reprogramming in mid-somitogenesis, and causes developmental abnormalities including reduced body size and organ specific defects. We demonstrate that knockdown of Dnmt1 rescues the phenotypic defects caused by H2afv deficiency, indicating that crosstalk between the histone code and DNA methylation factors pattern the embryonic methylome and are required for vertebrate development.
Results
H2afv is highly expressed during early zebrafish embryo development
Due to the genome duplication that occurred in teleosts, zebrafish have 2 H2A.Z orthologs, h2afva and h2afvb, which are 99% identical at the protein level and 83% identical at the nucleotide level (Figure S1A). H2a.z null mice die around gastrulationCitation49 but h2afva mutant zebrafish embryos die after 5 d post-fertilization (dpf),Citation47 suggesting that maternal sources of H2afva or compensation from H2afvb support early development of h2afva mutants. Although we were unable to generate primers that accurately distinguished between ha2afv paralogs, RNAseq suggests that h2afvb is expressed at higher expression levels than h2afva in early zebrafish embryos (Figure S1B). Using primers that detect both paralogs, we found high levels of h2afva/h2afvb mRNA throughout development (). Immunofluorescence using an antibody that recognizes a motif found in both paralogs shows high and ubiquitous distribution of H2afv protein from stages preceding zygotic genome activation through mid-somitogenesis (). These data show that there is substantial maternal contribution of h2afv mRNA and protein during the stages of zebrafish development when methylome reprogramming occurs.
Figure 1. H2afv expression successfully depleted by morpholino (MO) injection (A) RT-PCR using primers that detect both h2afva and h2afvb demonstrates high mRNA levels of expression across all stages of embryonic zebrafish development. (B) Whole mount immunofluorescence detects H2afv protein expression across stages of zebrafish embryonic development. Representative images from at least 5 embryos examined from 2 separate clutches are shown. (C) Western blot analysis demonstrates depletion of H2afv expression at 24 hpf in lysates from varying concentrations of h2afv MO injected embryos compared with control (uninjected) embryos. h2afv morphants (0.67 ng) were used for the rest of the study (marked*). Representative image of 2 different blots are shown. (D) Kaplan-Meier curve illustrates increased mortality of h2afv morphants compared with control (uninjected) embryos across 96 h post fertilization (hpf). MO injected at 3.3, 0.67 and 0.34 ng per embryo, n = number of clutches and number of total embryos analyzed per condition in parenthesis.
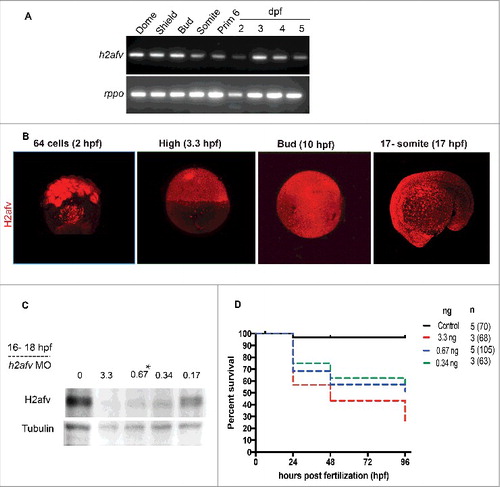
Since the h2afva mutants only show a phenotype well after somitogenesis, when the maternal stores are depleted,Citation47 we reasoned that h2afv zygotic mutants will not be useful for defining the role of H2afv in these processes due to protein derived from maternal mRNA. To address the requirement of H2afv at earlier stages of development, we used morpholino injection to block translation of the maternal and zygotic h2afv mRNA. We identified an h2afv morpholino that specifically targeted the start codon of both variants (Figure S1A) and effectively depleted total H2afv protein at a range of concentrations, with 0.67 ng injected per embryo as the lowest amount that achieved maximal depletion of H2afv protein (). Given that the entire H2afv protein band was depleted using this morpholino, we reasoned either that the morpholino effectively targeted the mRNAs encoding both paralogs or that only one of the paralogs was primarily responsible for generating the abundant amount of H2afv protein present in the embryo at the stages investigated. Given that we cannot distinguish which isoform is primarily responsible for the effects we observe, hereafter we refer to the mRNA and protein using the generic h2afv and H2afv, respectively.
Nearly 70% of embryos injected with 0.67 ng of h2afv morpholino survived to 24 hpf and over 45% survived to 96 hpf (). Since this amount allowed for maximal H2afv depletion and excellent survival through the developmental stages where methylation levels change, 0.67 ng was used for the remainder of our studies.
Transient H2afv depletion has long-lasting developmental impact
h2afv morphants displayed a range of developmental abnormalities, which were first apparent in some embryos during late somitogenesis, but were consistently observed in over 80% of morphants at 24 hpf and were never observed in uninjected embryos or those injected with 0.67 ng of a standard control morpholino (). Embryos were classified as having a mild phenotype if they were less than 80% of the length of control embryos, had fewer somites but retained the mid-brain/hindbrain boundary, and had formed all regions of the head, eye, and body axis and tail. Nearly all mildly affected embryos survived to 72–96 hpf ( and S2); however, they had defects in several organs, including the liver and pancreas (Figure S2A), and reduced stimuli-induced motility at 48 hpf (Figure S2B). Embryos classified as having a severe phenotype were smaller by nearly 50% than age-matched controls and had small, gray heads that were indicative of necrosis, notably reduced height of the hindbrain, and lacked a midbrain/hindbrain boundary. Some severely affected embryos lacked entire body parts, such as the eyes or tails (). Severely affected embryos rarely survived to 5 dpf. Nearly all embryos injected with 0.67 ng h2afv morpholino were abnormal at 24 hpf and 48 hpf, with the majority scored as having a severe phenotype compared with those with the mild phenotype (). The severity of the phenotype was directly correlated with the efficacy of H2afv knockdown, as severely affected embryos had no detectable H2afv at prim 4 stage (24 hpf) and the mildly affected embryos retained some H2afv protein ( and S3). We did not consistently detect a significant number of embryos with a prominent phenotype at somitogenesis stages in h2afv morphants (Figure S4), suggesting that the cause of the h2afv morphant phenotype may have occurred during the somitogenesis stage of development.
Figure 2. H2afv depletion causes significant phenotypic abnormality (A) Bright field images comparing Control (uninjected) and h2afv MO injected fish at 24 and 48 hpf, with the same embryo imaged at both time points. (B) Embryos were scored as Severe, Mild or Normal phenotype in a minimum of 5 clutches and displayed as percentages, with n = the number of total embryos and the number clutches analyzed per condition. (C) Western blot analysis of 24 hpf embryos demonstrates complete depletion of the H2afv protein in morphants exhibiting the severe phenotype and slight residual protein expression in h2afz morphants with mild phenotype while the control (uninjected) embryos express high levels of H2afv protein. (D) Bright field images of 48 hpf embryos injected with h2afv mRNA (50 ng), h2afv MO or h2afv mRNA plus h2afz MO. Phenotypic normality is restored in co-injected embryos. (E) Phenotype percentages of h2afv MO or h2afv mRNA injected with control morpholino-1 or with h2afz morpholino scored at 48 hpf. Scale bar: 1000 μm.
