
ABSTRACT
MeCP2 binds to methylated DNA in a chromatin context and has an important role in cancer and brain development and function. Histone deacetylase (HDAC) inhibitors are currently being used to palliate many cancer and neurological disorders. Yet, the molecular mechanisms involved are not well known for the most part and, in particular, the relationship between histone acetylation and MeCP2 is not well understood. In this paper, we study the effect of the HDAC inhibitor trichostatin A (TSA) on MeCP2, a protein whose dysregulation plays an important role in these diseases. We find that treatment of cells with TSA decreases the phosphorylation state of this protein and appears to result in a higher MeCP2 chromatin binding affinity. Yet, the binding dynamics with which the protein binds to DNA appear not to be significantly affected despite the chromatin reorganization resulting from the high levels of acetylation. HDAC inhibition also results in an overall decrease in MeCP2 levels of different cell lines. Moreover, we show that miR132 increases upon TSA treatment, and is one of the players involved in the observed downregulation of MeCP2.
Introduction
Methyl CpG binding protein 2 (MeCP2) is a basic DNA binding protein with preference for methylated DNACitation1,2 and is highly abundant in the brain.Citation3 This protein functions as a transcriptional regulator by virtue of its interaction with both transcription activator and repressor complexes.Citation4,5 In neurons, one MeCP2 molecule is present in approximately every two nucleosomes;Citation6 hence, it is a very important chromosomal protein whose alterations have dire neurological consequences,Citation1,7 for example, Rett syndrome.Citation8 In other cell types and tissues, the abundance of this protein is approximately 30- to 40-fold lower than in neurons.Citation3 Nevertheless, its functional role in these cells is also important. Indeed, MeCP2, by virtue of its specific binding to methylated DNA, plays a very important role in cancer,Citation9,10 where the dysregulation of this epigenetic mark is a hallmarkCitation11 that leads to important structural chromatin changes.Citation12 Moreover, MECP2 was recently shown to be a bona fide oncogene,Citation13 which is frequently amplified in several cancer types.
Histone acetylation affects inter- and intra-nucleosome interactionsCitation14,15 within the chromatin fiber as well as inter-chromatin fiber associationsCitation16 and has been extensively studied in our lab over the years.Citation17,18
The interaction of MeCP2 with methylated DNA on a chromatin template and the massive presence of MeCP2 in neuronal chromatin unavoidably makes it an important functional partner of histones and their posttranslational modifications (PTMs).Citation19 For instance, histone acetylation also plays a critical role in neurogenesis and neuronal plasticity,Citation20 and its levels are affected in many functional neuronal alterations.Citation1,21 However, the potential cross-talk between MeCP2 and histones is largely unknown and remains to be elucidated.
In particular, the regulation of MeCP2 mediated by HDAC inhibitors is very important, as such compounds are currently used both in cancer therapyCitation22-33 and for the treatment of several neurological disordersCitation34-40 to different levels of success.Citation41,42 Hence, understanding the molecular details of the effect(s) of such inhibitors on MeCP2 homeostasis is of major importance.
CREB and microRNAs arising from the miR132/212 cluster have also been shown to play important roles in MeCP2 homeostasis. CREB has been shown to stimulate miR132Citation43 and miR212Citation44 expression; miR212 conversely activates CREB.Citation44 At the same time, both mRNAs can block MeCP2 translationCitation43,45 and MeCP2 can repress pri-miR132/212 expression. In a similar fashion, CREB can repress MeCP2 and vice versa.Citation46
Several years ago, an intriguing observation was made in our lab that showed that treatment of HeLa cells with the HDAC inhibitor sodium butyrate in order to induce global genome histone acetylation resulted in a significant decrease in MeCP2.Citation47 While other labs have seen decreased MeCP2 with HDAC inhibition in other cell lines,Citation48,49 the observation remains puzzling as it is hard to explain by the ‘dogma’ ensuing from the initial association of MeCP2 to transcriptional repression.Citation50 This activity is mediated by the binding of the Sin3A-HDAC repressive complexCitation51 to MeCP2, and, hence, the presence of MeCP2 in the genome should result in an overall reciprocal decrease in histone acetylation, as was experimentally observed.Citation6 Yet, the opposite did not follow from said mechanism (i.e., HDAC inhibition did not lead to increased MeCP2). Similarly, an anti-correlation between DNA methylation and histone acetylation has been reported.Citation52 The current paper represents a follow up on our initial observation and it shows that the relation between histone acetylation, HDACs and MeCP2 is not as simple as originally envisaged.
To date, the most significant studies on MeCP2 have been carried out in the brain.Citation1 In this study, we took advantage of some mechanistic molecular information already available for MeCP2 from this tissue and analyzed it in the frame of non-neuronal cell lines. As mentioned, MeCP2 plays a critical role in cancer;Citation53,54 however, the detailed underlying mechanisms have not been widely studied.
Results
TSA treatment results in chromatin reorganization and MeCP2 redistribution within the nucleus without affecting the dynamics of MeCP2 association-dissociation. To follow up on our intriguing initial observation on the decrease of MeCP2 levels upon sodium butyrate treatment, and since we had originally shown that MeCP2 chromatin partitioning was minutely affected by the HDAC inhibitor treatment of the cell,Citation47 we decided to check first the effects of TSA (a commonly used HDAC inhibitor in recent studies)Citation55-57 on MeCP2 nuclear distribution and on its interactions with chromatin. To this end, we used mouse embryonic fibroblast NIH/3T3 cells because their chromatin is organized into dense pericentomeric domainsCitation58 (“chromocenters”) consisting of major and minor satellite DNA regionsCitation59 that can be easily visualized by optical microscopy and are associated with MeCP2.Citation60
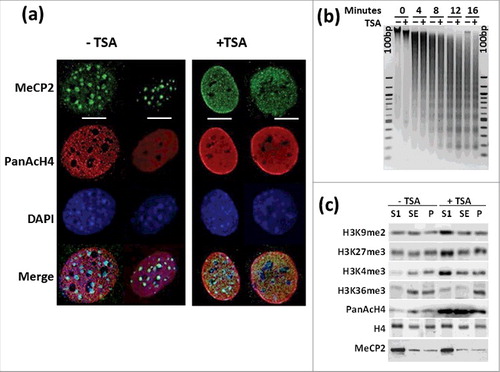
The chromatin reorganization in the nucleus resulting from TSA treatment has been extensively characterized and mouse NIH/3T3 cells have been shown to provide a unique system for these kinds of analyses.Citation61 shows an example of this in which we also monitored the distribution of MeCP2. As previously described, TSA treatment leads to chromatin decondensation that affects both the euchromatin and heterochromatin compartments. This chromatin rearrangement is mirrored by that of MeCP2, which exhibits a more diffuse distribution across the nucleus in which the chromocenters have been partially dispersed, as indicated by DAPI staining. This results in overall global opening of chromatin as visualized by the small but noticeable MNase digestion of chromatin observed in . Biochemically, TSA treatment results in a redistribution and increase of both repressive and activating histone PTMs in the MNase generated SI, SE, and P chromatin fractionationsCitation3 () without affecting their characteristic MeCP2 content.Citation3,47 The enrichment of these histone epigenetic marks in the nuclease-accessible SI fraction upon treatment with TSA is in agreement with our previous results.Citation62
Figure 1. TSA treatment results in chromatin rearrangement. (a) Immunofluorescence analysis of MeCP2 and acetylated H4 distribution in NIH/3T3 cells +TSA and -TSA. Scale bars: 10 μm. (b) Time course microccocal nuclease digestion and (c) Western blot analysis of several histone PTMs before and after TSA treatment of the same cells. “100 bp” corresponds to a 100 bp marker. SI, SE, and P represent the chromatin fractions obtained upon micrococcal nuclease digestion as described in 3.
