
ABSTRACT
Colorectal cancer is a major cause of cancer death and approximately 20% arises within serrated polyps, which are under-recognized and poorly understood. Human serrated colorectal polyps frequently exhibit both oncogenic BRAF mutation and widespread DNA methylation changes, which are important in silencing genes restraining neoplastic progression. Here, we investigated whether in vivo induction of mutant Braf is sufficient to result in coordinated promoter methylation changes for multiple cancer-related genes. The BrafV637E mutation was induced in murine intestine on an FVB;C57BL/6J background and assessed for morphological and DNA methylation changes at multiple time points from 10 days to 14 months. Extensive intestinal hyperplasia developed by 10 days post-induction of the mutation. By 8 months, most mice had murine serrated adenomas with dysplasia and invasive cancer developed in 40% of mice by 14 months. From 5 months onwards, Braf mutant mice showed extensive, gene-specific increases in DNA methylation even in hyperplastic mucosa without lesions. This demonstrates that persistent oncogenic Braf signaling is sufficient to induce widespread DNA methylation changes. This occurs over an extended period of time, mimicking the long latency followed by rapid progression of human serrated neoplasia. This study establishes for the first time that DNA methylation arises slowly in direct response to prolonged oncogenic Braf signaling in serrated polyps; this finding has implications both for chemoprevention and for understanding the origin of DNA hypermethylation in cancer generally.
Introduction
The serrated colorectal neoplasia pathway describes the progression of morphologically serrated polyps to cancer. The malignant potential of serrated polyps has only been understood in the last decade and this pathway is now accepted to account for approximately 20% of all colorectal cancers. Mutation of the BRAF oncogene has been exclusively associated with serrated polyp morphology [Citation1]. BRAF mutant polyps and cancers commonly methylate a defined subgroup of CpG islands, a phenomenon termed the CpG Island Methylator Phenotype (CIMP) [Citation2–5]. CIMP-specific methylation facilitates malignant progression of serrated polyps by targeting tumor suppressor genes involved in escape from oncogene induced senescence (e.g., CDKN2A), inappropriate activation of Wnt signaling (e.g., SFRP1,2,5), and mismatch repair deficiency (e.g., MLH1) [Citation6]. The cause of CIMP in colorectal cancer has been unknown. It is important to understand the mechanism by which CIMP develops, as this will impact chemoprevention and therapeutic strategies for this molecularly distinct subgroup of polyps and cancers.
Studies to date have been inconclusive regarding the timing of BRAF mutation and altered DNA methylation in colorectal cancer. Clinically, BRAF mutation occurs in the earliest identifiable serrated lesions, including aberrant crypt foci [Citation7], microvesicular hyperplastic polyps, and the vast majority of sessile serrated adenomas [Citation8]. Altered DNA methylation has been observed in histologically normal colorectal mucosa of patients with serrated polyps [Citation9]; however, CIMP marker panel methylation is not detected in normal mucosa but is common in sessile serrated adenomas and BRAF mutant cancers [Citation4,Citation5,Citation10]. In vitro knockdown of BRAF produced genome-wide alterations in DNA methylation profiles in two BRAF mutant thyroid [Citation11] and three BRAF mutant melanoma cell lines [Citation11,Citation12]. Introduction of the BRAFV600E mutation to a CIMP colonic cell line was reported to induce methylation of the MLH1 gene promoter [Citation13]; however, another report of stable expression of the BRAFV600E mutation in a CIMP-negative cell line for up to 27 passages failed to show increased methylation that differed from control cells [Citation14]. A recent study of BRAF mutant human organoids did not demonstrate development of CIMP; however, it is unclear how long the organoids were cultured [Citation15].
By inducing the murine equivalent of the human BRAFV600E mutation specifically in the intestine of adult mice, we have, for the first time, studied in detail, at multiple time points, DNA methylation alterations accompanying initiation and progression of serrated neoplasia in vivo. We demonstrate that induction of the BRAF mutation in normal intestinal cells results in the accumulation of DNA methylation defects slowly over time, mimicking progression of the serrated neoplasia pathway in humans.