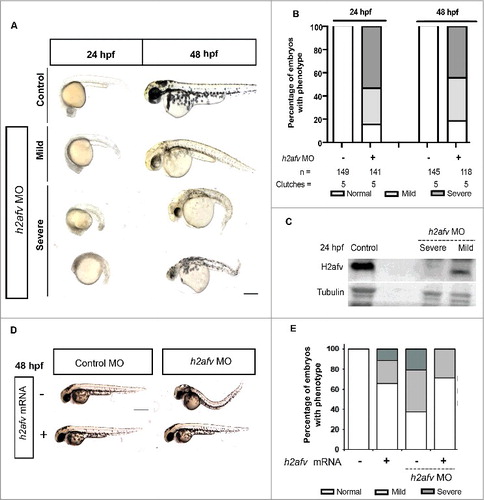
We demonstrated morpholino specificity through several controls. Most importantly, the severity and incidence of the h2afv morphant phenotypes was significantly reduced by co-injection of the h2afv morpholino with mRNA encoding H2afv-mCherry fusion protein, with mutations that prevent binding to the h2afv morpholino (, ). Both imaging (Figure S5A) and Western blotting (Figure S5B) show that the exogenous H2afva-mCherry was expressed in h2afv morphants and while co-injection of h2afva mRNA significantly reduced the percent of morphants with any phenotype, it did not restore expression of endogenous H2afv (Figure S5B). Notably, this shows that the H2afva paralog can compensate for depletion of both paralogs during development and also demonstrates that H2afva overexpression is well tolerated in zebrafish embryos, as has been reported by many other groups.Citation48,50 Additionally, we showed that 2 different control morpholino (standard controls 1 and 2) injected alone at 0.67 ng did not induce any phenotype (, and S6A-B) and, injecting a standard morpholino in combination with the h2afv morpholino to effectively increase the amount of total morpholino in the embryo did not enhance the h2afv morphant phenotype (Figure S6A-B). Finally, co-injecting a p53-targeting morpholino, which has been reported to alleviate some non-specific morpholino toxicity resulting in apoptosis,Citation51 did not alter the h2afv morphant phenotype (Figure S7). We thus conclude that the h2afv morpholino specifically targets h2afv mRNA and reveals an essential role for H2afv during late somitogenesis and embryonic survival.
H2afv is required for DNA methylation during somitogenesis
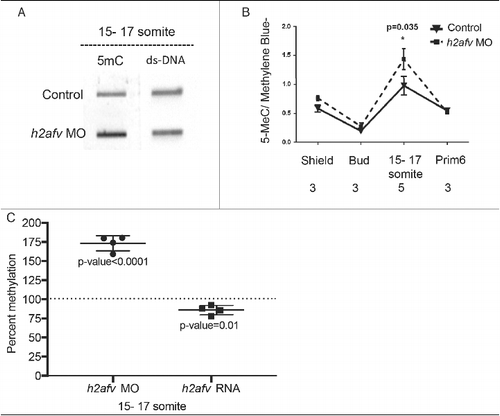
To understand the relationship between H2afv loss and DNA methylation, we analyzed total 5mC levels using genomic DNA slot-blots probed with anti-5mC normalized to total genomic DNA (ds-DNA; ). shows that, in control embryos, global 5mC levels decreased between shield (6 hpf) and bud stage (10 hpf), and then increased during somitogenesis. By prim 5 (24 hpf) stage, 5mC levels were equivalent to those detected at gastrulation. This pattern is consistent with other studies,,Citation44,Citation45,Citation52 and demonstrates that widespread and dynamic changes of DNA methylation occur throughout the stages of development when the axis elongates and the brain and organs are patterned. Strikingly, 5mC levels were higher in h2afv morphants compared with controls at the shield stage (P = 0.078) and mid-somitogenesis stages (P < 0.05; and ). Interestingly, 15–17 somite embryos overexpressing h2afv RNA had the opposite effect, with moderate, but significant, lower 5mC levels compared with controls (85% methylation, compared with a 100% in controls, P = 0.01; ). We therefore conclude that H2afv is required for restricting DNA methylation during development.
Figure 3. Differences in DNA methylation apparent in h2afv morphants at 15-17 somite stage (A) Genomic DNA was isolated from both control (uninjected) embryos h2afv morphants at 15-17 somite stage and levels of 5mC analyzed by slot blot. The morphants exhibit hypermethylation at this stage (n = 2) (B) Slot blot analysis for levels of methylated cytosine (5mC) analyzed at early stages of development. h2afv morphants exhibit hyper methylation at stages analyzed compared with control (standard morpholino control-1) embryos with statistically significant hyper methylation at the 15-17 somite stage corresponding to the complete loss of the H2afv protein in the morphants (C) Bulk DNA methylation of 15-17 somite embryos with h2afv depletion (MO) or over expressing h2afva-GFP RNA. Depletion results in an increase of 73% methylation (P < 0.0001) and overexpression results in a decrease to 85% methylation (P = 0.01), DNA methylation in control is depicted at 100% as a dotted line (n = 4). n = number of clutches.
While extensive analysis of the zebrafish methylome before and during gastrulation and at 24 hpf has been reported,Citation36-38,Citation43 only total 5mC has been examined in the intervening stages.Citation12 We assessed DNA methylation changes affected by H2afv loss using reduced representation bisulfite sequencing (RRBS) to map the CpG methylation state on a global scale. To avoid secondary effects due to phenotypic changes, we collected embryos between 13–15 somite stage, before the onset of any morphological phenotypes induced by H2afv depletion (see Figure S4). For both control and h2afv morphants samples, we obtained approximately 1.5 to 3 million total reads and 0.8 to 1.5 million CpGs that aligned to the zebrafish genome (GRCz10). Of these, 127,945 and 37,362 CpGs had >5X coverage in controls and morphants, respectively. We found that 50.72% of all mapped CpGs with >5X coverage in the controls were considered methylated (i.e., >75% of reads were methylated) compared with 48.73% of CpGs in h2afv morphants, which were scored as methylated.
The genomic distribution of the mapped CpG residues was similar between the controls and h2afv (). We examined the distribution of methylated and non-methylated CpGs across genomic elements in h2afz morphants compared with controls and found that there was a statistically significant difference between each gene element compared with total mapped CpGs methylated and non-methylated CpGs. The majority of methylated CpGs in both control and morphant samples are located in the intergenic region ( and ), which is similar to our previous analysis of methylation distribution in zebrafish tissues using RRBS.Citation3 We found some differences in the distribution of methylated CpGs in h2afv morphants compared with controls: methylated CpGs in h2afv morphants were significantly enriched in intergenic regions (P < 0.05) and introns (P < 0.05) and were depleted from exons (P < 0.05) compared with controls ( and ). CpG dense regions—i.e., CpG islands—, which are found in most promoters,Citation53 are typically protected from methylation.Citation54 There were fewer non-methylated CpGs mapped to promoters in h2afv morphants compared with control embryos (P < 0.05; and ).