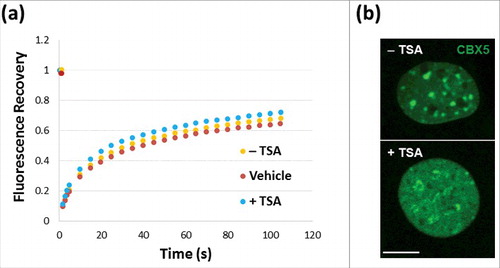
We then wanted to analyze how all of this affects the interaction of MeCP2 with chromatin. In this regard, it is noteworthy that despite the nuclear chromatin relocalization of MeCP2 observed in , fluorescence recovery after photobleaching (FRAP) () clearly indicates that TSA treatment does not affect the dynamics with which MeCP2 associates and dissociates from chromatin. To ensure that the cells being analyzed by FRAP had undergone TSA treatment, cells were visualized using a chromobox homolog 5 (CBX5) antibody. CBX5 binds to H3K9me present in heterochromatin and, as seen in , has undergone redistribution upon TSA treatment similar to that shown in and is in agreement with H3K9me2 redistribution.
Figure 2. FRAP analysis of NIH/3T3 cells. (a) Untransfected NIH/3T3 cells in the presence (red)/absence (orange) of DMSO controls or transfected with GFP-MeCP2 (blue). (a) Green fluorescence analysis of the distribution of chromobox homolog (CBX5) in the nuclei used for the analyses shown in (a), with (+) or without (−) TSA treatment. Scale bar: 10 μm.
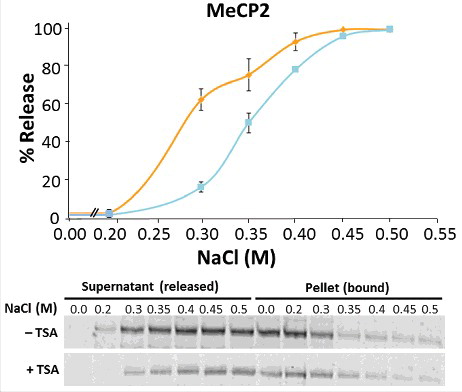
TSA treatment increases MeCP2 chromatin binding affinity and decreases its global phosphorylation in NIH/3T3 cells. To gain further molecular detail on the effects of TSA on the interaction of MeCP2 with chromatin we performed ionic-strength-dependent experiments.Citation3 Despite the little effect of TSA treatment on the dynamics of MeCP2 binding (), we decided to check whether its chromatin binding affinity was affected. Indeed, MeCP2 and histone H1, which bind and compete for similar linker DNA regions, have very different binding affinities [(see , in reference 3)], yet they exhibit almost identical chromatin binding dynamics (unpublished results andCitation63,64). For consistency, the analysis was performed in NIH/3T3 cells grown in the presence or absence of TSA (). Upon treatment with different NaCl concentrations, nuclear suspensions were pelleted and MeCP2 levels were measured in both supernatant and pellet fractions. The reciprocal increase and decrease of MeCP2, respectively, provided validation of experimental success. We found that TSA substantially increased the binding affinity of MeCP2 to chromatin. This was unexpected, as previous results from our lab had shown that, in vitro, histone acetylation did not affect the binding of recombinant MeCP2 to the nucleosome.Citation47
Figure 3. TSA treatment affects the binding of MeCP2 to chromatin. Salt (NaCl) extraction of MeCP2 from nuclei of untreated (orange line) and TSA treated (blue line) NIH/3T3 cells. Quantitative western blots were carried out on n = 2 independent experiments. MeCP2 levels were measured in both supernatant and pellet fractions, given that the reciprocal increase and decrease of MeCP2 represent the unbound (released) and bound fractions from cell nuclei, respectively. Data presented as mean ± SEM.
The obvious candidate for the intriguing in vivo results seen in became MeCP2 phosphorylation.Citation65-67 MeCP2 is an intrinsically disordered protein and, as such, is subject to many PTMs,Citation7 including phosphorylation.Citation67 Histone phosphorylation has been shown to globally decrease its chromatin binding affinityCitation68,69 and TSA has been shown to downregulate several cyclin dependent kinases (CDKs).Citation70
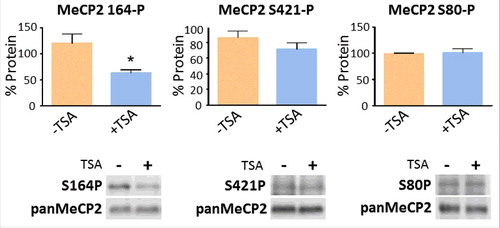
We decided to analyze two of the most intensively characterized phosphorylation sites to date, S80-P and S421-P.Citation71 In neurons, S80 is constitutively phosphorylated in the resting stateCitation72 and S421 is phosphorylated in response to neuronal activity.Citation69 We also decided to include S164-P, a recently described abundant developmental MeCP2 phosphorylation, which has been documented to lower its binding affinity to chromatin.Citation66 The results of such analyses are shown in . While we were not able to detect any significant change in S80 and only a relative change in S421 phosphorylation, the decrease observed in S164 suggests that MeCP2 phosphorylation reduction could be responsible for the TSA-driven increase in chromatin binding affinity ().
Figure 4. TSA exposure alters the levels of MeCP2 phosphorylation. Changes in the levels of MeCP2 phosphorylation at positions S80, S164, and S421, normalized to total MeCP2, both without (−) and with (+) exposure to TSA. Equal loadings of total MeCP2 (in control and treated samples) were used so that changes in the phosphorylated forms could be visualized. Westerns for total MeCP2 and each one of the corresponding phosphorylated forms were obtained from the same gel (membrane). Data represents mean ± SEM of 4 independent experiments. Mann-Whitney tests were used to calculate significance; * P value <0.05, **P value <0.01, ***P value <0.001. Representative blot images are shown in greyscale black on white format.
TSA treatment decreases the level of MeCP2. TSA has the same effect in decreasing the levels of MeCP2 as previously observed when we exposed HeLa cells to sodium butyrate. Different types of mouse and human cell lines were treated (). The cells chosen encompass different developmental origin: human HeLa (ectoderm) and HEK293 (mesoderm), as well as mouse embryonic fibroblast NIH/3T3 (mesoderm).
Figure 5. TSA decreases the levels of MeCP2 in different cell lines. (a) Comparison of the effects of TSA (330 nM) treatment for 24 h. on the levels of MeCP2 and histone H4 acetylation in NIH/3T3 mouse fibroblasts. (b) Effects of TSA treatment on HeLa S3 [40 ng/mL (132 nM), 12 h] and HEK293 [120 ng/mL (396 nM), 18 h]. Representative WB bands shown to be disconnected were non-adjacent signals visualized on the same membrane. (c) Quantification of MeCP2 transcripts by real-time RT-PCR of NIH/3T3 cells with and without TSA treatment. Data presented as mean ± SEM of 3–11 experiments. Unpaired two tailed t-tests were used for large sample sizes wherein normal distribution could be validated (using Shapiro-Wilk test) and Mann-Whitney tests were used for small sample sizes. * P value <0.05, **P value <0.01, ***P value <0.001. Representative blot images are shown in greyscale black on white format.
