
ABSTRACT
Homeobox (HOX) genes are frequently dysregulated in leukemia. Previous studies have shown that aberrant HOX gene expression accompanies leukemogenesis and affects disease progression and leukemia patient survival. Patients with acute myeloid leukemia (AML) bearing PML-RARα fusion gene have distinct HOX gene signature in comparison to other subtypes of AML patients, although the mechanism of transcription regulation is not completely understood. We previously found an association between the mRNA levels of HOX genes and those of the histone demethylases JMJD3 and UTX in PML-RARα- positive leukemia patients. Here, we demonstrate that the release of the PML-RARα-mediated block in PML-RARα-positive myeloid leukemia cells increased both JMJD3 and HOX gene expression, while inhibition of JMJD3 using the specific inhibitor GSK-J4 reversed the effect. This effect was driven specifically through PML-RARα fusion protein since expression changes did not occur in cells with mutated RARα and was independent of differentiation. We confirmed that gene expression levels were inversely correlated with alterations in H3K27me3 histone marks localized at HOX gene promoters. Furthermore, data from chromatin immunoprecipitation followed by sequencing broaden a list of clustered HOX genes regulated by JMJD3 in PML-RARα-positive leukemic cells. Interestingly, the combination of GSK-J4 and all-trans retinoic acid (ATRA) significantly increased PML-RARα-positive cell apoptosis compared with ATRA treatment alone. This effect was also observed in ATRA-resistant NB4 clones, which may provide a new therapeutic opportunity for patients with acute promyelocytic leukemia (APL) resistant to current treatment. The results of our study reveal the mechanism of HOX gene expression regulation and contribute to our understanding of APL pathogenesis.
Introduction
Acute myeloid leukemia (AML) represents 15-20% of childhood leukemia cases and is a highly heterogeneous disease with a worse outcome than other subtypes of leukemias [Citation1]. Acute promyelocytic leukemia (APL) is a subtype of AML with a PML (promyelocytic leukemia)-RARα (retinoic acid receptor α) fusion gene formed as a result of the chromosomal translocation t(15;17)(q22;q12-22) in majority of APL cases [Citation2]. PML-RARα blocks myeloid differentiation and enhances the proliferation of leukemic cells that are arrested in the promyelocytic stage [Citation3]. This differentiation block can be released by all-trans retinoic acid (ATRA), which binds to and transcriptionally activates the RARα moiety and induces the degradation of the PML-RARα fusion protein [Citation4–Citation7]. For this reason, ATRA is commonly used to treat PML-RARα-positive patients [Citation8–Citation10]. This therapeutic agent has transformed a previously fatal disease into a diagnosis with an excellent prognosis. However, approximately 15% of all APL patients still develop resistance to current therapies, including ATRA treatment (mostly due to de novo mutations in the PML-RARα moiety or the destruction of PML-RARα followed by the activation of other oncogenes [Citation11,Citation12].
The PML-RARα fusion protein modulates the expression of various target genes, including epigenetic modifiers that chemically alter nucleotides or amino acids in the chromatin structure and thus activate or repress target genes. JMJD3 (also known as KDM6B) is a lysine (K)-specific demethylase that contains a Jumonji C (JmjC) catalytic domain. JMJD3 catalyzes the demethylation of trimethylated histone 3 lysine 27 (H3K27me3) – a repressive histone mark. JMJD3 has previously been described as a direct target of PML-RARα [Citation13]. Interestingly, JMJD3 and another histone demethylase, UTX (KDM6A – lysine (K)-specific demethylase 6A), have been identified as regulators of homeobox (HOX) genes during embryogenesis [Citation14,Citation15].
In the present study, we determined the role of the H3K27me3 demethylases JMJD3 and UTX in HOX gene regulation in PML-RARα-positive leukemic cells. Our findings contribute to the understanding of the complexity of leukemogenesis in APL and the molecular mechanisms by which the response to ATRA affects the chromatin landscape of PML-RARα-positive leukemic cells.
Results
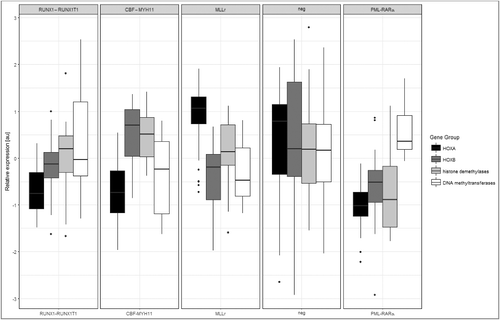
Expression of HOX genes and chromatin modifiers in genetically characterized AML subgroups
In our previous study [Citation16], we evaluated the mRNA levels of HOX genes and representative chromatin modifiers in 46 pediatric AML samples. In the current study, we further analyzed these data to determine how epigenetic modifications contribute to transcriptional regulation. We compared the expression of HOXA, HOXB, histone demethylases and de novo DNA methyltransferases (DNMTs) in genetically characterized AML subgroups (RUNX1-RUNX1T1 (n = 6), CBF-MYH11(n = 5), PML-RARα (n = 8), MLL translocations (MLLr; n = 9) and normal karyotype (neg; n = 18). Each AML subgroup as presented by graph displayed unique pattern represented by four sets of genes (). We turned our attention to PML-RARα-positive subgroup, which is characterized by overall low HOX gene expression [Citation16,Citation17]. In this subtype, levels of HOX genes and histone demethylases (JMJD3 and UTX) were in the same range, which predicts potential functional interaction. On the other hand gene levels of DNA methyltransferases (DNMT3a and DNMT3b) exhibited overall higher expression.