Figure 4. Significant increase in gross hypermethylation of promoters in h2afv morphants, with both hyper- and hypo-methylation observed at base pair resolution (A) The number of CpGs sites detected in the RRBS experiment is illustrated by gene elements annotation with different colors. Methylated CpG site is defined as >75% aligned reads at this locus are methylated, while non-methylated CpG site is defined as <25% aligned reads at this locus are not methylated. (B) Common CpGs bases are those detected in both control and h2afv morphants (MO) with minimum coverage 5. Hypermethylated CpGs are higher in h2afv MO and Hypomethylated are lower in h2afv MO, while the difference of methylation at each CpG site is larger that 10%. Different gene elements are marked with color, respectively. * denotes P < 0.05 using the Chi-squared test (C) A hypermethylated region of chromosome 18 in epigenome browser view. In the top panel, methylation difference at common CpG sites between h2afv MO and control, increased methylation in h2afv MO is displayed in blue, and in red for the control. The individual panels below display the overall view of methylation in h2afv MO (in blue) and control (in red). The sequence and annotation are also represented in this panel.
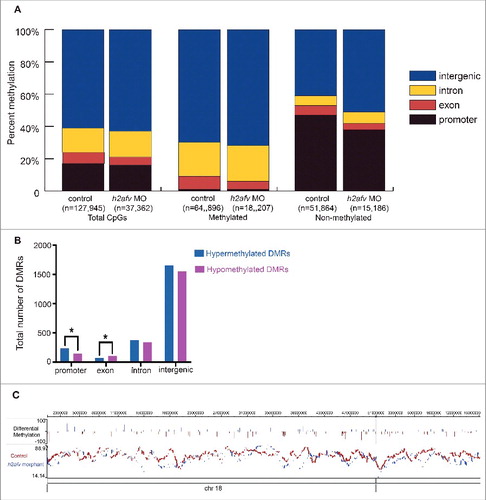
Table 1. Comparison of CpGs analyzed by RRBS and DMRs between control siblings and h2afv morphants at 13-15 somite stage. The methylation status of the CpGs (A) and differentially methylated regions (B) in controls and h2afv morphants. Hyper and hypomethylated DMRs reflect the change in methylation in h2afv morphants compared to controls.
We next identified differentially methylated regions (DMRs) defined as regions of the genome based on the bimodal normal distribution model of CpG distance between controls and morphants and mapped the genomic elements where these DMRs were located. We found a total of 4,462 DMRs, with both increased (hyper DMR) and decreased (hypo DMR) regions of methylation in h2afv morphants ( and ). Most of the DMRs were localized to the intergenic regions, likely reflecting the fact that the majority of the CpGs with sufficiently high coverage in both samples were located in the intergenic region (). Hypermethylated DMRs were significantly enriched in intergenic regions, introns, and promoters. There were a total of 383 DMRs in promoters, and 61% of these were hypermethylated, which indicates that significantly more DMRs are hypermethylated than hypomethylated ( and ; P = 0.0003).
We found the effects of H2afv depletion on methylation to be complex, as in the same region both loss and gain of methylation at distinct CpGs were identified. shows the DMRs across chromosome 18 and Figure S8 illustrates a base pair resolution image of a region of chromosome 14 where the trend is toward hypermethylation in h2afv morphants; however, specific CpGs are also hypomethylated. Together, these data suggest that bulk DNA methylation increases in h2afv morphants and that H2afv loss results in redistribution of 5mC at some loci.
H2afv is typically found mainly in promoters and to some extent in the gene bodies; in A. thaliana,Citation29,Citation30 and puffer fish,Citation31,Citation32 H2A.Z occupancy is inversely correlated with DNA methylation. H2A.Z is enriched in promoters as well as at the enhancer and insulator elements in mammalian cells;Citation55-57 hence, we examined the correlation between methylation and H2afv in zebrafish embryos using previously published data. While there are no available data sets describing H2afv occupancy during the somitogenesis stages of zebrafish development, we analyzed a previously published H2afv ChIP-seq data set from embryos at 30% epiboly (4.5 hpf) embryosCitation58 and found a similar trend with an inverse correlation of H2afv and the pattern of DNA methylation as determined by MeDipSeq data setsCitation59 from embryos at 30% epiboly (4.5 hpf) and prim 5 (24 hpf; Figure S9A). Interestingly, we found that some genes with H2afv peaks in the promoters are expressed early in development based on RNAseq data (Figure S9B), suggesting that H2afv occupancy may be one epigenetic factor that contributes to their expression. We asked whether loss of H2afv affects the expression of the selection of genes (in Figure S8A) whose promoters are associated with H2afv at 30% epiboly (prox1, ta, hoxd3a, irf7, smarcc1b, and sox17)Citation58,Citation60 using real-time PCR at a stage before and after the onset of the h2afv morphant phenotype at 30% epiboly and at 22 hpf. We selected genes that were expressed at low or undetectable levels (sox17, neurod1, hox3a, myod1, ta, spry1, prox1a) and those that were expressed throughout development (prox1a, irf7, snai1a, gapdh) based on RNAseq data deposited in The Expression Atlas (http://www.ebi.ac.uk/gxa/home/) (Figure S9B). Among the H2afv bound genes, prox1, ta, smarcc1b, sox17 (sox17 was neither expressed at 22 hpf in wild-type controls nor in h2afv morphants) were downregulated as early as 70% epiboly; hoxd3a was reduced in expression by 22 hpf, while irf7 expression was increased (Figure S9C, D). Other genes, which are important regulators of key events in early development, including myod1 and spry1, were downregulated by 70% epiboly while, surprisingly, neurod1 was upregulated upon H2afv loss at 22 hpf (Figure S9C, D). While the current analysis does not demonstrate that these genes are directly regulated by H2afv, these data indicate that H2afv loss results in gene expression changes before the onset of the morphological phenotype.
Dnmt1 loss partially rescues the phenotype of H2afv depleted embryos
Dnmt1 is essential in maintaining methylation patterns following DNA replication.Citation61 We therefore performed a genetic interaction experiment to determine whether DNA hypermethylation in h2afv morphants was the mechanism underlying the morphological defects that occurred in these embryos. To do this, we co-injected the h2afv morpholino with another morpholino targeting dnmt1, which we previously demonstrated effectively and transiently knocks down Dnmt1.Citation62 Consistent with our previous study,Citation62 dnmt1 morphants did not exhibit any overt morphological phenotypes at 24 and 48 hpf (). There was a significantly higher percent of embryos scored as severe vs. mild or normal when h2afv morpholino was injected alone (P < 0.0001; ). In 5 out of 5 individual experiments, co-injecting the dnmt1 and h2afv morpholino significantly shifted the phenotype distribution, such that there were more embryos with normal and mild phenotype at 24 and 48 hpf ( and S10). This rescue was not observed when the h2afv morpholino was co-injected with a standard control (Figure S6A-B). dnmt1 morpholino injection did not significantly rescue the mortality in h2afv morphants (). We tested the possibility that Dnmt1 knockdown served to stabilize H2afv protein and this being how the h2afv morphants were rescued, and found that this was not the case, as H2afv was completely depleted in embryos where the h2afv moprholino was injected, with or without dnmt1 morpholino (). Therefore, we conclude that depletion of Dnmt1 suppresses the h2afv morphant phenotype.