Results
Morphological progression of murine serrated neoplasia
Intestine-specific induction of the Braf mutation at 2 weeks of age resulted in MAPK pathway activation (Supplementary Figure 1) and extensive hyperplasia in all mice within 10 days (A–D, ), which persisted lifelong. In the colon, the hyperplasia resembled a human microvesicular hyperplastic polyp (B). No discrete serrated lesions developed in the colon of any mice. Murine serrated hyperplasia (mSH) of the small intestine varied by region. In the duodenum, the hyperplasia was exuberant and as the mice aged the villi displayed cellular crowding, pseudostratification, and occasional mitotic activity, enlarged nuclei, and a vesicular chromatin pattern. These changes gradually reduced in severity distally and were inconspicuous by the early jejunum where the villi, although still hyperplastic, displayed a more uniform cytology.
Figure 1. Morphological changes following intestinal Braf mutation. (A) Braf wild type colon and (B) Braf mutant colon 10 days after induction of the mutation. (C) Braf wild type small intestine and (D) Braf mutant small intestine 10 days after induction of the mutation. (E) A murine serrated precursor at 5 months. (F) A murine serrated adenoma with an overtly dysplastic zone at 14 months. (G) A moderately differentiated invasive cancer arising from a murine serrated adenoma at 14 months post-induction and (H) the same cancer showing retention of MLH1 protein.
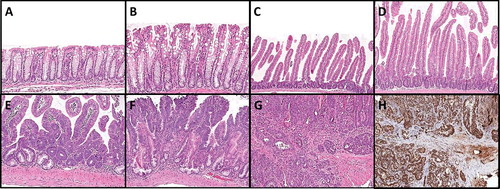
Table 1. Temporal accumulation of serrated morphology.
The earliest identifiable discrete lesions were seen in 50% of mice 10 weeks post-induction of the Braf mutation, consisting of dilatation and mucinous differentiation of some crypt bases (E). These increased in number and extent with age and were histologically reminiscent of human sessile serrated adenomas. We refer to these lesions as murine serrated precursors (mSP). These rarely involved more than 2–4 crypts. As the mice aged, some precursor lesions displayed a more eosinophilic cellular appearance towards the luminal aspect, developing dysplasia and forming large, macroscopically identifiable tumors (F). We refer to these lesions as murine serrated adenoma (mSA), as opposed to the mSA with high-grade dysplasia designation used by Rad et al. [Citation16] These were occasionally present at early time points (1/6 mice at 10 weeks, 1/12 mice at 5 months), but were present in the majority of mice at 8 months (8/10), 10 months (14/18), and 14 months (8/8) (). An invasive cancer was identified in a mouse 8 months post-induction of the Braf mutation as well as 3/8 mice at 14 months, one of which had metastasized to the liver (G, ). The average number and size of lesions per mouse increased during the study period, with an average of 11.9 lesions per mouse at 14 months, averaging 12.7 mm in diameter ().
Small lesions reminiscent of human conventional tubular adenomas were also observed in 6 Braf mutant mice but no control mice. We have designated these as murine tubular adenomas (mTA). These were present in the proximal colon in 4 mice (2 at 10 months, 2 at 14 months), and the jejunum of a 5-month and a 10-month-old mouse.