![Figure 5. TSA decreases the levels of MeCP2 in different cell lines. (a) Comparison of the effects of TSA (330 nM) treatment for 24 h. on the levels of MeCP2 and histone H4 acetylation in NIH/3T3 mouse fibroblasts. (b) Effects of TSA treatment on HeLa S3 [40 ng/mL (132 nM), 12 h] and HEK293 [120 ng/mL (396 nM), 18 h]. Representative WB bands shown to be disconnected were non-adjacent signals visualized on the same membrane. (c) Quantification of MeCP2 transcripts by real-time RT-PCR of NIH/3T3 cells with and without TSA treatment. Data presented as mean ± SEM of 3–11 experiments. Unpaired two tailed t-tests were used for large sample sizes wherein normal distribution could be validated (using Shapiro-Wilk test) and Mann-Whitney tests were used for small sample sizes. * P value <0.05, **P value <0.01, ***P value <0.001. Representative blot images are shown in greyscale black on white format.](/cms/asset/8c248322-da9b-4db1-8203-78ed5ab6ba59/kepi_a_1380760_f0005_oc.jpg)
As seen in , TSACitation73 treatment resulted in a decrease of MeCP2 and an increase in the levels of histone acetylation, thus recapitulating our earlier observation.Citation47 Treatment with TSA resulted in an approximate 30–40% decrease in MeCP2, while the level of histone acetylation increase was 2–2.5-fold. Moreover, the lower levels of MeCP2 observed are the result of a similar decrease in the levels of MeCP2 mRNA (). Therefore, we decided to analyze the potential molecular mechanism involved.
Exposure of NIH/3T3 cells to HDAC inhibition increases the expression of miR132/212. The observed decrease of MeCP2 in NIH/3T3 cells upon exposure to TSA was intriguing. Homeostasis of MeCP2 is critical, particularly in neurons, where a tight regulation of its levels is required for their proper function.Citation74 cAMP responsive element binding protein (CREB)-binding protein (CREB-CBP) have been involved in this process through a CREB-induced microRNA (miR132/212).Citation43 CBP is a highly abundant histone acetyl transferase (HAT) subunit that interacts with CREB. We reasoned that TSA treatment could decrease the levels of MeCP2 by upregulating the expression of miR132/212.
shows that TSA-treated NIH/3T3 cells exhibit a significant upregulation of the pri-miR132/212, therefore resulting in the presence of higher levels of mature miR132. To verify if this upregulation directly affects MeCP2 protein levels we blocked the miR132 recognition element (MRE) in the MeCP2 3′ UTR as described before.Citation43 To this end, we transfected NIH/3T3 with a locked nucleic acid (LNA) oligonucleotide complementary to the MRE and an LNA control, complementary to a non-MRE sequence, also present in the MeCP2 3′ UTR.Citation43 Cells transfected with the specific LNA showed increased MeCP2 levels upon TSA treatment as compared to untransfected and control LNA cells (). The control LNA could possibly be affecting MeCP2 mRNA stability in our system, since we observed higher levels of MeCP2 compared with untransfected TSA-treated cells. Nevertheless, our results are in agreement with a role of miR132 in the downregulation of MeCP2. Hence, overexpression of miR132 upon TSA treatment appears to be responsible for the decreased MeCP2 protein levels through a mechanism that would resemble that described in neuronal systemsCitation43 (). However, we cannot rule out the possibility of other additional regulatory mechanisms also involved.
Figure 6. TSA treatment increases miR132 levels and affects MeCP2 protein levels in NIH/3T3 cells (a) Unprocessed pri-miR132/212 and mature miR132 transcription increases in TSA treated NIH/3T3 cells. Values were collected via quantitative RT-PCR. Data presented as mean ± SEM of 3–5 independent experiments. Mann-Whitney tests were used to calculate significance; * P value <0.05, **P value <0.01, ***P value <0.001. (b) Densitometric analysis and representative WB showing MeCP2 protein levels after transfection of NIH/3T3 with an LNA oligonucleotide against miR132 MRE located in the 3′UTR of MeCP2 mRNA and a LNA oligonucleotide control upon TSA treatment.
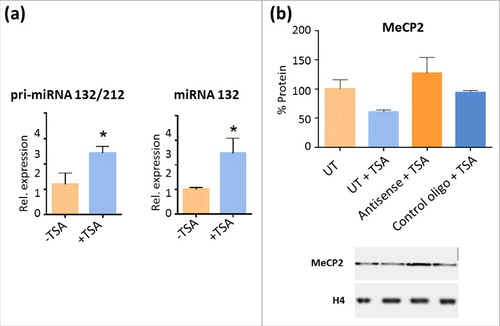
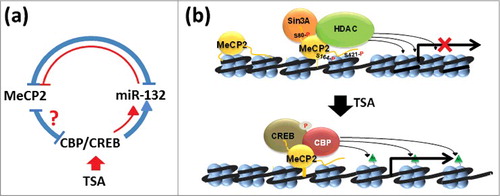
Figure 7. Model representation of the effects of HDAC inhibitors (TSA) on MeCP2 metabolism. (a) In the brain, MeCP2 expression is regulated by a complex regulatory loop 94 (shown in blue; see text for a detailed description). In red we are highlighting the regions of this network that we have been able to confirm in NIH/3T3 non-neuronal cells. (b) Based on early models of MeCP2 function, interaction of MeCP2 with an HDAC-Sin3A complex results in deacetylation of histones on surrounding nucleosomes leading to chromatin organization repressive to transcription 51. Under physiological conditions, MeCP2 is phosphorylated at several residues (S80),72 (S164),66, 72 and (S421)65 that fine-tune its transcriptional regulatory nature67, 71. Upon exposure to HDAC inhibitors such as TSA, the levels of MeCP2 phosphorylation decrease and so do the levels of the protein itself. This decrease is likely mediated by the action of CREB-CBP binding to MeCP2, which, upon acetylation of the neighboring nucleosomes (green triangles), creates a relaxed/transcriptionally-permissive chromatin organization that enhances transcription of miR132/212 4 and creates a regulatory loop of MeCP2 expression,43 shown in (a).
Discussion
Because of their peculiar chromatin organization in well-defined dense nuclear chromocenters, mouse NIH/3T3 cells provide a very good model to study the nuclear distribution of MeCP2.Citation60 Under normal conditions, MeCP2 concentrates in these condensed heterochromatin regions.Citation60,75 These regions correspond to pericentromeric domains and arise from the coalescence of major satellite DNA regions with minor satellites associated with their periphery and exhibit different micrococcal nuclease sensitivity. Moreover, both regions contain the methylated forms of H3K9 and the minor satellites exhibit a relaxed chromatin organization compared to the highly compacted major type, which interacts with heterochromatin protein 1α (HP1α).Citation59 It is likely that the minor satellite domains are more prone to become disorganized by TSA treatment further enhancing their nuclease sensitivity in agreement with the results observed in .