Figure 5. Co-injection of dnmt1 MO partially rescues the h2afv morphant phenotype (A) Bright field images comparing embryos injected with control morpholino-2 (0.67 ng) or with h2afv (0.67 ng), dnmt1 (0.43 ng) or dnmt1-h2afv co injected embryos exhibiting the severe phenotype at 24 and 48 hpf. The dnmt1 morphants do not exhibit any significant phenotype either at 24 hpf or 48 hpf (n = 5) (B) Phenotype scored in a minimum of 5 clutches as Severe, Mild or Normal and displayed as percentages show that the percentage of h2afv morphants exhibiting the severe phenotype is significantly reduced in a dnmt MO background. (C) Percent survival of h2afv morphants with and without addition of dnmt1 MO at 24 hpf and 48 hpf. The survival of the h2afv morphants is not significantly affected with knockdown of Dnmt1 protein (p-value between the dead vs. alive numbers of h2afv and dnmt1- h2afv double morphants at 24 hpf = 0.4855 and at 48 hpf = 0.05. (D) Western blot analysis of protein lysates of 16-18 hpf embryos demonstrates complete depletion of the H2afv protein in both h2afv morphants and dnmt1- h2afv morphants while the expression of H2afv protein is unaffected in control and dnmt1 morphant embryos (n = 2). (E) Slot blot analysis of 5-MeC levels using genomic DNA from control, h2afv morphants and dnmt1- h2afv morphants at 14-18 somite stage (16-18 hpf). The h2afv morphants exhibit significant hypermethylation and the inhibition of dnmt1 in this background does not have a significant effect on the global DNA hypermethylation. One way ANOVA was performed comparing each of the morphants with the uninjected controls. Number of clutches (n) = 4. Scale bar: 1000 μm.
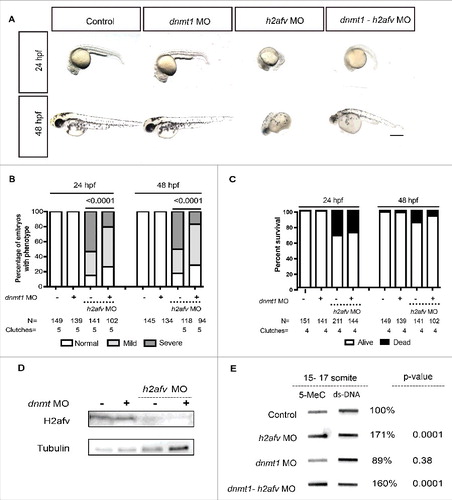
We next examined if Dnmt1 depletion also suppressed bulk DNA hypermethylation in h2afv morphants. We found that dnmt1 morphants displayed slightly but not significantly lower levels of total 5mC in 14–18 somite stage embryos (average 89%; n = 3, P = 0.38). Co-injection with the h2afv morpholino resulted in 5mC levels that were not significantly different to those detected in embryos with h2afv morpholino injection alone (171% vs. 160%; n = 4; ). Hence, knockdown of dnmt1 partially rescued the h2afv morphant phenotype, but was not able to significantly reduce the global hypermethylation caused by H2afv loss. This epistatic analysis demonstrates that dnmt1 is required for the embryonic defects caused by H2afv loss but it is not clear whether this is due to locus specific changes in DNA methylation or to other epigenetic modifications.
Discussion
The zygotic epigenome is substantially re-patterned during early development, but little is known about the dynamics of the methylome during later development or how the pattern of methylation is set in embryos. We report a functional relationship between the histone variant H2afv and DNA methylation, such that transient depletion of H2afv in zebrafish embryos causes DNA hypermethylation during embryonic patterning and organ specification. This suggests that a primary function of H2afv in zebrafish is to restrict DNA methylation, as suggested for other organisms. We conclude that the zebrafish H2A.Z orthologs are essential for epigenetic programing and development of the early vertebrate embryo.
These findings confirm previous studies showing that H2A.Z is critical for normal animal development, as genetic deletion causes early developmental arrest and death across species.Citation49,Citation63 In H2az−/− mice, complete absence of H2A.Z is tolerated in early embryogenesis, indicating no critical requirement for the early differentiation events leading up to formation of the inner cell mass and trophoectoderm.Citation49 However, with the onset of proliferation and the need for complex tissue differentiation from day 4.5 onwards, H2az−/− embryos die. This is markedly different from zebrafish embryos, where h2afva mutants survive early development due to the abundant maternal stores and possibly due to redundancy with the h2afvb paralog. h2afva mutants develop severe abnormalities and die later in development when these stores are depleted or when H2afvb is no longer able to compensate for the loss of H2afva.Citation47 We took advantage of the ability of morpholino to target mRNA of both paralogs, and thus deplete the early embryo of H2afv derived from maternally provided and zygotically derived mRNA generated after genome activation. The technical advantage of this approach is the ability to examine the effects of depleting H2afva and H2afvb from the early embryo, which is not feasible with standard zygotic mutants as these only show a phenotype after the maternally derived product is depleted. Since we achieved partial H2afv knockdown, which was only transient due to the nature of morpholino-mediated translation attenuation, we were able to assess longer-term developmental consequences of transient depletion of H2afv in the post-fertilization embryo. We observed dramatic multi-organ and functional defects at later developmental stages including misshapen organs and motility defects, with many similarities to h2afva mutants.Citation47 Thus, the morphants both recapitulate the mutant phenotype at later stages and also provide an opportunity to interrogate the requirement for H2afv during methylome patterning in early embryos.
Analysis of bulk 5mC showed an anti-correlative relationship between H2afv and DNA methylation, consistent with previous studies.Citation29,64 Moreover, in 3 out of 4 experiments, H2afva overexpression led to a modest reduction of the bulk DNA methylation levels compared with controls. Base pair resolution methylome mapping revealed that there were both hypo- and hyper-methylated CpGs in h2afv morphants; however, more DMRs were hypermethylated, confirming a trend toward increased methylation in morphants. The site-specific hypo- and hyper-methylation in ha2afv morphants differs from the unidirectional gain of methylated CpG sites that was anticipated from previous studies in A. thalianaCitation29 and from our highly reproducible finding by analyzing total 5mC (). Since RRBS sequencing is confined primarily to CpG islands, which are largely unmethylated, the majority of the CpGs detected are unmethylated in both controls and morphants. Moreover, since comparison of distinct CpG sites was restricted to those sites that were sequenced with sufficient depth in both samples, this represented only a small fraction of the total CpGs in the genome. In contrast, blotting for 5mC detects all CpGs in the genome. Therefore, the global differences in methylation levels detected by these 2 approaches are not readily comparable. Nevertheless, these complementary approaches to assessing the methylome point to an essential role for H2afv in restricting DNA methylation, but also indicate that it might redirect DNA methylation to new regions. Further analysis using deeper sequencing and more comprehensive CpG coverage could resolve how H2afv impacts methylome patterning.