BRAF mutation induces widespread, gene-specific DNA hypermethylation in vivo
Mucosa from the proximal small intestine was sampled in BrafV637E and age-matched control BrafCA littermates at 10 days, 10 weeks, 5 months, 8 months, 10 months, and 14 months after injection of tamoxifen or vehicle, respectively. As the mice aged, there was a dramatic and gene-specific increase in DNA methylation in Braf mutant compared to Braf wild type intestinal mucosa (a). Regression lines were fit to the data for the control Braf wild type and the hyperplastic Braf mutant tissue groups versus time (b). The slope for the Braf wild type samples was significantly greater than 0 (P = 0.0008), demonstrating age-related increases in DNA methylation for a subset of genes (Supplementary Table 1). The accumulation of methylation changes in these and additional genes was accelerated by induction of the Braf mutation (). The difference in the slopes between the Braf wild type and mutant mucosal samples was highly significant (P<0.0001), reflecting the temporal accumulation of DNA methylation events following induction of the Braf mutation. To confirm that the increase in methylation observed in hyperplastic mucosa was not a result of unidentified microscopic lesions, we also assessed the samples from the jejunum 10 cm distal to the gastro-duodenal junction and from the proximal colon, two locations where hyperplasia was present but no discrete lesions were observed. A similarly dramatic increase in DNA methylation was observed at both these sites in Braf mutant samples compared to Braf wild type samples (C).
Figure 2. Temporal and gene-specific accumulation of DNA hypermethylation following Braf mutation. (A) Heat map showing hypermethylated genes in yellow, compared to unmethylated genes in blue. There was a dramatic age-related and gene-specific increase in DNA methylation in Braf mutant proximal small intestine and lesions compared to Braf wild type mucosa. (B) The average percentage methylation is plotted across time-points for Braf wild type proximal small intestine (blue), Braf mutant small intestine (yellow) and lesions (red). (C) At 12 months the proximal jejunum and colon were significantly methylated in Braf mutant (yellow) compared to Braf wild type (blue) mucosa. ****P<0.0001.
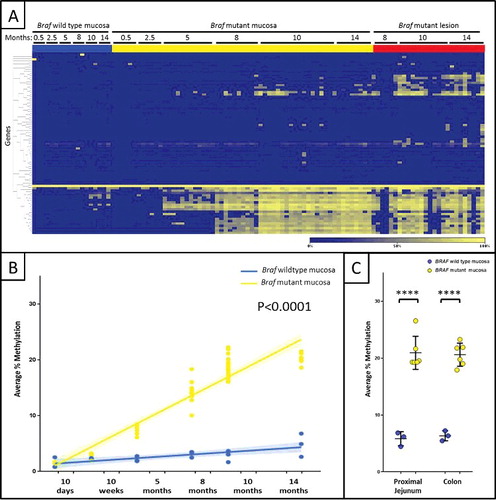
Table 2. Temporal methylation changes in Braf mutant mice.
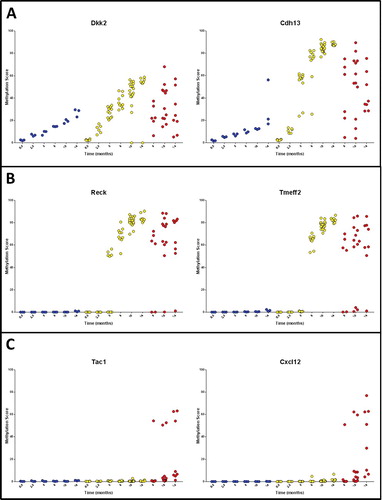
There were three major patterns of methylation observed for the genes examined: Type A, in which methylation accumulated with age but was accelerated by mutant Braf (A); Type B, in which hypermethylation was limited to Braf mutant samples (B); and Type C, where methylation was largely limited to murine serrated adenomas and cancers (C). This model may be investigated to identify epigenetic events associated with progression of serrated neoplasia. Genes differentially methylated between Braf mutant hyperplastic mucosa and murine serrated adenomas and/or cancers are shown in Supplementary Table 2. Significantly hypermethylated genes that may have a role in progression of serrated neoplasia include Sfrp5 (P = 9.3 × 10−9), Hs3st2 (P = 2.1 × 10−5), and Cxcl12 (P = 5.0 × 10−4). The most striking examples were Sfrp5, which had a methylation level of >50% in 18/21 (86%) lesions compared to <0.5% methylation in 18/18 control samples and 59/60 Braf mutant hyperplastic mucosa samples. Similarly, Hs3st2 had a methylation level >50% in 14/21 (66.7%) lesions compared to a single Braf mutant, hyperplastic sample, and no control samples.