Figure 1. HOX and epigenetic modifiers gene expression detected in AML patients.
Effect of the PML-RARα-mediated inhibition of HOX and epigenetic modifier gene expression
Since ATRA has been shown to release the PML-RARα-mediated differentiation block, we treated NB4 (PML-RARα-positive) leukemic cells with this agent. Upon 8 h administration of ATRA, we observed significantly increased expression of HOX genes (HOXA1, A3, A4, A5, A6, A7, B5 and B6, A, D) and the histone demethylase JMJD3, while the expression of the DNMT3b gene decreased. Upregulated JMJD3 expression was detected at both the mRNA and protein level, while the expression of the histone demethylase UTX remained unchanged (A, C). Interestingly, in concordance with Thompson et al., we did not observe HOX gene expression increase after longer exposure to ATRA (48 h, time of differentiation effect) even though JMJD3 levels remained increased ([Citation18], Fig. S2C). We did not observe anymore increase in HOX expression after ATRA when compared to 8 h treatment, even though levels of JMJD3 remained increased. We did not observe changes in other epigenetic modifiers either, such as EZH2, MLL, BMI-1 or DNMT3a, upon ATRA treatment (Fig. S1). To exclude non-specific, PML-RARα-independent effects of ATRA in leukemic cells, we also analyzed two previously described clones of the NB4 cell line (NB4-LR2 and NB4-MR2) that are resistant to ATRA due to mutations in the retinoic acid binding domain of RARα, which disables the ATRA-mediated release of the PML-RARα differentiation block. In this model of ATRA-resistant leukemia, ATRA administration did not change the mRNA level of any of the studied genes (B), indicating that ATRA acts specifically through the RARα moiety. We also used phorbol myristate acetate (PMA), a pan-differentiation agent, to determine if ATRA-resistant cell lines are capable of PML-RARα-independent differentiation and, if so, how it affects HOX gene expression. Both ATRA-sensitive and ATRA-resistant cell lines differentiated into the monocytic stage upon PMA treatment (Fig. S2A). Interestingly, HOX gene expression in all three cell lines decreased upon PMA treatment (Fig. S2B) and the effect was preserved even after 48 h (Fig. S2C).
Figure 2. Expression of specific HOX genes and related epigenetic modifiers in ATRA-sensitive and ATRA-resistant cell lines.
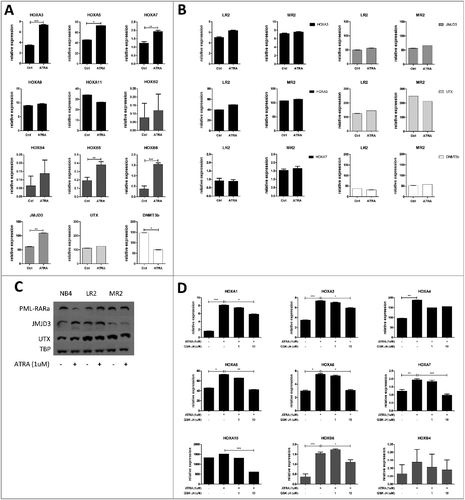
Role of JMJD3 in the regulation of HOX gene expression in PML-RARα-positive cells
To study the role of JMJD3 in HOX gene expression regulation, we employed GSK-J4, inhibitor of the Jumonji demethylases JMJD3 and UTX [Citation19]. We have chosen concentrations of inhibitor that did not interfere with cell viability (Fig. S3). We determined the level of specific HOX genes after 8 h treatment with ATRA alone or in combination with GSK-J4. The HOX genes chosen for this analysis were the most downregulated genes in PML-RARα-positive patients compared with other genetically characterized AML subgroups described in our previous publication [Citation16]. We found a significant increase in HOX gene levels (HOXA1, P = 0.0003; HOXA3, P = 0.0013; HOXA4, P = 0.0054; HOXA5, P = 0.0118; HOXA7, P = 0.004; HOXB5, P = 0.0049 and HOXB6, P<0.0001). Changes in the HOXA10 gene expression were in the same trend even milder. HOXA9, HOXA11 and HOXB2, HOXB4 remained unchanged. The effect of ATRA was reversed by JMJD3/UTX inhibition for majority tested HOX genes. Lower concentration of GSK-J4 (1uM) showed non-significant trend to reverse the effect of ATRA. This observation was more profound and statistically significant when higher concentration of GSK-J4 (10μm) was used (HOXA1, P = 0.0305; HOXA3, P = 0.0152; HOXA4, P = 0.1118; HOXA5, P = 0.0092; HOXA6, P = 0.0315; HOXA7, P = 0.0017; HOXA10, P = 0.0003; and HOXB6, P = 0.0029, D).