The factors that dictate what regions of DNA become methylated and stay methylated during development are not known. Several histone marks and H2afv have been shown to be anti-correlated with DNA methylation, but the functional relationships between these have yet to be defined in developing embryos. High CpG density in promoters is inversely correlated with methylation,Citation65,Citation66 and a major question is how promoters are protected from methylation. Our data demonstrates that although there was some increase in the hypermethylated DMRs in promoter regions, these regions remain largely unmethylated in h2afv morphants. As the RRBS technique has a bias toward CG-rich regions, including CpG islands found in promoters, it is unlikely that this reflects a technical limitation of the DNA methylation sequencing protocol. Instead, our data are consistent with a model whereby several mechanisms collaborate to restrict DNA methylation at CG-rich promoters, including the activities of other histone modifications, such as H3K4me3, which has a strong anti-correlative relationship with DNA methylation.Citation67-69
Data from Arabidopsis has suggested that the anti-correlative nature of H2A.Z and DNA methylation occupancy is based on DNA methylation preventing incorporation of H2A.Z into chromatin, rather than vice versa.Citation33 In support of this, very few changes in promoter methylation were observed following the substantial redistribution of H2A.Z from transcriptional active promoters to gene bodies during mouse B-cell tumorogenesis.Citation64 Treatment of human colon cancer cells with the demethylating drug 5-Aza-deoxycytidine results in genome-wide increased occupancy of H2A.Z and H3K4me3, indicative of DNA methylation having the dominant instructive role in methylome/chromatin remodeling.Citation70 In this same tumor cell model, depletion of SRCAP, an ATP-dependent chromatin remodeler that mediates H2A.Z incorporation into chromatin, had no effect on promoter DNA methylation but did inhibit gene expression.Citation70 Together, this data suggest that lack of DNA methylation in promoters is required for H2A.Z enrichment and, in turn, high rates of transcription. Thus, the major role of H2A.Z at promoters may be to maintain a transcriptionally permissive state of chromatin that is reinforced by the local DNA methylation landscape. H2afv/H2A.Z has structural roles that, when disrupted, may alter genome accessibility to the DNA methylation machinery.Citation63,Citation71 Thus, both the structural properties of H2afv and the interaction with other epigenetic factors may dictate how the rest of the epigenome is patterned.
A widely held assumption is that DNA methylation serves as a global and potent transcriptional repressor; however, much recent data have generated a shift in this dogma surrounding DNA methylation and gene expression. While DNA methylation clearly functions as a major mechanism to repress the expression of transposons, repeat sequences, and the inactive X chromosome, whether DNA methylation directly regulates the expression of differentially expressed genes is intensely debated.Citation72 In fact, only a few genes have been clearly shown to be directly impacted by DNA methylation. In support of this, RNAseq on h2afv morphants collected at the same time when DNA methylation was increased showed virtually no change in gene expression (not shown) and, conversely, we found only modest changes in gene expression in zebrafish mutants with global loss of DNA methylation.Citation73,Citation74 The emerging view is that multiple modifications are required to modulate gene expression. Our finding that some genes with promoter occupancy of H2afv at 30% epiboly are significantly downregulated in h2afv morphants (prox1a, ta, hoxd3a; Figure S8A) suggests that H2afv may be required for their expression. In contrast, irf7, which shows a strong H2afv peak in its promoter, is not, possibly because irf7 is maternally provided and expressed throughout development (Figure 10 or S8). Moreover, some genes that play key roles in development, such as sox17, which were not found to have promoter occupancy of H2afv at 30% epiboly, are affected at the developmental stage when h2afv morphants show a phenotype, which could potentially reflect the disruption of the tissues that typically express high levels of these genes. We conclude that H2afv loss may affect the expression of genes that are highly occupied by H2afv in the promoter, but it is not yet clear whether this change in expression reflects any change in the level of DNA methylation.
In summary, we discovered that the zebrafish orthologs of H2A.Z are required for limiting DNA methylation levels during somitogenesis and are essential for embryonic development after this stage. Moreover, the phenotype caused by H2afv requires Dnmt1, demonstrating that these important epigenetic modifiers work together to ensure proper embryonic development.
Materials and methods
Zebrafish maintenance
Adult zebrafish were maintained on a light dark cycle of 14:10 h at 28°C. Embryos were collected immediately after spawning and incubated at 28°C in fish water [0.6 g/L salt (Crystal Sea Marinemix; Marine Enterprises International, Cat. No. AQ02210] containing methylene blue (0.002 g/L) (MilliporeSigma, Cat. No. M9140). The Icahn School of Medicine at Mount Sinai and the New York University Abu Dhabi Institutional Animal Care and Use Committee approved all protocols. Embryos were staged using standard landmarks and no stage differences in gene expression, phenotypic penetrance, or methylation were observed in stage-matched embryos raised at different temperatures.
Morpholino and mRNA injection
A morpholino was designed targeting the ATG of the h2afv transcript (CTTTTCCTGCTTTGCCTCCTGCCAT) and obtained from Gene Tools (Philomath, OR, USA). Either uninjected embryos or standard control morpholino 1 (CCTCTTACCTCAGTTACAATTTATA) or standard control morpholino 2 (CTCCATCATGAGTGTACATGCACCC) with no known targets in the zebrafish genome served as a control. Morpholino was injected before the 2-cell stage using needles calibrated to inject on average 4 nl per embryo. Final amounts of morpholino were 6.7, 3.3, 0.67, 0.34, and 0.17 ng. Injection of 0.67 ng was identified as optimal. Viable larvae were scored as ‘normal’, ‘mildly affected’—embryos were smaller than uninjected control, exhibited reduced somites, and had delayed development—, or ‘severely affected’—embryos had smaller and gray necrotic heads, reduced distance between the head and yolk, curved body with some missing eyes or even full heads; these embryos usually did not survive beyond 48 hpf. A morpholino targeting the ATG site of dnmt1 (5′-ACAATGAGGTCTTGGTAGGCATTTC-3′) was used as previously reported,Citation62 with no evident abnormal phenotype either at 24 or 48 hpf.
mRNA encoding H2afva tagged with either mCherry or GFP was injected into 1-cell stage embryos in the presence or absence of the h2afz morpholino, according to established protocols,Citation50 and the resulting phenotype and DNA methylation levels were assessed.