Figure 3. Patterns of DNA methylation accumulation included (A) Type A methylation that accumulated with age but was accelerated by mutation of Braf, (B) Type B methylation changes specific to Braf mutant tissue and (C) Type C methylation changes specific to murine serrated adenomas and/or cancers.
Bhlhb9 was the only gene to exhibit gender-specific methylation. This appeared to be imprinted, with control female murine mucosa averaging 48.7% methylation across all time points, compared to 0.2% for male animals (P<0.0001). In Braf mutant hyperplasia samples, Bhlhb9 methylation significantly increased over time from 5 months in both males and females. In males Bhlhb9 methylation averaged 4.6% at 5 months compared to 63–73% at 8 months (Supplementary Figure 2).
Murine colonic organoids were cultured for 4 weeks post-induction of the Braf mutation in vitro. Complete conversion to the Braf mutant allele was observed (Supplementary Figure 3); however, at this time point there was not significant increase in DNA methylation in any of the 94 genes examined.
Mismatch repair status of Braf mutant mSA and cancers
Microsatellite instability status was examined in the 5 mSA and 3 cancers sampled at 14 months, supplemented with an additional 3 mSA and 4 cancers from 7 additional mice at 14 months. Of these 15 samples, none met the criteria for microsatellite instability based on the seven microsatellite markers examined. Two cancers and two mSA had a 20–30 bp deletion in the mBat67 marker; however, all other markers were stable. These lesions all retained immunohistochemical expression of MLH1 (H). Based on the PCR methylation array data, no sample was methylated for Mlh1.
Discussion
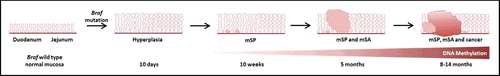
A significant subset of colorectal cancers are characterized by oncogenic BRAF mutation and widespread, coordinate DNA hypermethylation. The unique origin of these cancers in serrated polyps has been described over the last decade. However, the temporal relationship between BRAF mutation and altered DNA methylation in early serrated tumorigenesis is not well understood. Here, we present an in vivo model that morphologically mimics human serrated neoplasia and that slowly accumulates DNA methylation changes over many months, congruent with the slow development of serrated adenomas which then transition to high grade dysplasia and ultimately cancer ().
Figure 4. Model of murine serrated neoplasia initiated by intestinal Braf mutation. At 10 days post-induction of the Braf mutation, unstable hyperplasia is evident in the proximal small intestine. Murine serrated polyps (mSP) develop by 10 weeks and by 5 months murine serrated adenomas (mSA) are seen. mSP, mSA and cancer can be seen by 8–14 months. This is accompanied by the accumulation of gene-specific DNA methylation changes over time.
Intestine-specific induction of the Braf mutation in adult mice rapidly induced prolonged hyperplasia throughout the small and large intestine, consistent with the constitutive Villin-Cre model reported by Rad et al. [Citation16]. In contrast to the mSA with low-grade dysplasia described by Rad et al., we defined subtle areas of crypt dilatation and mucinous differentiation as murine serrated precursors (mSP). These were diminutive and were reminiscent of small human sessile serrated adenomas. By 8 months the majority of animals had developed murine serrated adenomas (mSA) that are likely comparable to the mSA with high-grade dysplasia described by Rad et al. By 14 months, mice developed invasive cancer. Thus, the present study, incorporating careful examination of the morphology by a gastrointestinal pathologist, confirms that induction of an oncogenic Braf mutation in murine intestine is sufficient to create a mouse model that closely mimics human serrated neoplasia.