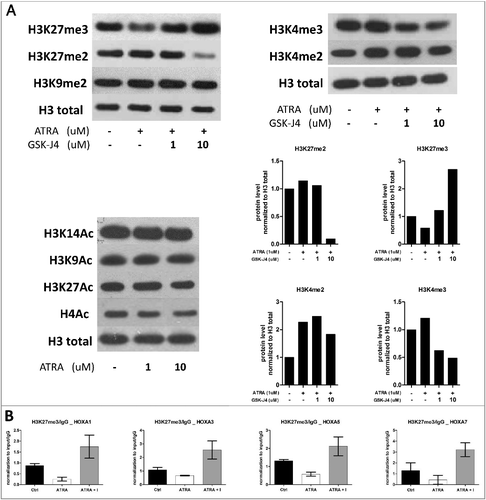
Global histone code changes upon ATRA treatment in PML-RARα-positive cells
ATRA releases the PML-RARα-dependent differentiation block, which is accompanied by extensive epigenetic landscape alterations. To identify these changes, we investigated posttranslational histone modifications such as methylation and acetylation in NB4 cells treated with ATRA. We also used combination treatment with GSK-J4 to evaluate the role of histone demethylases in the ATRA-driven changes. The level of the repressive histone methylation mark H3K27me3 decreased after ATRA treatment and increased after ATRA/GSK-J4 co-treatment. In the case of dimethylated H3 lysine 27 (H3K27me2), a decrease after combination treatment with ATRA and GSK-J4 was observed (A). The opposite effect was detected for the active histone methylation marks di- and trimethylated histone 3 lysine 4 (H3K4), which were increased after ATRA treatment (H3K4me2) and decreased after ATRA/GSK-J4 co-treatment. Methylated H3 lysine 9, acetylated H4, acetylated H3 lysine 9 and acetylated H3 lysine 14, as representatives of other active/repressive histone marks, were not changed (A).
Figure 3. Histone mark changes upon ATRA and GSK-J4 inhibitor.
Histone mark changes at HOX gene promoters upon treatment of PML-RARα-positive cells with ATRA
We used chromatin immunoprecipitation (ChIP) to examine the changes in histone marks at selected HOX (HOXA1, HOXA3, HOXA5, HOXA7) gene promoters. We focused on H3K27me3 modification after ATRA treatment as well as combination treatment with GSK-J4 as described above. ATRA significantly reduced the level of the repressive histone mark H3K27me3 (HOXA1, P = 0.007; HOXA3, P = 0.006; HOXA5, P = 0.033 and HOXA7, P = 0.049) and GSK-J4 reversed this effect (HOXA1, P = 0.046; HOXA3, P = 0.036; HOXA5, P = 0.007 and HOXA7, P = 0.007; B). These results correspond to the changes observed by western blot.
To further investigate the histone methylation pattern of HOX gene promoters, we performed ChIP with an H3K27me3 antibody followed by next-generation sequencing (ChIP-seq). We identified regions bound by the H3K27me3 antibody (peaks) and retained only the peaks that were present in both replicates of each sample. The total number of detected peaks decreased after ATRA treatment compared with the control sample and then increased back after the co-treatment of ATRA with GSK-J4 inhibitor (Ctrl ∼ 92 738, ATRA ∼ 42 650, ATRA+I ∼ 117 751) (A). Similar trend was also visible for the number of genes with peaks in their promoter region [less than 2 kb from their transcription start site (TSS)] (Ctrl ∼ 4057, ATRA ∼ 1677, ATRA+I ∼ 4926) (A). These results are in line with the changes observed by ChIP-qPCR.
Figure 4. Enrichment of histone mark H3K27me3 on HOX gene promoters upon changes in JMJD3 expression.
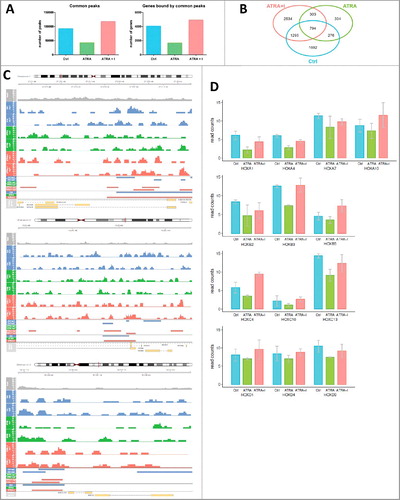
Next, we compared the list of genes with H3K27me3 peaks in their promoters under all three conditions (control, ATRA treatment and combination treatment with ATRA and GSK-J4) and presented it as a Venn diagram (B). The intersection between ‘ATRA+I’ and ‘Ctrl’ (n = 1295) contains genes whose methylation pattern predicts that their transcription is regulated by JMJD3. HOXA3, HOXA10, HOXB3, HOXC9 and HOXC11 were included in that intersection (B). Visualization of peak calling analysis of HOXA10, HOXB3 and HOXC9 is represented in C.