SDS-PAGE and immunoblotting
Protein lysates were prepared from embryos snap frozen on dry-ice and collected in lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1% NP-40, 2 mM EDTA, 10% glycerol, and protease inhibitor cocktail AEBSF (Aprotinin, E-64, Bestatin and Leupeptin; Amresco, Cat. No. M221). Samples were homogenized by sonication for 3 × 1 sec pulses. Equal amounts of lysates were mixed with 5X Laemmli buffer and denatured at 95°C for 5 min. Proteins were separated on 12% SDS polyacrylamide gels and transferred onto a nitrocellulose membrane in buffer containing 25 mM Tris, 192 mM glycine, and 20% methanol. Blots were blocked with TBS/Tween 20 (0.1% T-TBS) containing 5% bovine serum albumin (BSA) before overnight incubation with primary antibody in 5% BSA. Primary antibodies used were Histone H2A.Z (1:1000; Cell Signaling Technology, Cat. No. 2718S) and α-Tubulin (1:5000; Abcam, Cat. No. ab27671). Secondary HRP conjugated antibody was used at 1:2000 dilution (anti-rabbit IgG (H+L) HRP conjugate; Promega, USA, Cat. No. W4011). Antibody complexes were detected by chemiluminescence using Pierce ECL Western Blotting Substrate (Thermo Scientific, USA, Cat. No. 32106).
Genomic DNA isolation and slot blot
DNA was isolated from 15–20 embryos, which were homogenized in lysis buffer (10 mM TRIS, 100 mM EDTA, 0.5% SDS) and incubated with 10 ng/ml Proteinase K at 55°C for 2 h. Samples were centrifuged at maximum speed for 5 min and supernatant used for DNA isolation via Phenol/chloroform extraction and ethanol precipitation. The 5mC modification was detected in 600 ng per sample using slot blot analysis as described previously.Citation75 Antibodies used were anti-5mC (1:2000; Eurogentech, Cat. No. BI-MECY), anti-ds-DNA (1:2000; Abcam, Cat. No. ab27156), and rabbit anti-mouse IgG (H+L) HRP conjugate (1:5000; Promega, Cat. No. W4021). Samples blotted in parallel were stained with either 0.2% Methylene blue in 0.3 M NaOAc or anti-ds-DNA primary antibody (1:2000; Abcam, Cat. No. ab27156) to detect total DNA. Antibody complexes were detected by chemiluminescence using Pierce ECL Western Blotting Substrate and the Gel Analyzer was used to quantify 5mC, ds-DNA; methylene blue intensity and 5mC levels were determined by normalizing to ds-DNA or methylene blue.
Reduced representation bisulfite sequencing and methylation analysis
The NEBNext Ultra DNA Library Prep Kit (New England Biolabs, Cat. No. E7370S) for Illumina platform was used for bisulfite sequencing library prep. Isolated genomic DNA was digested overnight with MspI (New England Biolabs, Cat. No. R0106S), followed by end-repair and ligation of methylated adaptors (New England Biolabs, Cat. No. E7350). Bisulfite conversion was performed using the EZ DNA methylation Gold Kit (Zymo Research, Cat. No. 5005) following manufacturer's instructions, then bisulfite-converted libraries were amplified by PCR. These amplified libraries were sequenced on Illumina Miseq platform. Image capture, analysis, and base calling were performed using Illumina's CASAVA 1.8. RRBS reads were mapped on the zebrafish genome (GRCz10) using Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/) integrated with bowtie2 aligner. To increase CpG coverage of genome, 2 biologic replicates were integrated. All CpGs sites covered by at least 5 reads were kept for further differential methylation analysis, which was conducted by R (http://www.r-project.org/) using the package methylKit.Citation76 DMRs were further detected by the weighted optimization algorithm eDMR.Citation77
Whole-mount immunofluorescence
Embryos were staged based on time post-fertilization as well as the somite number. After collection, the embryos were washed in 1X Phosphate Buffered Saline (PBS) and fixed for 3–4 h in 4% paraformaldehyde (PFA), washed and stored in 1X PBS at 4°C. Fixed embryos were permeabilized in ice-cold acetone for 10 min followed by 20 μg/ml proteinase K in 1% Triton-X PBS for 10 min. Following 3 washes with 1X PBS, embryos were blocked with 5% BSA for 1 h before incubation with primary antibodies in the blocking solution overnight at 4°C. Binding of primary antibodies was detected using the appropriate secondary antibodies (Alexa Fluor 546 goat anti-rabbit IgG, Alexa Fluor 488 donkey anti-mouse IgG; Invitrogen). Negative controls consisted of permeabilized embryos incubated in the blocking buffer with no primary antibodies (not shown). Samples were counterstained with Hoechst stain (1:1000; Thermo Fisher) and photomicrographs were captured on an Olympus FV1000 laser scanning multi-photon confocal system at NYU Abu Dhabi microscopy facility.
Disclosure of potential conflicts of interest
The authors have no conflicts of interest.
SUPPLEMENTAL
Download Zip (13.6 MB)Acknowledgments
The authors wish to acknowledge Brandon Kent for his technical assistance. The work was funded by grants awarded by the UK Medical Research Council (MK/K001949/1 to D.A.M) and NIH (R01DK80789 to K.C.S and UO1AA018663 to D.A.M and J.M). We thank John Parant for providing the constructs encoding H2afva-GFP.
Additional information
Funding
Related Research Data
References
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315-22. doi:10.1038/nature08514. PMID:19829295
- Long HK, King HW, Patient RK, Odom DT, Klose RJ. Protection of CpG islands from DNA methylation is DNA-encoded and evolutionarily conserved. Nucleic Acids Res. 2016;44(14):6693-706. doi:10.1093/nar/gkw258. PMID:27084945
- Zhang C, Hoshida Y, Sadler KC. Comparative epigenomic profiling of the DNA methylome in mouse and zebrafish uncovers high interspecies divergence. Front Genet. 2016;7:110. doi:10.3389/fgene.2016.00110. PMID:27379160
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20(3):320-31. doi:10.1101/gr.101907.109. PMID:20133333
- Lee HJ, Lowdon RF, Maricque B, Zhang B, Stevens M, Li D, Johnson SL, Wang T. Developmental enhancers revealed by extensive DNA methylome maps of zebrafish early embryos. Nat Commun. 2015;6:6315. doi:10.1038/ncomms7315. PMID:25697895
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10(8):2709-21. doi:10.1093/nar/10.8.2709. PMID:7079182
- Branco MR, King M, Perez-Garcia V, Bogutz AB, Caley M, Fineberg E, Lefebvre L, Cook SJ, Dean W, Hemberger M, et al. Maternal DNA methylation regulates early trophoblast development. Dev Cell. 2016;36(2):152-63. doi:10.1016/j.devcel.2015.12.027. PMID:26812015
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321-6. doi:10.1038/nature14192. PMID:25592537
- Smith ZD, Meissner A. DNA methylation: Roles in mammalian development. Nat Rev Genet. 2013;14(3):204-20. doi:10.1038/nrg3354. PMID:23400093
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117(1-2):15-23. doi:10.1016/S0925-4773(02)00181-8. PMID:12204247
- Mayer W, Fundele R, Haaf T. Spatial separation of parental genomes during mouse interspecific (Mus musculus x M. spretus) spermiogenesis. Chromosome Res. 2000;8(6):555-8. doi:10.1023/A:1009227924235. PMID:11032324
- Mhanni AA, McGowan RA. Global changes in genomic methylation levels during early development of the zebrafish embryo. Dev Genes Evol. 2004;214(8):412-7. doi:10.1007/s00427-004-0418-0. PMID:15309635