The present study is the first to demonstrate the accumulation over time of multiple gene-specific DNA methylation events, seemingly driven by prolonged oncogenic Braf signaling in this rapidly proliferating tissue. This methylation was apparent even in tissue where the only morphological change was stable hyperplasia and thus is unlikely to be secondary to other genetic changes associated with the progression of malignancy. Consistent with a previous study, methylation was not observed in Braf mutant organoids cultured for 1 month, highlighting the additive value of long term in vivo models [Citation15]. Despite using a murine-specific PCR array that included 94 genes previously reported to be methylated in human colorectal cancer, the DNA methylation that occurred following Braf mutation was limited to a defined subset of these gene promoters, consistent with the specific nature of human CIMP [Citation2,Citation5]. By 5 months, there was a highly significant, global increase in DNA methylation in Braf mutant epithelium compared to age-matched control samples. Significant and persistent increases were seen in several genes, including Igfbp7, which is a human CIMP target gene that, when silenced, may facilitate escape from oncogene induced senescence [Citation14]. By 8 months, significant methylation had been accumulated, including in Crabp1, which is highly specific for human CIMP [Citation5,Citation17]. Maximum global methylation changes were reached by 10 months, including specific hypermethylation of the Wnt pathway gene Sfrp2.
Differential methylation of a number of genes was significantly associated with development of mSA or cancer, including the Wnt pathway genes Wnt5a [Citation18] and Sfrp5 [Citation19]. Two studies have reported methylation of defined sites within the p16INK4a gene promoter in murine mSA or cancer induced by intestinal Braf mutation [Citation16,Citation20]. At the CpG sites assayed in the present study, p16INK4a was not significantly methylated at any time point assessed for any sample. This may reflect strain-specific sequence differences mediating p16 promoter hypermethylation, or may be due to the different CpG sites assessed in these studies. The study by Rad et al. showed an increase in average p16INK4a transcript expression with progression to high-grade dysplasia. The average p16INK4a expression level remained elevated in cancers, but was reduced somewhat, likely reflecting silencing due to acquired promoter hypermethylation and silencing in only a proportion of mSAs and cancers. This is consistent with what has been observed during progression of human serrated neoplasia [Citation4].
A surprising finding was that in contrast to the model presented by Rad et al., we did not observe microsatellite instability in the current study, despite using the same marker panel [Citation16]. Our findings were consistent with a lack of MLH1 methylation and retained MLH1 protein expression in all samples examined. Rad et al. showed MSI in 9/23 mSA with high-grade dysplasia and 3/8 cancers, but did not present data for MLH1 methylation or protein expression. We expanded our study numbers to investigate a total of 8 mSA and 7 invasive cancers from 14-month-old mice but still did not observe microsatellite instability or MLH1 protein loss in any of these lesions. In humans, single nucleotide polymorphisms have been associated with increased risk of site-specific DNA methylation, particularly for the -93 SNP in the MLH1 promoter [Citation21]. It is possible that strain differences between the current (FVB/BL6) and Rad et al. models may have impacted such a site in mice. These mice may therefore present an excellent model to study the development BRAF mutant, microsatellite stable cancer in vivo, which is a subgroup of human colorectal cancers with a particularly poor prognosis [Citation22].
The specificity of gene methylation occurring in stable hyperplasia at defined time points is intriguing and may relate to a sequence-specific threshold of MAPK signaling required to trigger DNA methylation. This is consistent with the model proposed by Fang et al., in which MAPK signal transduction increases phosphorylation of MAFG and sequence-specific promoter binding and hypermethylation by DNMT3b [Citation23]. It is possible that prolonged exposure to elevated MAPK signaling is required for efficient phosphorylation of MAFG (or other) transcription factors involved in this process. Promoters with multiple binding sites for relevant transcription factors may be more susceptible to DNA hypermethylation by this mechanism. We did not observe an over-representation of MAFG consensus binding sites in silico for genes significantly hypermethylated in this model. Consistent with this was the lack of promoter methylation for MLH1 in any samples in this study, despite being the classic example of MAFG-mediated methylation presented by Fang. This may be due to different MLH1 CpG sites being assessed in the present study; however, the lack of MLH1 methylation was consistent with the retention of mismatch repair efficiency indicated by microsatellite stability and retention of the MLH1 protein. An alternate hypothesis is that methylation of MLH1 may be mediated by DNA sequences that vary between humans and mice, or even different strains of mice.