In the regions of HOX genes in which there were not enough peaks called (due to suboptimal ChIP-seq outputs), we subsequently used normalized numbers of reads (ncounts) to identify methylation enrichment on other HOX gene promoters. D (and Fig. S5) show HOX genes with reduced H3K27me3 upon ATRA treatment, with the effect reversed by the addition of the GSK-J4 inhibitor (n = 10). Moreover, we show HOX genes that did not follow this pattern (Fig. S4A). Neither did the gene expression of HOXA9 and HOXA11 change upon ATRA and combination with GSK-J4 (Fig. S4B).
Effect of JMJD3 inhibition on the ATRA-mediated differentiation of PML-RARα-positive cells
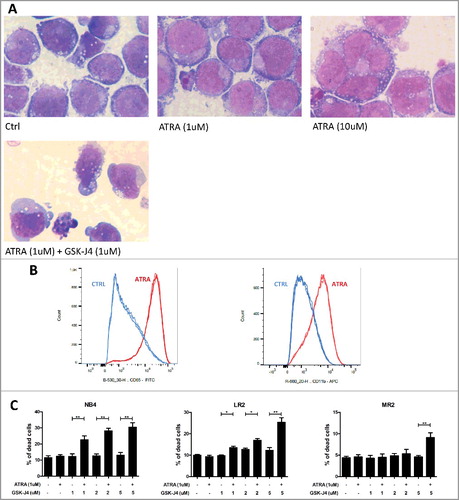
Next, we studied the impact of JMJD3 inhibition on ATRA-driven differentiation. The differentiation of NB4 cells from promyelocytes to an abnormal type of neutrophil granulocyte stage after ATRA and GSK-J4 treatment (48 h) was determined morphologically. The cells did not show any additional differentiation changes (such as increased nuclear segmentation) upon treatment with a combination of the drugs compared with ATRA treatment alone (A). However, since we observed apoptotic signs in a proportion of the treated cells, we also measured cell death markers using flow cytometry. Indeed, the combination of ATRA and GSK-J4 significantly increased cell death compared with ATRA or GSK-J4 treatment alone (B). Interestingly, we also observed a significant apoptotic effect with higher doses of GSK-J4 in combination with ATRA in the ATRA-resistant clones of the NB4 cell line (B).
Figure 5. Determination of differentiation and apoptotic status of APL cells upon ATRA and ATRA/GSK-J4 combination.
Role of DNA methylation in the regulation of HOX gene expression in PML-RARα-positive cells
Since ATRA treatment decreased the mRNA level of DNMT3b in NB4 cells, we also investigated the role of DNA methylation in the PML/RARα-mediated regulation of HOX genes. We performed bisulfite sequencing of specific HOX gene promoter sites (two sites in the HOXA5 promoter [−400 – (−223) bp upstream of the TSS; and −233 bp upstream – (+15) bp downstream of the TSS] and the HOXA7 promoter [−186 bp upstream – (+2) bp downstream of the TSS; and −427 – (−186) bp upstream of the TSS)] upon treatment of NB4 cells with ATRA (24, 48 and 72 h time points). We did not observe any differences in CpG dinucleotide methylation upon ATRA treatment compared with control cells (Fig. S6).
Discussion
Our previous study suggested that AML-specific fusion oncoproteins affect pathways that deregulate the HOX genes, thereby acting as one of underlying factors of their characteristic expression patterns. Specifically, AML patients with the PML-RARα fusion protein exhibited the lowest HOXA and HOXB gene expression among all AML patients, and this pattern was not disturbed even by the presence of the FLT3-ITD (internal tandem duplication) mutation known to be associated with overall higher HOX gene levels [Citation16,Citation17]. These findings imply that the presence of PML-RARα influences the regulation of HOX gene expression, and we therefore aimed to explore the regulation of HOX gene expression in this particular genetic subtype of AML.
HOX genes are regulated by complex molecular mechanisms, including chromatin accessibility. HOX gene promoters consist of so-called bivalent domains where the repressive histone mark methylated H3K27 and active histone mark methylated H3K4 co-localize [Citation20,Citation21]. The primary enzymes involved in chromatin structure modifications of these domains are (1) polycomb repressive complex 2 (PRC2), which is responsible for H3K27 methylation; (2) polycomb repressive complex 1 (PRC1), which maintains the silencing; (3) the trithorax complex (Trx), which coordinates H3K4 methylation; and (4) DNA methyltransferases, which inhibit transcription [Citation22–Citation28]. H3K27 methylation can be reversed by another class of histone modifiers erasers, the Jumonji C histone demethylases JMJD3 and UTX. Croce et al. showed that UTX regulates HOX gene levels, and its binding to HOXB1 was also described by Agger et al. [Citation14,Citation29]. UTX has been shown to establish functional interactions with the TrxG member MLL2 (KMT2D) [Citation30], suggesting coordinated action of H3K4me3-promoting and H3K27me3-removing activities on the HOX gene cluster.