Our previous analyses on the chromatin distribution of MeCP2 in cells treated with sodium butyrate indicated that the chromatin reorganization that takes place upon such treatment does not affect the partitioning of MeCP2,Citation47 as observed in the MeCP2 lane of (). This means that, despite the loss of chromocenter compaction upon TSA exposure [ TSA (C)], the remaining MeCP2 stays bound to the same chromatin territories and it exchanges from them with very similar dynamics (). In addition, the elevated presence of MeCP2 in the highly nuclease accessible SI fraction of different cell types and tissues (seeCitation3 and ) makes it tempting to speculate a particular type of association of MeCP2 to the minor satellite regions of the chromocenters. This may provide a hint to the massive presence of MeCP2 in the SI chromatin fraction of all organisms and cell types studied to dateCitation76 and whose molecular explanation remains a mystery.Citation1 It would be interesting to know which regions of DNA are partially depleted of MeCP2 as a result of TSA treatment. Interestingly, the changes in chromatin reorganization and the more relaxed chromatin conformation adopted upon TSA treatment () are accompanied by a seemingly counterintuitive increase in the levels of repressive marks, such as H3K9me2 and H3K27me3, as well as the H3K4me3 activating mark, which is most likely the result of changes in gene expression resulting from TSA treatment.Citation77,78 In agreement with this observation, a very recent paper has shown that the increase in nuclear histone acetylation upon treatment of barley pollen with TSA also results in a nuclear accumulation of H3K27me3 and H3K4me2.Citation79 Such indiscriminate accumulation of histone methylation marks is most likely the result of the upregulation of histone methyltransferases by TSA treatment.Citation80
Besides increasing global histone acetylation, TSA exposure alters the binding characteristics of MeCP2. It is interesting to compare the results obtained here with those obtained for histone H1 under similar TSA treatment conditions.Citation81 There is a striking similarity with which both chromosomal proteins compete with each otherCitation7,82 for presumably similar in vivo binding sitesCitation6 and for chromatinized DNA templates in vitro.Citation50,83 This is despite the preference of MeCP2 for methylated CpN (where N = G, A, T or C)Citation2 DNA regions, which are prevalent at chromatin linker regionsCitation84,85 where linker histones (H1) bind. Our data () clearly shows enhanced ionic strength-dependent binding affinity of MeCP2 to chromatin upon TSA treatment, which contrasts with the unchanged binding affinity observed under the same conditions for histone H1.Citation16 Nevertheless, contrarily to histone H1,Citation86 TSA did not affect the binding kinetics of MeCP2 to chromatin (). This may simply reflect the fact that while histone acetylation has been shown to alter histone H1 binding back to acetylated chromatin once it has dissociated,Citation87 that of MeCP2 remains unchanged.Citation47 The increased binding affinity of MeCP2 upon treatment with HDAC inhibitors cannot be attributed to the overall increase in histone acetylation of nucleosomes as the binding of MeCP2 to nucleosomes in vitro has been shown to be unaffected by their extent of histone acetylation.Citation47
One of the potential contributors to the TSA-induced increase in binding affinity of MeCP2 just described could be the result of a decrease in the phosphorylation of this protein due to kinase dysregulation ensuing from the HDAC inhibitors treatment. Global MeCP2 phosphorylation has been described to decrease its chromatin binding affinity.Citation69,88 and TSA has been shown to downregulate cdk4, cdk6, and cdk2Citation70 and Calcium/calmodulin-dependent protein kinase II (CaMKII).Citation89 MeCP2 S421-P is mediated by CaMKIICitation72,90 and MeCP2 S80-P is mediated by homeodomain-interacting protein kinase 2 (HIPK2).Citation91 shows that in our hand, neither one of these two phosphorylations were significantly affected by the treatment of NIH/3T3 cells with TSA. However, phosphorylation of S164 was reduced by half. Phosphorylation of S164 is a very abundant MeCP2 PTM recently described to decrease the binding affinity of MeCP2 for chromatin and to be involved in the control of dendrite patterning.Citation66 Although the kinase responsible is still not known, it is plausible to think that it could be a member of the cdk family. For instance, alterations in the kinase cdkl5 lead to Rett syndrome phenotypes, as is the case for MeCP2.Citation92 Moreover, cdkl5 has been involved in dendrite development.Citation93 While in the context of NIH/3T3 cells these functions may lack relevance, the fact that CDKs are also downregulated by TSA treatment and that they share neurological pathways with S164 phosphorylation, suggest that CDKL5 could be a likely candidate.
Regardless of the above chromatin structural considerations, we found that treatment of cells with TSA lead to a decrease in the overall content of nuclear MeCP2 (), as previously observed via treatment of HeLa cells with sodium butyrate.Citation47 Human HeLa, HEK293, and mouse NIH/3T3 cell lines were used () to corroborate those preliminary results using different germ layers and/or species.
In the brain, where most information is available for MeCP2 metabolism, CREB-CBP, MeCP2, and miR132/212 have been shown to be connected through a regulatory network () that is responsible for maintaining MeCP2 homeostasis.Citation94 HDAC inhibitors enhance the activation of genes regulated by the CREB-CBP transcriptional complex,Citation95 including miR132,Citation43 which represses MeCP2 levels in rat neuronsCitation46 [see blue loop in ]. The results obtained here () clearly support the notion of a similar regulatory feedback loop [red lines in ] occurring in non-neuronal cells.
As stated in the introduction, our work shows that the relationship between histone acetylation and MeCP2 is much more complex than originally anticipated. In earlier studies, mainly repressive models for the function of MeCP2,Citation50 antagonism was observed between the presence of MeCP2 and histone acetylation.Citation6 However, there is now abundant evidence in support of a transcriptional regulatory role, wherein MeCP2 can also bind to activator complexes such as CREB-CBPCitation4 [see ]. TSA, as well as other HDAC inhibitors, by decreasing the phosphorylation of MeCP2 may enhance the binding of these activator complexes to promoters (), underscoring, at any rate, the larger than envisioned complexity of the relationship between histone acetylation and MeCP2 on their shared chromatin substrate.
To conclude, this work was based on an observation made in our lab several years ago: that HDAC inhibitor-induced hyperacetylation of histones in non-neuronal cells resulted in significant decrease in the levels of MeCP2.Citation47 A follow up to that observation here shows that HDAC inhibitor treatment results in increased MeCP2 chromatin binding affinity, which appears to be affected by a global decrease in MeCP2-S164 phosphorylation and is accompanied by significant decrease in the levels of MeCP2, in part involving miR132.
Materials and methods
Cell culture
NIH/3T3, HEK293, and HeLa S3 cells were grown at 37°C in 5% CO2 atmosphere in Dulbecco's modified essential medium (DMEM) (Sigma-Aldrich, St. Louis, MO, USA) containing 10% bovine growth serum (BGS) (VWR, Radnor, PA, USA), 1% penicillin and streptomycin (10,000 UI and 100 mg/mL, respectively) (Sigma-Aldrich, St. Louis, MO, USA), and 1X Gibco GlutaMAX (ThermoFisher Scientific Inc., Waltham, MA, USA).
Cells were grown to high density according to ATCC standards, media was removed, plates washed in phosphate buffered saline (PBS) and cells were either treated with Trichostatin A (TSA) (Sigma-Aldrich, St. Louis, MO, USA) or new medium was added to control plates. Treatment concentrations and times used were based on both literature and/ or time-course experiments (data not shown)Citation61,96 and were as follows: NIH/3T3: 100 ng/mL (330 nM) for 24 hours; HeLa S3: 40 ng/mL (132 nM) for 12 hours; and HEK293: 120 ng/mL (396 nM) for 18 hours.
Sample preparation
Cells were harvested by removing media from plates on ice, washing with cold PBS then dislsodged by trypsinization. Trypsin was neutralized with medium (see “Cell Culture”), and samples were pelleted at 4°C for 2 minutes at 1,000 × g. Cell pellets were homogenized in 4 volumes (w/v) of cell lysis buffer (0.25 M Sucrose, 60 mM KCl, 15 mM NaCl, 10 mM MES, 5 mM MgCl2, 1 mM CaCl2, 0.8% Triton X-100), incubated on ice for 5 minutes, then centrifuged at 4°C for 10 minutes at 1,000 × g. The supernatant was discarded and pellets were then resuspended in a half volume of nuclei resuspension buffer (50 mM NaCl, 10 mM Pipes pH 6.8, 5 mM MgCl2, 1 mM CaCl2). Complete protease cocktail inhibitor (Roche Molecular Biochemicals, Laval, Quebec, Canada) was added at each step at a dilution of 1:100, as was phosphatase inhibitor cocktail (Roche Molecular Biochemicals, Laval, Quebec, Canada) at a dilution of 1:10 in the case of probing for protein phosphorylation. SDS-PAGE sample buffer (125 mM Tris HCl pH 6.8, 2% SDS. 20% glycerol, 1.43 M β-mercaptoethanol, 0.2% bromophenol blue) was added to samples, which were then sonicated in 10-second intervals for approximately 1 minute and then boiled for 60–90 seconds at 95°C. Samples were flash frozen in liquid nitrogen and stored at −80°C until use.