Despite the high rate of conversion to the mutant Braf allele and development of stable hyperplasia throughout the intestine, the neoplastic phenotype was predominantly localized to the duodenum and proximal jejunum. This is a common feature of murine models of colorectal carcinogenesis and is also observed in models of conventional adenomas driven by Apc mutation. Despite this morphological gradient, there was not a concomitant DNA methylation gradient, based on the 94 genes assessed in this study.
This study substantially advances our understanding of the role of BRAF mutation in orchestrating multiple DNA methylation changes that underlie progression of serrated neoplasia. We have presented a conditional murine model that faithfully recapitulates human serrated polyp development, both morphologically and in terms of accumulation of gene-specific DNA methylation changes. For the first time, we have demonstrated in vivo that Braf mutation is sufficient to induce a methylator phenotype and that this is associated with a protracted time frame, consistent with the long dwell time observed for human serrated neoplasia. This model will provide a valuable tool for development of chemoprevention and therapeutic strategies for BRAF mutant colorectal neoplasia, as well as to further probe the mechanism by which CIMP is established.
Materials and methods
BRAF mutant murine model
Mice homozygous for the conditionally active BrafV637E mutant allele (BrafCA/CA, FVB background) (analogous to the human BRAFV600E mutation) [Citation24] were crossed with heterozygous Villin-CreERT2/wt mice (C57BL/6J background) [Citation25] and genotyped at 10–12 days old (Supplementary Methods). Induction of the Braf mutation was directed to the intestine at 2 weeks of age using a single 75 mg/kg intraperitoneal injection of tamoxifen (Sigma-Aldrich). Control mice had the same genotype but were injected with vehicle only. At defined time points the gastrointestinal tract from esophagus to rectum was opened longitudinally and mucosal scrapings were sampled from the proximal small intestine, the jejunum 10 cm distal to the gastroduodenal junction and the proximal colon. Macroscopic lesions were bisected for molecular and histological assessment. The entire remaining intestine was examined histologically. Animal breeding and experimental protocols were approved by the QIMR Berghofer Animal Ethics Committee (P1208).
Morphological assessment of murine intestinal epithelium
Hematoxylin and eosin stained sections were examined by an anatomical pathologist (CL). BrafV637E samples were compared with age-matched Braf wild type littermates. Histological type and number of lesions were recorded for all mice. Lesions were characterized predominantly using the terminology of Rad et al. [Citation16] as murine serrated adenomas with either low grade or high grade dysplasia (mSA-LGD or mSA-HGD), dependent on the degree of architectural and cytological abnormality. As murine small bowel contains very little muscularis mucosae and submucosa, it was difficult to distinguish between intramucosal carcinoma and submucosal invasive carcinoma. We therefore designated invasive carcinoma only in unequivocal cases that invaded the muscularis propria or beyond. Closely located lesions were counted separately if at least one layer of normal epithelial cells was seen between the lesions.
DNA methylation profiling in murine tissue
Proximal small intestinal epithelium was assessed for DNA methylation changes at 10 days, 10 weeks, 5 months, 8 months, 10 months and 14 months post-induction of the Braf mutation (minimum 5 samples per time point) with age-matched Braf wild type littermates (n = 3 for each time point). Mucosal scrapings were snap frozen in liquid nitrogen and genomic DNA was extracted using AllPrep Kits (Qiagen). DNA methylation was assessed from a total of 2 μg DNA using Epitect Methyl II Complete PCR Arrays (Qiagen) that include 94 genes that have previously been found to be methylated in colorectal cancer. Following digestion with either a methylation-sensitive or methylation-dependent restriction enzyme, DNA was amplified using a BioRad CFX384 real-time cycler and the percentage of gene methylation was calculated using the Epitect data analysis template. Heatmaps were generated using hierarchical clustering with a Pearson correlation measurement for similarity.