- Sanz LA, Kota SK, Feil R. Genome-wide DNA demethylation in mammals. Genome Biol. 2010;11(3):110. doi:10.1186/gb-2010-11-3-110. PMID:20236475
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101-5. doi:10.1038/nature08829. PMID:20098412
- Siklenka K, Erkek S, Godmann M, Lambrot R, McGraw S, Lafleur C, Cohen T, Xia J, Suderman M, Hallett M, et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science. 2015;350(6261):aab2006. doi:10.1126/science.aab2006. PMID:26449473
- Rodgers AB, Morgan CP, Leu NA, Bale TL. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci U S A. 2015;112(44):13699-704. doi:10.1073/pnas.1508347112. PMID:26483456
- Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng GH, Peng H, Zhang X, Zhang Y, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351(6271):397-400. doi:10.1126/science.aad7977. PMID:26721680
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351(6271):391-6. doi:10.1126/science.aad6780. PMID:26721685
- Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16(9):519-32. doi:10.1038/nrm4043. PMID:26296162
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133(3):523-36. doi:10.1016/j.cell.2008.03.029. PMID:18423832
- Rea M, Zheng W, Chen M, Braud C, Bhangu D, Rognan TN, Xiao W. Histone H1 affects gene imprinting and DNA methylation in Arabidopsis. Plant J. 2012;71(5):776-86. doi:10.1111/j.1365-313X.2012.05028.x. PMID:22519754
- Saze H, Tsugane K, Kanno T, Nishimura T. DNA methylation in plants: Relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol. 2012;53(5):766-84. doi:10.1093/pcp/pcs008. PMID:22302712
- Wollmann H, Stroud H, Yelagandula R, Tarutani Y, Jiang D, Jing L, Jamge B, Takeuchi H, Holec S, Nie X, et al. The histone H3 variant H3.3 regulates gene body DNA methylation in Arabidopsis thaliana. Genome Biol. 2017;18(1):94. doi:10.1186/s13059-017-1221-3. PMID:28521766
- Zemach A, Grafi G. Methyl-CpG-binding domain proteins in plants: Interpreters of DNA methylation. Trends Plant Sci. 2007;12(2):80-5. doi:10.1016/j.tplants.2006.12.004. PMID:17208509
- Nekrasov M, Amrichova J, Parker BJ, Soboleva TA, Jack C, Williams R, Huttley GA, Tremethick DJ. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat Struct Mol Biol. 2012;19(11):1076-83. doi:10.1038/nsmb.2424. PMID:23085713
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD. Histone variant H2A.Z Marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005;123(2):233-48. doi:10.1016/j.cell.2005.10.002. PMID:16239142
- Soboleva TA, Nekrasov M, Ryan DP, Tremethick DJ. Histone variants at the transcription start-site. Trends Genet. 2014;30(5):199-209. doi:10.1016/j.tig.2014.03.002. PMID:24768041
- Nashun B, Yukawa M, Liu H, Akiyama T, Aoki F. Changes in the nuclear deposition of histone H2A variants during pre-implantation development in mice. Development. 2010;137(22):3785-94. doi:10.1242/dev.051805. PMID:20943707
- Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456(7218):125-9. doi:10.1038/nature07324. PMID:18815594
- Kobor MS, Lorincz MC. H2A.Z and DNA methylation: Irreconcilable differences. Trends Biochem Sci. 2009;34(4):158-61. doi:10.1016/j.tibs.2008.12.006. PMID:19282182
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328(5980):916-9. doi:10.1126/science.1186366. PMID:20395474
- Zemach A, Zilberman D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol. 2010;20(17):R780-5. doi:10.1016/j.cub.2010.07.007. PMID:20833323
- Coleman-Derr D, Zilberman D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 2012;8(10):e1002988. doi:10.1371/journal.pgen.1002988. PMID:23071449
- Yang X, Noushmehr H, Han H, Andreu-Vieyra C, Liang G, Jones PA. Gene reactivation by 5-aza-2'-deoxycytidine-induced demethylation requires SRCAP-mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genet. 2012;8(3):e1002604. doi:10.1371/journal.pgen.1002604. PMID:22479200
- Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, Oakley F, Burt AD, Wilson CL, Anstee QM, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18(9):1369-77. doi:10.1038/nm.2893. PMID:22941276
- Jiang L, Zhang J, Wang JJ, Wang L, Zhang L, Li G, Yang X, Ma X, Sun X, Cai J, et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell. 2013;153(4):773-84. doi:10.1016/j.cell.2013.04.041. PMID:23663777
- Potok ME, Nix DA, Parnell TJ, Cairns BR. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell. 2013;153(4):759-72. doi:10.1016/j.cell.2013.04.030. PMID:23663776
- Lindeman LC, Winata CL, Aanes H, Mathavan S, Alestrom P, Collas P. Chromatin states of developmentally-regulated genes revealed by DNA and histone methylation patterns in zebrafish embryos. Int J Dev Biol. 2010;54(5):803-13. doi:10.1387/ijdb.103081ll. PMID:20336603
- Andersen IS, Reiner AH, Aanes H, Alestrom P, Collas P. Developmental features of DNA methylation during activation of the embryonic zebrafish genome. Genome Biol. 2012;13(7):R65. doi:10.1186/gb-2012-13-7-r65. PMID:22830626
- Bogdanovic O, Smits AH, de la Calle Mustienes E, Tena JJ, Ford E, Williams R, Senanayake U, Schultz MD, Hontelez S, van Kruijsbergen I, et al. Active DNA demethylation at enhancers during the vertebrate phylotypic period. Nat Genet. 2016;48(4):417-26. doi:10.1038/ng.3522. PMID:26928226
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8(1):e1002440. doi:10.1371/journal.pgen.1002440. PMID:22242016
- Kobayashi H, Sakurai T, Miura F, Imai M, Mochiduki K, Yanagisawa E, Sakashita A, Wakai T, Suzuki Y, Ito T, et al. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res. 2013;23(4):616-27. doi:10.1101/gr.148023.112. PMID:23410886
- Lee HJ, Lowdon RF, Maricque B, Zhang B, Stevens M, Li D, Johnson SL, Wang T. Developmental enhancers revealed by extensive DNA methylome maps of zebrafish early embryos. Nat Commun. 2015;6:6315. doi:10.1038/ncomms7315. PMID:25697895
- Martin CC, Laforest L, Akimenko MA, Ekker M. A role for DNA methylation in gastrulation and somite patterning. Dev Biol. 1999;206(2):189-205. doi:10.1006/dbio.1998.9105. PMID:9986732
- Rai K, Jafri IF, Chidester S, James SR, Karpf AR, Cairns BR, Jones DA. Dnmt3 and G9a cooperate for tissue-specific development in zebrafish. J Biol Chem. 2010;285(6):4110-21. doi:10.1074/jbc.M109.073676. PMID:19946145
- Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452(7189):877-81. doi:10.1038/nature06714. PMID:18354397
- Sivasubbu S, Balciunas D, Davidson AE, Pickart MA, Hermanson SB, Wangensteen KJ, Wolbrink DC, Ekker SC. Gene-breaking transposon mutagenesis reveals an essential role for histone H2afza in zebrafish larval development. Mech Dev. 2006;123(7):513-29. doi:10.1016/j.mod.2006.06.002. PMID:16859902
- Pauls S, Geldmacher-Voss B, Campos-Ortega JA. A zebrafish histone variant H2A.F/Z and a transgenic H2A.F/Z:GFP fusion protein for in vivo studies of embryonic development. Dev Genes Evol. 2001;211(12):603-10. doi:10.1007/s00427-001-0196-x. PMID:11819118
- Faast R, Thonglairoam V, Schulz TC, Beall J, Wells JRE, Taylor H, Matthaei K, Rathjen PD, Tremethick DJ, Lyons I. Histone variant H2A.Z is required for early mammalian development. Curr Biol. 2001;11(15):1183-7. doi:10.1016/S0960-9822(01)00329-3. PMID:11516949
- Percival SM, Parant JM. Observing mitotic division and dynamics in a live zebrafish embryo. J Vis Exp. 2016;(113). PMID:27501381.
- Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, Ekker SC. p53 activation by knockdown technologies. PLoS Genetics. 2007;3(5):e78. doi:10.1371/journal.pgen.0030078. PMID:17530925
- Fang X, Corrales J, Thornton C, Scheffler BE, Willett KL. Global and gene specific DNA methylation changes during zebrafish development. Comp Biochem Physiol B Biochem Mol Biol. 2013;166(1):99-108. doi:10.1016/j.cbpb.2013.07.007. PMID:23876386
- Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103(5):1412-7. doi:10.1073/pnas.0510310103. PMID:16432200
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010-22. doi:10.1101/gad.2037511. PMID:21576262
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823-37. doi:10.1016/j.cell.2007.05.009. PMID:17512414
- Bruce K, Myers FA, Mantouvalou E, Lefevre P, Greaves I, Bonifer C, Tremethick DJ, Thorne AW, Crane-Robinson C. The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res. 2005;33(17):5633-9. doi:10.1093/nar/gki874. PMID:16204459
- Thambirajah AA, Dryhurst D, Ishibashi T, Li A, Maffey AH, Ausio J. H2A.Z stabilizes chromatin in a way that is dependent on core histone acetylation. J Biol Chem. 2006;281(29):20036-44. doi:10.1074/jbc.M601975200. PMID:16707487
- Nepal C, Hadzhiev Y, Previti C, Haberle V, Li N, Takahashi H, Suzuki AM, Sheng Y, Abdelhamid RF, Anand S, et al. Dynamic regulation of the transcription initiation landscape at single nucleotide resolution during vertebrate embryogenesis. Genome Res. 2013;23(11):1938-50. doi:10.1101/gr.153692.112. PMID:24002785
- Wu SF, Zhang H, Hammoud SS, Potok M, Nix DA, Jones DA, Cairns BR. DNA methylation profiling in zebrafish. Methods Cell Biol. 2011;104:327-39. doi:10.1016/B978-0-12-374814-0.00018-5. PMID:21924171
- Wu S-F. Epigenomes of zebrafish mature sperm and early embryos. ProQuest: The University of Utah; 2013.
- Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805-11. doi:10.1038/nrg2651. PMID:19789556
- Kent B, Magnani E, Walsh MJ, Sadler KC. UHRF1 regulation of Dnmt1 is required for pre-gastrula zebrafish development. Dev Biol. 2016;412(1):99-113. doi:10.1016/j.ydbio.2016.01.036. PMID:26851214
- Ridgway P, Brown KD, Rangasamy D, Svensson U, Tremethick DJ. Unique Residues on the H2A.Z containing nucleosome surface are important for xenopus laevis development. J Biol Chem. 2004;279(42):43815-20. doi:10.1074/jbc.M408409200. PMID:15299007
- Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010;20(10):1383-90. doi:10.1101/gr.106542.110. PMID:20709945
- Boyes J, Bird A. Repression of genes by DNA methylation depends on CpG density and promoter strength: Evidence for involvement of a methyl-CpG binding protein. EMBO J. 1992;11(1):327-33. PMID:1310933
- Klose RJ, Bird AP. Genomic DNA methylation: The mark and its mediators. Trends Biochem Sci. 2006;31(2):89-97. doi:10.1016/j.tibs.2005.12.008. PMID:16403636
- Okitsu CY, Hsieh C-L. DNA methylation dictates histone H3K4 methylation. Mol Cell Biol. 2007;27(7):2746-57. doi:10.1128/MCB.02291-06. PMID:17242185
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766-70. doi:10.1038/nature07107. PMID:18600261
- Ke XS, Qu Y, Cheng Y, Li WC, Rotter V, Oyan AM, Kalland KH. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics. 2010;11:669. doi:10.1186/1471-2164-11-669. PMID:21108828
- Yang X, Noushmehr H, Han H, Andreu-Vieyra C, Liang G, Jones PA. Gene reactivation by 5-Aza-2′-Deoxycytidine–induced demethylation requires SRCAP–Mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genetics. 2012;8(3):e1002604. doi:10.1371/journal.pgen.1002604. PMID:22479200
- Abbott DW, Ivanova VS, Wang X, Bonner WM, Ausió J. Characterization of the stability and folding of H2A.Z chromatin particles: Implications for transcriptional activation. J Biol Chem. 2001;276(45):41945-9. doi:10.1074/jbc.M108217200. PMID:11551971
- Bestor TH, Edwards JR, Boulard M. Notes on the role of dynamic DNA methylation in mammalian development. Proc Natl Acad Sci U S A. 2014. PMID:25368180.
- Jacob V, Chernyavskaya Y, Chen X, Tan PS, Kent B, Hoshida Y, Sadler KC. DNA hypomethylation induces a DNA replication-associated cell cycle arrest to block hepatic outgrowth in uhrf1 mutant zebrafish embryos. Development. 2015;142(3):510-21. doi:10.1242/dev.115980. PMID:25564650
- Chernyavskaya Y, M R, Tokarz D, Jacob V, Gopinath S, Zhang C, Sun X, Wang S, Magnani E, Madakashira BP, et al. Loss of DNA methylation in zebrafish embryos activates retrotransposons to trigger an antiviral response. Development. 2017 (in press); doi:10.1242/dev.147629. PMID:28698226
- Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K, Thung S, Bronson Roderick T, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25(2):196-209. doi:10.1016/j.ccr.2014.01.003. PMID:24486181
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87. doi:10.1186/gb-2012-13-10-r87. PMID:23034086
- Li S, Garrett-Bakelman FE, Akalin A, Zumbo P, Levine R, To BL, Lewis ID, Brown AL, D'Andrea RJ, Melnick A, et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 2013;14 Suppl 5:S10. doi:10.1186/1471-2105-14-S5-S10. PMID:23735126