Chromatin fractionation
Nuclei were prepared as described in “Sample preparation” for the MNase digestions. MNase digestion for chromatin fractionation (30 U/mg of DNA) was carried out for 15 minutes on pre-warmed samples at 37°C with shaking (in a water bath) and for time-course chromatin accessibility (5 U/mg of DNA) was performed for 0, 4, 8, 12, and 16-minutes time intervals. The activity of MNase was stopped using 20 mM EDTA (pH 8.0). Chromatin fractionation was then carried out according to.Citation3 In brief, samples were centrifuged at 10,000 × g for 10 minutes at 4°C. Supernatants (SI) were removed and pellets were resuspended harshly and vortexed in 0.25 mM EDTA (volume equal to Buffer B), then tumbled at 4°C for 60 minutes. The pellets containing 0.25 mM EDTA were centrifuged at 16,000 × g for 10 minutes at 4°C. Supernatants (SE) were recovered and appropriate volumes of SDS sample buffer were added to the resulting pellets (P), S1 and SE, and samples were sonicated before electrophoresis.
DNA extraction
Time course MNase digested samples were supplemented with a 1:1 ratio of Protein K digestion buffer (600 mM NaCl, 40 mM EDTA, 20 mM Tris pH 7.5, 1% SDS), treated with 50 μg/ml of RNase (10 mg/ml) at 37°C for 1 h and subsequently with Proteinase K (1 μg/10 μg of DNA) for another hour at 37°C. An equal volume of Phenol:Chloroform:Isoamylic alcohol (PCI) (25:24:1) was added and samples were vortexed and centrifuged at 16,000 × g at room temperature for 10 minutes. The top aqueous layer was removed and the PCI step was repeated once more. One volume of Chloroform:Isoamyl alcohol was added to the aqueous layer, vortexed well, and centrifuged as above. The aqueous phase was removed and 0.1 volume of 3 M sodium acetate (pH 5.2) was added and vortexed. Overnight precipitation was then carried out in 2.5 volumes of ice-cold 95% ethanol. Samples were centrifuged at 16,000 × g at room temperature for 10 minutes and the supernatant was discarded. Pellets were then gently washed with same volume of 75% ethanol and centrifuged as above. The resulting pellet was then allowed to air dry and dissolved in a small volume of Tris-EDTA (TE) buffer (pH 8.0) (10 mM Tris-HCl, 1 mM EDTA). DNA concentrations were determined using NanoDrop and analyzed on 1.5% agarose gel. DNA band intensities were measured using Image Studio Lite version 5.2 to confirm an equal amount of DNA for each time interval.
Gel electrophoresis
SDS-polyacrylamide gel electrophoresis (PAGE) (10% and 15% acrylamide) were prepared as previously described.Citation97
Western blot (WB)
SDS-PAGE gels were transferred for 2–3 hours at 400 mA on ice onto 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, CA, USA) in 40 mM:192 mM Tris:glycine, 20% methanol or 20 mM sodium phosphate (pH 6.8) buffer, 15% ethanol (for the analyses of PTMs). Membranes were blocked in 3% skim milk, PBS-Tween 0.1%, or 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS)-Tween 0.1% (for phosphorylation) for 45 minutes at room temperature with shaking. Primary antibodies incubation was done overnight at 4°C. Primary antibodies and dilutions used were as follows: MeCP2 1:1,000 (M9317, Sigma-Aldrich, St. Louis, MO, USA), PanAcetyl H4 1:1,000 (06–866, EMD Millipore, Temecula, CA, USA), H4 1:30,000 (rabbit serum produced in-house). MeCP2 pSer80 1:1,000 (AP31635SU-N, Acris, Dunwoody Park, Atlanta GA), MeCP2 pSer164 1:1,000 (rabbit serum produced in houseCitation66), MeCP2 pSer421, β-actin 1:5,000 (A2228, Sigma-Aldrich, St. Louis, MO, USA), H3K9me2 1:1,000 (13969P, Cell Signaling Technologies, Danvers, MA, USA), H3K27me3 1:1,000 (07–449, EMD Millipore), H3K4me3 1:1,000 (5326P, Cell Signaling Technologies), and H3K36me3 1: 2,000 (4909P Cell Signaling Technologies). Secondary antibody incubation was carried out at room temperature for 1 hour, with three 5 minute PBS-Tween 0.1% or TBS-Tween 0.1% (for phosphorylation) washes before and afterwards. Secondary antibodies used were enhanced chemiluminescent (ECL) Anti-rabbit IgG 1:2,000 (NA934, GE Healthcare, Piscataway, NJ, USA), Donkey Anti-Sheep/Goat IgG 1:2,000 (AB324P, EMD Millipore Corporation), IRDye 800 Anti-rabbit IgG 1:10,000 (611-132-122, Rockland Antibodies & Assays, Gilbertsville, PA, USA), IRDye 680LT Anti-mouse IgG 1:20,000 (926–68020, LI-COR, Lincoln, NK, USA). The latter two secondary incubations were carried out in darkness. Western blots were acquired using Li-Cor C-digit for chemiluminescent imaging or Li-Cor Odyssey (LI-COR Biosciences) for fluorescent imaging. Images were analyzed using Li-Cor Image Studio version 5.2 software.
Fluorescence recovery after photobleaching
MeCP2-GFP plasmid and fluorescence recovery after photobleaching (FRAP) was carried out as described elsewhere.Citation98,99
Salt extraction
Equal amounts of solutions of 10 mM Tris (pH 7.5), 1 mM EDTA (pH 8.0) with increasing NaCl concentration (0.4, 0.6, 0.7, 0.8, 0.9, and 1 M) were added to aliquots of TSA-treated and untreated control sample preparations (see “Sample preparation”), vortexed well, and incubated on ice for 20 minutes. Samples were then centrifuged at 4°C for 10 minutes at 16,000 × g. The supernatant was removed and appropriate volumes of SDS-PAGE sample buffer were added to both supernatants and pellets and stored at −80°C. Normalizations for WB experiments were based on equal suspension and loading volumes, determined via spectrophotometric DNA quantity analysis as well as total histone protein visualization in Coomassie-stained SDS-PAGE gels. Western blots were acquired using Li-Cor Odyssey (LI-COR Biosciences). Images were analyzed using Li-Cor Image Studio Version 5.2 software.
Immunofluorescence
NIH/3T3 cells were plated on glass coverslips (12–541B, Fisher Scientific, Pittsburgh, PA, USA) during incubation. At 24 hours TSA treatment, immunofluorescence protocol was carried out as follows: Media was removed and coverslips were washed three times in PBS-T 0.1%. Fixation was carried out in 4% paraformaldehyde for 15 min. Primary antibodies were added as follows: 1:500 for both MeCP2 and PanAcH4 (see “Western blot”) in PBS-T 0.1%. This was followed by an overnight incubation at 4°C and subsequent wash 3 times in PBS-T 0.1% prior to the addition of secondary antibodies, donkey anti-rabbit Alexa 555 and donkey anti-mouse Alexa 488, both at 1:1000. The secondary antibodies were incubated for 1 hour at room temperature followed by 3 washes in PBS-T 0.1% and mounted in Immumount to view.