Murine colonic organoid culture
Organoids were cultured from heterozygous BrafCA/wt / Villin-CreERT2/wt colonic epithelial cells [Citation26] and plated in a two-layer matrigel format [Citation27]. Established organoids were treated with vehicle or 1 μM 4-dyroxytamoxifen to generate BRAFV637E/wt mutant organoids. PCR genotyping confirmed efficient recombination of the floxed allele (Supplementary Figure 2). Braf mutant organoids were passaged every 5–7 days and harvested at day 30 for genomic DNA isolation using a Qiagen AllPrep Kit columns.
Mismatch repair status
Immunostaining for the MLH1 mismatch repair protein was performed using the BD Pharmingen G168-15 MLH1 primary antibody at a 1/100 dilution for 60 minutes at room temperature. Antigen retrieval was at pH 9.0 using Target Retrieval Solution (Agilent Technologies). Background Sniper (Biocare Medical) and 5% peroxidase blocks were applied for 30 minutes each to minimize background staining in the murine tissue. Microsatellite instability was assessed as a functional readout of mismatch repair activity using a panel of 7 murine microsatellite markers (mBat26, mBat37, mBat67, TG27, GA29, A33, and A27) [Citation16]. PCR products were separated on an ABI PRISM 3100 Genetic Analyzer to assess altered fragment size in tumors compared to matched intestinal tissue for each sample. Methylation status of the MLH1 mismatch repair gene promoter was extracted from the Epitect Methyl II PCR Array.
Statistical analysis
Continuous variables were analysed using Student's T-test. Linear regressions were used to analyze continuous variables versus time. Parallelism of regression lines models was assessed using multiple regression, which included slope by group interaction terms. The family wise error rates for comparisons of large sets of genes were controlled using the Benjamini-Hochberg procedure for adjusting P values to control the false discovery rate. P values ≤0.05 were considered statistically significant.
Supplementary methods
Genotyping
DNA for genotyping was extracted from the tail tissue using QuickExtract (EpiBio). The Villin-Cre-ERT2 allele was genotyped by qPCR (2.5 mM MgCl2, 0.25 mM dNTP, 0.5 mM forward primer (5’-CAA GCC TGG CTC GAC GGC C-3’), 0.5 mM reverse primer (5’-CGC GAA CAT CTT CAG GTT CT-3’), 0.5 mM forward bthal positive control primer (5’-TGA GAA GGC TGC TGT CTC TTG-3’) and reverse bthal positive control primer (5’-CAG AGG ATA GGT CTC CAA AGC TA-3’), 0.25 uM SYTO9, 4 uL 5X GoBuffer (Promega), 0.5 U GoTaq (Promega) under the following thermal cycling conditions: 94⁰C for 120 s; 40 cycles of: 94⁰C for 30 s, 55⁰C for 30 s, 72⁰C for 45 s followed by 72⁰C for 300 s, 50⁰C for 120 s and high resolution melt from 80⁰C to 92⁰C ramping by 0.2⁰C/step) and subsequent high resolution melt profile analysis. BRAFCA was similarly genotyped via qPCR (2.5 mM MgCl2, 0.25 mM dNTP, 0.5 mM forward primer, 0.5 mM reverse primer, 4 uL 5X GoBuffer, 0.5 U GoTaq under the following thermal cycling conditions: 94⁰C for 120 s; 40 cycles of: 94⁰C for 30 s, 58⁰C for 30 s, 72⁰C for 40 s followed by 72⁰C for 300 s, 50⁰C for 120 s and high resolution melt from 72⁰C to 92 ramping by 0.2⁰C/step) and subsequent high resolution melt profile analysis.
Disclosure of potential conflicts of interest
The authors have no conflicts to disclose.
supp_data_1411446.zip
Download Zip (132.8 KB)Acknowledgements
We are grateful to Prof Martin McMahon for providing the BRAFV600E conditional mouse and to Prof Greg Anderson and Sarah Wilson for providing the VillinCre mouse with the permission of Prof Sylvie Robine.