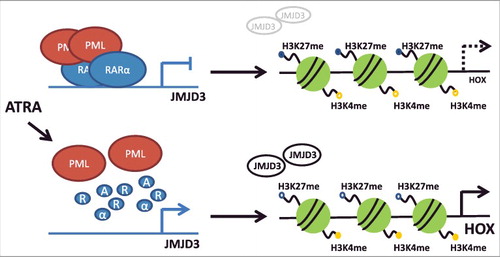
Genetic aberrations found in hematopoietic stem cells or progenitor cells initiate leukemogenesis through disruption of normal hematopoietic development and impairment of physiological functions of blood cells. The PML-RARα fusion gene is an example of a genetic lesion that blocks differentiation and leads to leukemia development [Citation31]. PML-RARα induces a unique chromatin landscape by co-recruiting various epigenetic modifiers such as DNMTs, histone methyltransferases and histone deacetylase complexes [Citation18,Citation32–Citation35]. A genome-wide binding analysis showed that PML-RARα targets different chromatin modifiers including JMJD3 [Citation13]. In the current study, we explored the role of histone demethylases in the regulation of HOX gene transcription in PML-RARα-positive leukemia. Our data demonstrated that disruption of the PML-RARα block by ATRA increased JMJD3 accompanied by an increase in the levels of specific HOXA and HOXB genes, and this effect was reversed by a JMJD3/UTX inhibitor (). In agreement with this, we observed H3K27me3 demethylation upon ATRA treatment and reverse methylation upon ATRA/GSK-J4 treatment, which was accompanied by a reduction in H3K27me2. We assume that this outcome is caused by dynamics between di- and tri-methylation, where one modification is accumulated at the expense of the other. Changes in other histone modifications upon ATRA were not observed supporting unique effect of H3K27me3 in HOX gene regulation. Levels of the repressive H3K27me3 mark localized at chosen HOXA promoters corresponded to mRNA expression levels upon ATRA and ATRA/GSK-J4 treatment. These results were obtained using ChIP-qPCR, and to widen the target detection, we also used ChIP-seq. We confirmed the ChIP-qPCR data and extended the list of HOX genes regulated by histone demethylation in PML-RARα-positive leukemic cells. HOXB6 was one of the genes which gene expression changed but we found opposite enrichment of H3K27me3 on its promoter site. One of the explanation could be that HOXB6 has different regulatory machinery [Citation35]. Importantly, we demonstrated that the observed effect of ATRA was dependent on RARα signaling, since there was no change in the expression of either histone demethylases or HOX genes in ATRA-resistant cells carrying a RARα mutation. Thus, we excluded any non-specific, RARα-independent effects of ATRA on the expression of the studied genes. This supports the notion that HOX gene expression is regulated specifically by PML-RARα. Moreover, ATRA-induced increase of JMJD3 or HOX gene expression seems to be independent of the PML-RARa-dependent differentiation block. We showed that differentiation driven by pan–differentiation agent PMA in cell lines carrying either wild-type or mutated PML-RARα actually led to an overall decrease in HOX gene expression. Decreased HOX gene expression and increased JMJD3 expression have been shown to occur during physiological hematopoietic differentiation [Citation16,Citation36]. Furthermore, treatment with ATRA for 48 h (sufficient time for ATRA-driven differentiation) led to no change or decrease of HOX gene levels. This is in concordance with already published data of Thompson et al. [Citation18]. Finally, DNA methylation has been shown to participate in the establishment of HOX gene expression patterns, further supporting the role of epigenetics in the regulation of these genes [Citation22]. Nevertheless, we did not observe any changes in DNA methylation in our studied model. This could be explained by the fact that PML-RARα has been shown to physically interact with DNMT3a, but not DNMT3b [Citation37], and we observed changes only in DNMT3b in our model cell line.
Figure 6. Scheme representing the HOX gene regulation by histone demethylation in PML-RARα- positive leukemia.
The role of JMJD3 in the PML-RARα-dependent differentiation block was further examined by studying morphological changes. We did not find any obvious change in differentiation in ATRA-treated samples with or without GSK-J4 treatment. However, we observed a significant cytotoxic effect of treatment with ATRA and the JMJD3/UTX inhibitor together. Moreover, GSK-J4 significantly enhanced cell death in both ATRA-sensitive NB4 cells and ATRA-resistant clones. This effect requires further exploration, but we hypothesize that it is independent of the mechanism underlying HOX gene regulation upon ATRA/GSK-J4 co-treatment. So far, the cytotoxic effect of GSK-J4 has only been reported in T-ALL, and no other types of acute leukemia responded to the treatment [Citation38].