RNA extraction and RT-qPCR
NIH/3T3 total RNA purification was performed using TRIzol reagent (Invitrogen) following manufacturer instructions. To assess pri-miR132/212 expression, RNA was reverse-transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Each PCR was carried out in triplicate using SYBR Green PCR Master Mix (Applied Biosystems). Primers used are: Mecp2Citation98; pri-miR132/212 (fwd: CGGTGACTCAGCCTAGATGG, rv: GGACGGGACAGGGAAGGGCitation100). To confirm reproducibility of the observed changes in transcripts levels, three different housekeeping genes were analyzed: Gapdh (fwd: AAGGTCATCCCTGAGCTGAACGGG, rv: CCAGGAAATGAGCTTGACAAAGTG), Ppia (fwd: CAAATGCTGGACCAAACACAAACG, rv: GTTCATGCCTTCTTTCACCTTCCC) and Tbp (fwd: CCCCACAACTCTTCCATTCT, rv: GCAGGAGTGATAGGGGTCAT). Representative figures used were Mecp2/Gapdh () and pri-miR132/212/Tbp ().
Mature miR132 reverse-transcription and RT qPCR were carried out using the TaqMan microRNA Assays #000457 (miR132-3p) and #001973 (endogenous control U6 snRNA), TaqMan microRNA Reverse Transcription kit and TaqMan Universal Master Mix II (Applied Biosystems). In all cases, thermocycling conditions were 10 minutes at 95°C, then 40 cycles of 15 seconds at 95°C and 1 minute at 60°C. Fluorescent signals were acquired by the Stratagene Mx3005P qPCR System (Agilent Technologies). Relative mRNA levels were calculated using the comparative Ct method (ΔΔCt), considering PCR efficiency. Fold change is equal to 10ΔΔCt/m, where ‘m’ is the average slope of the calibration curves for the gene of interest and the endogenous control.
Locked nucleic acid (LNA) assay
In a 6-well plate, NIH/3T3 cells at 70–80% confluency were transfected with control and antisense LNA oligos [Antisense, 5′- TAACAGTCCTGGTGATATTTGGTCA-3′; Control, 5′- TGTAGACAATAATGTCCATGGCCTT-3′ (modified bases are underlined)] at a concentration of 10 nM using lipofectamine 3000 plus (Invitrogen), as per manufacturer's protocol. After 4 hours of transfection, the cells were treated with 100 ng/mL (330 nM) of TSA for 24 hours. This was followed by SDS preparation of protein lysate for immunoblot assays as mentioned above (see “Sample preparation”).
Author contributions
KVG, AMDP, AAT, GS, MSC, MT, OK, TLG, and KM performed the experiments. BC, AU and MH designed the experiments and contributed to the discussions in the paper. NL provided reagents and contributed to the writing of the manuscript. JA conceived and wrote the manuscript.
Competing financial interests
All authors declare no competing financial interests.
Acknowledgments
This research was supported by a Canadian Institutes of Health Research (CIHR) grant MOP -130417 to JA.
Additional information
Funding
References
- Ausio J. MeCP2 and the enigmatic organization of brain chromatin. Implications for depression and cocaine addiction. Clin Epigenetics. 2016;8:58. doi:10.1186/s13148-016-0214-5. PMID:27213019
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522:89-93. doi:10.1038/nature14319. PMID:25762136
- Thambirajah AA, Ng MK, Frehlick LJ, Li A, Serpa JJ, Petrotchenko EV, Silva-Moreno B, Missiaen KK, Borchers CH, Adam Hall J, et al. MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 2011;40:2884-97. doi:10.1093/nar/gkr1066. PMID:22144686
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224-9. doi:10.1126/science.1153252. PMID:18511691
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416-21. doi:10.1073/pnas.0707442104. PMID:18042715
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457-68. doi:10.1016/j.molcel.2010.01.030. PMID:20188665
- Ausió J, de Paz A, Esteller M. MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol Med. 2014;20:487-98. doi:10.1016/j.molmed.2014.03.004. PMID:24766768
- Zoghbi HY. Rett Syndrome and the ongoing legacy of close clinical observation. Cell. 2016;167:293-7. doi:10.1016/j.cell.2016.09.039. PMID:27716498
- Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J, Cigudosa JC, Huang TH, Esteller M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. Embo J. 2003;22:6335-45. doi:10.1093/emboj/cdg604. PMID:14633992
- Lopez-Serra L, Ballestar E, Fraga MF, Alaminos M, Setien F, Esteller M. A profile of methyl-CpG binding domain protein occupancy of hypermethylated promoter CpG islands of tumor suppressor genes in human cancer. Cancer Res 2006;66:8342-6. doi:10.1158/0008-5472.CAN-06-1932. PMID:16951140
- Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 2016;8(9). doi:10.1101/cshperspect.a019505. PMID:27194046
- Leonhardt H, Cardoso MC. DNA methylation, nuclear structure, gene expression and cancer. J Cell Biochem Suppl. 2000;Suppl 35:78-83. doi:10.1002/1097-4644(2000)79:35+<78::AID-JCB1129>3.0.CO;2-J. PMID:11389535
- Neupane M, Clark AP, Landini S, Birkbak NJ, Eklund AC, Lim E, Culhane AC, Barry WT, Schumacher SE, Beroukhim R, et al. MECP2 Is a Frequently Amplified Oncogene with a Novel Epigenetic Mechanism that Mimics the Role of Activated RAS in Malignancy. Cancer Discov. 2015;6(1):45-58. PMID:26546296
- Garcia Ramirez M, Rocchini C, Ausió J. Modulation of chromatin folding by histone acetylation. J Biol Chem. 1995;270:17923-8. doi:10.1074/jbc.270.30.17923. PMID:7629098
- Wakamori M, Fujii Y, Suka N, Shirouzu M, Sakamoto K, Umehara T, Yokoyama S. Intra- and inter-nucleosomal interactions of the histone H4 tail revealed with a human nucleosome core particle with genetically-incorporated H4 tetra-acetylation. Sci Rep. 2015;5:17204. doi:10.1038/srep17204. PMID:26607036
- Wang X, He C, Moore SC, Ausió J. Effects of histone acetylation on the solubility and folding of the chromatin fiber. J Biol Chem. 2001;276:12764-8. doi:10.1074/jbc.M100501200. PMID:11279082
- Calestagne-Morelli A, Ausio J. Long-range histone acetylation: biological significance, structural implications, and mechanisms. Biochem Cell Biol. 2006;84:518-27. doi:10.1139/o06-067. PMID:16936824
- Ausió J, Abbott DW. The role of histone variability in chromatin stability and folding. Amsterdam, The Netherlands: Elsevier; 2004.p. 241-90.