Additional information
Funding
References
- Kambara T, Simms LA, Whitehall VL, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53:1137–1144. doi:10.1136/gut.2003.037671. PMID:15247181
- Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686. doi:10.1073/pnas.96.15.8681. PMID:10411935
- Yang S, Farraye FA, Mack C, et al. BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol. 2004;28:1452–1459. doi:10.1097/01.pas.0000141404.56839.6a. PMID:15489648
- Bettington M, Walker N, Rosty C, et al. Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut. 2017;66:97–106. doi:10.1136/gutjnl. PMID:26475632
- Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi:10.1038/ng1834. PMID:16804544
- Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–2100. doi:10.1053/j.gastro.2009.12.066. PMID:20420948
- Rosenberg DW, Yang S, Pleau DC, et al. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer Res. 2007;67:3551–3554. doi:10.1158/0008-5472.CAN-07-0343. PMID:17440063
- Spring KJ, Zhao ZZ, Karamatic R, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology. 2006;131:1400–1407. doi:10.1053/j.gastro.2006.08.038. PMID:17101316
- Worthley DL, Whitehall VL, Buttenshaw RL, et al. DNA methylation within the normal colorectal mucosa is associated with pathway-specific predisposition to cancer. Oncogene. 2010;29:1653–1662. doi:10.1038/onc.2009.449. PMID:19966864
- Fernando WC, Miranda MS, Worthley DL, et al. The CIMP phenotype in BRAF mutant serrated polyps from a prospective colonoscopy patient cohort. Gastroenterol Res Pract. 2014;2014:374926. doi:10.1155/2014/374926. PMID:24812557
- Hou P, Liu D, Xing M. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells. Endocr Relat Cancer. 2011;18:687–697. doi:10.1530/ERC-11-0212. PMID:21937738
- Hou P, Liu D, Dong J, et al. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012;11:286–295. doi:10.4161/cc.11.2.18707. PMID:22189819
- Minoo P, Moyer MP, Jass JR. Role of BRAF-V600E in the serrated pathway of colorectal tumourigenesis. J Pathol. 2007;212:124–133. doi:10.1002/path.2160. PMID:17427169
- Hinoue T, Weisenberger DJ, Pan F, et al. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS One. 2009;4:e8357. doi:10.1371/journal.pone.0008357. PMID:20027224
- Fessler E, Drost J, van Hooff SR, et al. TGFbeta signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol Med. 2016;8:745–760. doi:10.15252/emmm.201606184. PMID:27221051
- Rad R, Cadinanos J, Rad L, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell. 2013;24:15–29. doi:10.1016/j.ccr.2013.05.014. PMID:23845441
- Ogino S, Cantor M, Kawasaki T, et al. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–1006. doi:10.1136/gut.2005.082933. PMID:16407376
- Ying J, Li H, Yu J, et al. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res. 2008;14:55–61. doi:10.1158/1078-0432.CCR-07-1644. PMID:18172252
- Linhart HG, Lin H, Yamada Y, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110–3122. doi:10.1101/gad.1594007. PMID:18056424
- Carragher LA, Snell KR, Giblett SM, et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med. 2010;2:458–471. doi:10.1002/emmm.201000099. PMID:20941790
- Chen H, Taylor NP, Sotamaa KM, et al. Evidence for heritable predisposition to epigenetic silencing of MLH1. Int J Cancer. 2007;120:1684–1688. doi:10.1002/ijc.22406. PMID:17230510
- Whitehall V, Burge M. Management of BRAF-muant metastatic colorectal cancer. Colorectal Cancer. 2016;5:131–133. doi:10.2217/crc-2016-0012.
- Fang M, Ou J, Hutchinson L, et al. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol Cell. 2014;55:904–915. doi:10.1016/j.molcel.2014.08.010. PMID:25219500
- Dankort D, Filenova E, Collado M, et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–384. doi:10.1101/gad.1516407. PMID:17299132
- el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi:10.1002/gene.20042. PMID:15282745
- Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–1772. doi:10.1053/j.gastro.2011.07.050. PMID:21889923
- Onuma K, Ochiai M, Orihashi K, et al. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc Natl Acad Sci U S A. 2013;110:11127–11132. doi:10.1073/pnas.1221926110. PMID:23776211