This study explored the regulation of HOX genes in PML-RARα-positive patients who present significantly lower HOX gene levels than other AML patients. The presence of PML-RARα blocks differentiation, accompanied by epigenetic changes that lead to leukemia progression. We show that the release of the PML-RARα-mediated block leads to H3K27 demethylation at specific HOX gene promoters, a process regulated by JMJD3. Our findings described the molecular mechanism responsible for the distinct HOX gene expression pattern in PML-RARα-positive patients. Additionally, we show the combinational treatment of ATRA and GSK-J4 which could be beneficial for ATRA-resistant patients.
Methods
Cell culture
Human APL cell lines (NB4, NB4-LR2 [Citation39,Citation40], and NB4-MR2 [Citation41]) were cultured in RPMI 1640 medium supplemented with 10% inactivated fetal bovine serum (BioSera) and 1% penicillin, streptomycin, and amphotericin B (Gibco). All cell lines were tested for mycoplasma, and only mycoplasma-free cells were used for experiments.
In vitro treatment
The APL cell lines were treated with different concentrations of ATRA (R2625, Sigma Aldrich) and the inhibitor GSK-J4 (SML0701, Sigma Aldrich). Both of these chemicals were dissolved in DMSO. DMSO was also used to treat control cells.
qRT-PCR
Patient samples were processed according to previous publication [Citation16]. After 8 h of treatment (ATRA – 1 µM; GSK-J4 – 1, 10 µM; PMA – 20, 200 nM), RNA was isolated from cell lines using an RNeasy Plus Mini Kit (74 134, Qiagen), and 1 µg of RNA was reverse transcribed to cDNA using an iScript cDNA Synthesis Kit (170-8891, BioRad). For HOXB genes expression detection cDNA was preamplified using TaqMan PreAmp Master Mix Kit (4384 267, Thermo Fisher Scientific). Gene expression was quantified using the iCycler iQ System (BioRad, Hercules, California, US). The primer design and qPCR conditions for amplification of the HOXA1, A3, A4, A5, A6, A7, A9, A10, A11 and HOXB2, B4, B5, B6 genes as well as the chromatin modifier genes (DNMT3a, DNMT3b and the histone demethylases JMJD3 and UTX) were performed as described previously [Citation42–Citation46]. Gene expression levels were normalized to that of the ABL1 gene.
Western blot
After 8 h of treatment (ATRA – 1 µM; GSK-J4 – 1, 10 µM), nuclear and cytoplasmic protein fractions were isolated from cells with NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific). Histones were isolated by acid extraction (Nature Protocols). The following antibodies were used: polyclonal rabbit anti-JMJD3 (3457, Cell Signaling Technology), anti-UTX (A302-374A, Bethyl), polyclonal rabbit anti-PML-RARα (ab43152, Abcam), polyclonal rabbit anti-TBP (ab63766, Abcam), anti-trimethyl-histone H3 (Lys27) (07-449, Millipore), anti-dimethyl-histone H3 (Lys27) (07-452, Millipore), anti-dimethyl-histone H3 (Lys4) (07-030, Millipore), anti-trimethyl-histone H3 (Lys4) (07-473, Millipore), anti-histone H3 (06-755, Millipore), anti-dimethyl-histone H3 (Lys9) (07-441, Millipore), anti-acetyl-histone H3 (Lys27) (07-360, Millipore), anti-acetyl-histone H3 (Lys9) (07-352, Millipore), anti-acetyl-histone H3 (Lys14) (07-353, Millipore), and anti-acetyl-histone H4 (06-598, Millipore).
Chromatin immunoprecipitation followed by qRT-PCR
After 8 h of treatment (ATRA – 1 µM; GSK-J4 – 1, 10 µM), 1% (v/v) formaldehyde was added to cells to crosslink DNA and proteins. After 1 min of crosslinking, 0.125 M glycine was added for 5 min. The cells were then lysed in lysis buffer (1% (v/v) SDS, 10 mM EDTA, 50 mM Tris-HCl (pH∼8)), and the crosslinked chromatin was fragmented by sonication (22 cycles – 30 sec intervals). The sonicated chromatin was immunoprecipitated with antibodies bound to magnetic beads (Dynabeads A and G in a 1:1 ratio). The following antibodies were used: polyclonal rabbit anti-H3K27me3 (07-449, Millipore), polyclonal rabbit anti-H3K4me2 (07-030, Millipore) and rabbit anti-gamma-globulin (011-000-002, Jackson ImmunoResearch) as a negative control. After a 16 h incubation, magnetic beads with bound chromatin were washed with RIPA buffer (50 mM HEPES, 1 mM EDTA, 0.7% (v/v) Na-deoxycholate, 1% (v/v) NP-40, 0.5 M LiCl) and TE buffer (pH 7.6) (10 mM Tris-HCl, 1 mM EDTA). The DNA/protein crosslinks were reversed by incubating in buffer (0.1 M NaHCO3, 1% SDS) for 5 min at room temperature and 6 h at 65 °C. DNA was purified using a QIAquick PCR Purification Kit (Qiagen). Diluted DNA was used for qRT-PCR quantification using PowerSYBR Green PCR Master Mix (4368 702, Life Technologies) and HOX gene (HOXA1, A3, A5, A7) primers designed to amplify sites upstream of the TSS in the 5´-UTR regions of the genes. The final data were normalized to input (total chromatin before immunoprecipitation) and to the IgG negative control.