- Zago V, Pinar-CabezaDeVaca C, Vincent JB, Ausio J. Histone variants and composition in the developing brain: should MeCP2 care? Curr Top Med Chem. 2016;17(7):829–42. PMID:27086783
- Mitrousis N, Tropepe V, Hermanson O. Post-Translational Modifications of Histones in Vertebrate Neurogenesis. Front Neurosci. 2015;9:483. doi:10.3389/fnins.2015.00483. PMID:26733796
- Bonnaud EM, Suberbielle E, Malnou CE. Histone acetylation in neuronal (dys)function. Biomol Concepts. 2016;7:103-16. doi:10.1515/bmc-2016-0002. PMID:27101554
- Yoshida M, Furumai R, Nishiyama M, Komatsu Y, Nishino N, Horinouchi S. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother Pharmacol. 2001;48 Suppl 1:S20-6. doi:10.1007/s002800100300. PMID:11587361
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287-99. doi:10.1038/nrd772. PMID:12120280
- La Thangue NB. Histone deacetylase inhibitors and cancer therapy. J Chemother. 2004;16 Suppl 4:64-7. doi:10.1179/joc.2004.16.Supplement-1.64. PMID:15688613
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981-9. doi:10.1158/1541-7786.MCR-07-0324. PMID:17951399
- Noureen N, Rashid H, Kalsoom S. Identification of type-specific anticancer histone deacetylase inhibitors: road to success. Cancer Chemother Pharmacol. 2010;66:625-33. doi:10.1007/s00280-010-1324-y. PMID:20401613
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85-94. doi:10.1038/icb.2011.100. PMID:22124371
- Delcuve GP, Khan DH, Davie JR. Targeting class I histone deacetylases in cancer therapy. Expert Opin Ther Targets. 2013;17:29-41. doi:10.1517/14728222.2013.729042. PMID:23062071
- Hrabeta J, Stiborova M, Adam V, Kizek R, Eckschlager T. Histone deacetylase inhibitors in cancer therapy. A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:161-9.
- Zhang L, Han Y, Jiang Q, Wang C, Chen X, Li X, Xu F, Jiang Y, Wang Q, Xu W. Trend of histone deacetylase inhibitors in cancer therapy: isoform selectivity or multitargeted strategy. Med Res Rev. 2015;35:63-84. doi:10.1002/med.21320. PMID:24782318
- Turtoi A, Peixoto P, Castronovo V, Bellahcene A. Histone deacetylases and cancer-associated angiogenesis: current understanding of the biology and clinical perspectives. Crit Rev Oncog. 2015;20:119-37. doi:10.1615/CritRevOncog.2014012423. PMID:25746107
- Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6. doi:10.1101/cshperspect.a026831. PMID:27599530
- Damaskos C, Valsami S, Kontos M, Spartalis E, Kalampokas T, Kalampokas E, Athanasiou A, Moris D, Daskalopoulou A, Davakis S, et al. Histone deacetylase inhibitors: an attractive therapeutic strategy against breast cancer. Anticancer Res. 2017;37:35-46. doi:10.21873/anticanres.11286. PMID:28011471
- Ganai SA, Ramadoss M, Mahadevan V. Histone Deacetylase (HDAC) Inhibitors – emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol. 2016;14:55-71. doi:10.2174/1570159X13666151021111609. PMID:26487502
- Penney J, Tsai LH. Histone deacetylases in memory and cognition. Sci Signal. 2014;7:re12. doi:10.1126/scisignal.aaa0069. PMID:25492968
- Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673-91. doi:10.1038/nrd4360. PMID:25131830
- Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605-17. doi:10.1016/j.tips.2010.09.003. PMID:20980063
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591-601. doi:10.1016/j.tins.2009.06.002. PMID:19775759
- Echaniz-Laguna A, Bousiges O, Loeffler JP, Boutillier AL. Histone deacetylase inhibitors: therapeutic agents and research tools for deciphering motor neuron diseases. Curr Med Chem. 2008;15:1263-73. doi:10.2174/092986708784534974. PMID:18537606
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41-50. doi:10.2174/1568007053005091. PMID:15723612
- Bojang P, Jr, Ramos KS. The promise and failures of epigenetic therapies for cancer treatment. Cancer Treat Rev. 2014;40:153-69. doi:10.1016/j.ctrv.2013.05.009. PMID:23831234
- Halsall JA, Turner BM. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. Bioessays. 2016;38(11):1102-1110. doi:10.1002/bies.201600070. PMID:27717012
- Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513-4. doi:10.1038/nn2010. PMID:17994015
- Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, Willoughby D, Wahlestedt C, Conkright MD, Kenny PJ. Striatal microRNA controls cocaine intake through CREB signalling. Nature. 2010;466:197-202. doi:10.1038/nature09202. PMID:20613834
- Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120-7. doi:10.1038/nn.2615. PMID:20711185
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422-37. doi:10.1016/j.neuron.2007.10.001. PMID:17988628
- Ishibashi T, Thambirajah AA, Ausio J. MeCP2 preferentially binds to methylated linker DNA in the absence of the terminal tail of histone H3 and independently of histone acetylation. FEBS Lett. 2008;582:1157-62. doi:10.1016/j.febslet.2008.03.005. PMID:18339321
- Host L, Dietrich JB, Carouge D, Aunis D, Zwiller J. Cocaine self-administration alters the expression of chromatin-remodelling proteins; modulation by histone deacetylase inhibition. J Psychopharmacol. 2011;25:222-9. doi:10.1177/0269881109348173. PMID:19939859
- Ghoshal K, Datta J, Majumder S, Bai S, Dong X, Parthun M, Jacob ST. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol Cell Biol. 2002;22:8302-19. doi:10.1128/MCB.22.23.8302-8319.2002. PMID:12417732
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471-81. doi:10.1016/S0092-8674(00)81887-5. PMID:9038338
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386-9. doi:10.1038/30764. PMID:9620804
- Eden S, Hashimshony T, Keshet I, Cedar H, Thorne AW. DNA methylation models histone acetylation. Nature. 1998;394:842. doi:10.1038/29680. PMID:9732866
- Ballestar E, Esteller M. The epigenetic breakdown of cancer cells: from DNA methylation to histone modifications. Prog Mol Subcell Biol. 2005;38:169-81. doi:10.1007/3-540-27310-7_7. PMID:15881895
- Ballestar E, Esteller M. Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem Cell Biol. 2005;83:374-84. doi:10.1139/o05-035. PMID:15959563
- Ou JN, Torrisani J, Unterberger A, Provencal N, Shikimi K, Karimi M, Ekstrom TJ, Szyf M. Histone deacetylase inhibitor Trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem Pharmacol. 2007;73:1297-307. doi:10.1016/j.bcp.2006.12.032. PMID:17276411
- Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166-79. PMID:21416059
- Vanhaecke T, Papeleu P, Elaut G, Rogiers V. Trichostatin A-like hydroxamate histone deacetylase inhibitors as therapeutic agents: toxicological point of view. Curr Med Chem. 2004;11:1629-43. doi:10.2174/0929867043365099. PMID:15180568
- Mateos-Langerak J, Brink MC, Luijsterburg MS, van der Kraan I, van Driel R, Verschure PJ. Pericentromeric heterochromatin domains are maintained without accumulation of HP1. Mol Biol Cell. 2007;18:1464-71. doi:10.1091/mbc.E06-01-0025. PMID:17314413
- Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004;166:493-505. doi:10.1083/jcb.200403109. PMID:15302854
- Hu K, Nan X, Bird A, Wang W. Testing for association between MeCP2 and the brahma-associated SWI/SNF chromatin-remodeling complex. Nat Genet. 2006;38:962-4; author reply 4–7. doi:10.1038/ng0906-962. PMID:16940996
- Felisbino MB, Gatti MS, Mello ML. Changes in chromatin structure in NIH 3T3 cells induced by valproic acid and trichostatin A. J Cell Biochem. 2014;115:1937-47. PMID:24913611