Chromatin immunoprecipitation followed by next-generation sequencing
After 8 h of treatment (ATRA – 1 µM; GSK-J4 – 10 µM), the same protocol for chromatin immunoprecipitation was used. For this purpose, we used an antibody against H3K27me3 verified for ChIP-seq: monoclonal mouse anti-H3K27me3, ChIP Grade (ab6002, Abcam). An NGS library was prepared from the final DNA using an NEBNext ChIP-Seq Library Prep Reagent Set for Illumina (E6200S, New England BioLabs) with Index Primers Set 1/2 (E7335S/E7500S, New England BioLabs). The library was sequenced on an Illumina NextSeq using the 70-base-pair single-read method.
ChIP-seq data analysis
H3K27me3 ChIP-seq primary single-end data were aligned by bowtie2 [Citation47] against the human reference genome hg19. All reads with a mapping quality of 10 and below were discarded from the subsequent analysis. Peak calling was performed by macs2 [Citation48] with the settings ‘–broad –nomodel –extsize 300 –to-large’ on 30%-down-sampled aligned data to reduce the very high non-specific background noise in ChIP-seq samples. A common ChIP-seq control sample for peak calling was created by merging aligned individual control samples and down-sampling to (100% / the number of individual samples) %. Peaks present in all replicates were obtained by merging the peaks from individual replicates that were less than 2 kb apart from each other. Peaks present in all replicates were then assigned to gene promoters (+/- 2 kb from the TSS). Gene promoter read counts were calculated by the bedtools suite [Citation49] in gene promoters defined as +/- 500 bp from the TSS and were normalized to the library size.
Cell morphology
After 48 h of treatment (ATRA – 1 µM; GSK-J4 – 1, 10 µM; PMA – 20 nM, 200 nM), cells were stained with May-Grunwald (MS500, Sigma Aldrich) and Giemsa stain (GS500, Sigma Aldrich) solution. Cell morphology was evaluated using an optical microscope.
Flow cytometry
We detected changes in differentiaton upon ATRA treatment using typical markers CD11b and CD65. NB4 cells were incubated 15 min with antibodies – CD65-FITC (B36299, Beckman Coulter) and CD11b-APC (333 143, BD International). Apoptosis was determined after 24 and 48 h of treatment (ATRA – 1 µM; GSK-J4 – 1, 2, 5 µM), cells were stained with Annexin V-Dy647 (RC-ANXD-T100, Exbio) and propidium iodide (PI) solution (130-093-233, Miltenyi Biotec). The percentages of apoptotic and necrotic cells were measured using an LSR II instrument (BD Biosciences, USA).
Bisulfite sequencing
After 24, 48 and 72 h of treatment (ATRA – 1 µM), DNA was isolated from cells using a DNA Mini Kit (Qiagen). Then, 500 µg of DNA was bisulfite converted (C→T) using an EZ DNA Methylation-Gold Kit (D 5005, Zymo Research). The converted DNA was used for PCR amplification of two different CpG-rich regions of the HOXA5 and HOXA7 promoters. The PCR products were cloned into the TOPO TA vector. The reaction products were transformed into One Shot Top 10 chemically competent bacterial cells (K450001, Life Technologies Czech Republic s.r.o.). From each construct, 10 ampicillin-resistant bacterial colonies were transferred from an agar plate to liquid Luria Broth medium containing ampicillin. After 16 h of bacteril growth, plasmids were isolated using a Presto Mini Plasmid Kit (PDH300, KRD). Each plasmid was sequenced by Sanger sequencing methods using M13 primers.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Authorship contributions
KR, KSK and MAJ performed experiments. AM, KF performed statistical analysis and analysis of ChIP-seq. JS designed the project and coordinated the study. JS, KR, JT and MZ wrote the paper and analyzed the data. All authors revised the manuscript and approved the final version.
Acknowledgement
We would like to thank Professor Eric So kindly providing us with resistant cell lines. This work was supported by the Grant Agency of the Czech Republic (P304/12/2214) (JS, JT); Supported by MH CZ – DRO, Motol University Hospital, Prague, Czech Republic 00064 203; KR was supported by the Grant Agency of Charles University GAUK 196 616.
Sup-mat-LOW_HOX_GENE_EXPRESSION_IN_PML-RAR_-POSITIVE_-Rejlova.rar
Download (602 KB)Additional information
Funding
Related Research Data
References
- Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004 Apr 08;350(15):1535–1548. doi:10.1056/NEJMra023001.
- Manno CS, Chew AJ, Hutchison S, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003 Apr 15;101(8):2963–2972. doi:10.1182/blood-2002-10-3296.