- Ishibashi T, Dryhurst D, Rose KL, Shabanowitz J, Hunt DF, Ausio J. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry. 2009;48:5007-17. doi:10.1021/bi900196c. PMID:19385636
- Kumar A, Kamboj S, Malone BM, Kudo S, Twiss JL, Czymmek KJ, LaSalle JM, Schanen NC. Analysis of protein domains and Rett syndrome mutations indicate that multiple regions influence chromatin-binding dynamics of the chromatin-associated protein MECP2 in vivo. J Cell Sci. 2008;121:1128-37. doi:10.1242/jcs.016865. PMID:18334558
- Lever MA, Th'ng JP, Sun X, Hendzel MJ. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000;408:873-6. doi:10.1038/35048603. PMID:11130728
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255-69. doi:10.1016/j.neuron.2006.09.037. PMID:17046689
- Stefanelli G, Gandaglia A, Costa M, Cheema MS, Di Marino D, Barbiero I, Kilstrup-Nielsen C, Ausio J, Landsberger N. Brain phosphorylation of MeCP2 at serine 164 is developmentally regulated and globally alters its chromatin association. Sci Rep. 2016;6:28295. doi:10.1038/srep28295. PMID:27323888
- Bellini E, Pavesi G, Barbiero I, Bergo A, Chandola C, Nawaz MS, Rusconi L, Stefanelli G, Strollo M, Valente MM, et al. MeCP2 post-translational modifications: a mechanism to control its involvement in synaptic plasticity and homeostasis? Front Cell Neurosci. 2014;8:236. doi:10.3389/fncel.2014.00236. PMID:25165434
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72-85. doi:10.1016/j.neuron.2011.08.022. PMID:21982370
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885-9. doi:10.1126/science.1086446. PMID:14593183
- Milton SG, Mathew OP, Yatsu FM, Ranganna K. Differential cellular and molecular effects of butyrate and trichostatin a on vascular smooth muscle cells. Pharmaceuticals (Basel). 2012;5:925-43. doi:10.3390/ph5090925. PMID:24280698
- Chao HT, Zoghbi HY. The yin and yang of MeCP2 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:4577-8. doi:10.1073/pnas.0901518106. PMID:19293386
- Tao J, Hu K, Chang Q, Wu H, Sherman NE, Martinowich K, Klose RJ, Schanen C, Jaenisch R, Wang W, et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci U S A. 2009;106:4882-7. doi:10.1073/pnas.0811648106. PMID:19225110
- Sekhavat A, Sun JM, Davie JR. Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem Cell Biol. 2007;85:751-8. doi:10.1139/O07-145. PMID:18059533
- Chao HT, Zoghbi HY. MeCP2: only 100% will do. Nat Neurosci. 2012;15:176-7. doi:10.1038/nn.3027. PMID:22281712
- Nan X, Tate P, Li E, Bird A. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol. 1996;16:414-21. doi:10.1128/MCB.16.1.414. PMID:8524323
- Thambirajah AA, Ausio J. A moment's pause: putative nucleosome-based influences on MeCP2 regulation. Biochem Cell Biol. 2009;87:791-8. doi:10.1139/O09-054. PMID:19898528
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929-35. doi:10.1126/science.1132939. PMID:17008526
- Yeakley JM, Shepard PJ, Goyena DE, VanSteenhouse HC, McComb JD, Seligmann BE. A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling. PLoS One. 2017;12:e0178302. doi:10.1371/journal.pone.0178302. PMID:28542535
- Pandey P, Daghma DS, Houben A, Kumlehn J, Melzer M, Rutten T. Dynamics of post-translationally modified histones during barley pollen embryogenesis in the presence or absence of the epi-drug trichostatin A. Plant Reprod. 2017;30:95-105. doi:10.1007/s00497-017-0302-5. PMID:28526911
- Sanders YY, Tollefsbol TO, Varisco BM, Hagood JS. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts. Am J Respir Cell Mol Biol. 2011;45:16-23. doi:10.1165/rcmb.2010-0154OC. PMID:20724553
- Raghuram N, Carrero G, Stasevich TJ, McNally JG, Th'ng J, Hendzel MJ. Core histone hyperacetylation impacts cooperative behavior and high-affinity binding of histone H1 to chromatin. Biochem. 2010;49:4420-31. doi:10.1021/bi100296z.
- iClaveria-Gimeno R, Abian O, Velazquez-Campoy A, Ausió J. MeCP2…Nature's wonder protein or medicine's most feared one. Curr Genet Med Rep. 2016;4:180-94. doi:10.1007/s40142-016-0107-0.
- Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol. 2010;30(19):4656-70. doi:10.1128/MCB.00379-10. PMID:20679481
- Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246-53. doi:10.1093/nar/gkq147. PMID:20223770
- Felle M, Hoffmeister H, Rothammer J, Fuchs A, Exler JH, Langst G. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 2011;39(16):6956-69. doi:10.1093/nar/gkr263. PMID:21622955
- Raghuram N, Carrero G, Th'ng J, Hendzel MJ. Molecular dynamics of histone H1. Biochem Cell Biol. 2009;87:189-206. doi:10.1139/O08-127. PMID:19234534
- Ridsdale JA, Hendzel MJ, Delcuve GP, Davie JR. Histone acetylation alters the capacity of the H1 histones to condense transcriptionally active/competent chromatin. J Biol Chem. 1990;265:5150-6. PMID:2318888
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890-3. doi:10.1126/science.1090842. PMID:14593184
- Eickhoff B, Germeroth L, Stahl C, Kohler G, Ruller S, Schlaak M, van der Bosch J. Trichostatin A-mediated regulation of gene expression and protein kinase activities: reprogramming tumor cells for ribotoxic stress-induced apoptosis. Biol Chem. 2000;381:1127-32. doi:10.1515/BC.2000.138. PMID:11154071
- Buchthal B, Lau D, Weiss U, Weislogel JM, Bading H. Nuclear calcium signaling controls methyl-CpG-binding protein 2 (MeCP2) phosphorylation on serine 421 following synaptic activity. J Biol Chem. 2012;287:30967-74. doi:10.1074/jbc.M112.382507. PMID:22822052
- Bracaglia G, Conca B, Bergo A, Rusconi L, Zhou Z, Greenberg M, Landsberger N, Soddu S, Kilstrup-Nielsen C. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 2009;10:1327-33. doi:10.1038/embor.2009.217. PMID:19820693
- Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935-46. doi:10.1093/hmg/ddi198. PMID:15917271
- Zhu YC, Li D, Wang L, Lu B, Zheng J, Zhao SL, Zeng R, Xiong ZQ. Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc Natl Acad Sci US A. 2013;110:9118-23. doi:10.1073/pnas.1300003110.
- Feng J, Nestler EJ. MeCP2 and drug addiction. Nat Neurosci. 2010;13:1039-41. doi:10.1038/nn0910-1039. PMID:20740030
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128-40. doi:10.1523/JNEUROSCI.0296-07.2007. PMID:17553985
- Brown CR, Kennedy CJ, Delmar VA, Forbes DJ, Silver PA. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev. 2008;22:627-39. doi:10.1101/gad.1632708. PMID:18316479
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680-5. doi:10.1038/227680a0. PMID:5432063
- Stuss DP, Cheema M, Ng MK, Martinez de Paz A, Williamson B, Missiaen K, Cosman JD, McPhee D, Esteller M, Hendzel M, et al. Impaired in vivo binding of MeCP2 to chromatin in the absence of its DNA methyl-binding domain. Nucleic Acids Res. 2013;41:4888-900. doi:10.1093/nar/gkt213. PMID:23558747
- Carrero G, McDonald D, Crawford E, de_Vries G, Hendzel MJ. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods (San Diego, Calif). 2003;29:14-28. doi:10.1016/S1046-2023(02)00288-8. PMID:12543068
- Remenyi J, van den Bosch MW, Palygin O, Mistry RB, McKenzie C, Macdonald A, Hutvagner G, Arthur JS, Frenguelli BG, Pankratov Y. miR-132/212 knockout mice reveal roles for these miRNAs in regulating cortical synaptic transmission and plasticity. PLoS One. 2013;8:e62509. doi:10.1371/journal.pone.0062509. PMID:23658634