- Ferrucci PF, Grignani F, Pearson M, et al. Cell death induction by the acute promyelocytic leukemia-specific PML/RARalpha fusion protein. Proc Natl Acad Sci U S A. 1997 Sep 30;94(20):10901–10906. doi:10.1073/pnas.94.20.10901.
- Collins SJ, Robertson KA, Mueller L. Retinoic acid-induced granulocytic differentiation of HL-60 myeloid leukemia cells is mediated directly through the retinoic acid receptor (RAR-alpha). Mol Cell Biol. 1990 May;10(5):2154–2163. doi:10.1128/MCB.10.5.2154.
- Zhu J, Gianni M, Kopf E, et al. Retinoic acid induces proteasome-dependent degradation of retinoic acid receptor alpha (RARalpha) and oncogenic RARalpha fusion proteins. Proc Natl Acad Sci U S A. 1999 Dec 21;96(26):14807–14812. doi:10.1073/pnas.96.26.14807.
- Nervi C, Ferrara FF, Fanelli M, et al. Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARalpha fusion protein. Blood. 1998 Oct 01;92(7):2244–2251.
- Degos L, Dombret H, Chomienne C, et al. All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia. Blood. 1995 May 15;85(10):2643–2653.
- Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood. 2011 Jun 02;117(22):5795–5802. doi:10.1182/blood-2011-02-329367.
- Grignani F, Ferrucci PF, Testa U, et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 1993 Aug 13;74(3):423–431. doi:10.1016/0092-8674(93)80044-F.
- Vitaliano-Prunier A, Halftermeyer J, Ablain J, et al. Clearance of PML/RARA-bound promoters suffice to initiate APL differentiation. Blood. 2014 Dec 11;124(25):3772–3780. doi:10.1182/blood-2014-03-561852.
- Imaizumi M, Suzuki H, Yoshinari M, et al. Mutations in the E-domain of RAR portion of the PML/RAR chimeric gene may confer clinical resistance to all-trans retinoic acid in acute promyelocytic leukemia. Blood. 1998 Jul 15;92(2):374–382.
- Ding W, Li YP, Nobile LM, et al. Leukemic cellular retinoic acid resistance and missense mutations in the PML-RARalpha fusion gene after relapse of acute promyelocytic leukemia from treatment with all-trans retinoic acid and intensive chemotherapy. Blood. 1998 Aug 15;92(4):1172–1183.
- Martens JH, Brinkman AB, Simmer F, et al. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell. 2010 Feb 17;17(2):173–185. doi:10.1016/j.ccr.2009.12.042.
- Agger K, Cloos PA, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007 Oct 11;449(7163):731–734. doi:10.1038/nature06145.
- Lee MG, Villa R, Trojer P, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007 Oct 19;318(5849):447–450. doi:10.1126/science.1149042.
- Skvarova Kramarzova KFK, Mejstrikova E, Rejlova K, et al. Homeobox gene expression in acute myeloid leukemia is linked to typical underlying molecular aberrations. J Hematol Oncol. 2014;7:94. doi:10.1186/s13045-014-0094-0. PMID:25539595
- Roche J, Zeng C, Baron A, et al. Hox expression in AML identifies a distinct subset of patients with intermediate cytogenetics. Leukemia. 2004 Jun;18(6):1059–1063. doi:10.1038/sj.leu.2403366.
- Thompson A, Quinn MF, Grimwade D, et al. Global down-regulation of HOX gene expression in PML-RARalpha + acute promyelocytic leukemia identified by small-array real-time PCR. Blood. 2003 Feb 15;101(4):1558–1565. doi:10.1182/blood.V101.4.1558.
- Kruidenier L, Chung CW, Cheng Z, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012 Aug 16;488(7411):404–408. doi:10.1038/nature11262.
- Azuara V, Perry P, Sauer S, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006 May;8(5):532–538. doi:10.1038/ncb1403.
- Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006 Apr 21;125(2):315–326. doi:10.1016/j.cell.2006.02.041.
- Tsumagari K, Baribault C, Terragni J, et al. DNA methylation and differentiation: HOX genes in muscle cells. Epigenetics Chromatin. 2013 Aug 02;6(1):25. doi:10.1186/1756-8935-6-25.
- Wei J, Zhai L, Xu J, et al. Role of Bmi1 in H2A ubiquitylation and Hox gene silencing. J Biol Chem. 2006 Aug 11;281(32):22537–22544. doi:10.1074/jbc.M600826200.
- Beuchle D, Struhl G, Muller J. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development. 2001 Mar;128(6):993–1004.
- Wang J, Mager J, Schnedier E, et al. The mouse PcG gene eed is required for Hox gene repression and extraembryonic development. Mamm Genome. 2002 Sep;13(9):493–503. doi:10.1007/s00335-002-2182-7.
- Khan SN, Jankowska AM, Mahfouz R, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013 Jun;27(6):1301–1309. doi:10.1038/leu.2013.80.
- Milne TA, Briggs SD, Brock HW, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002 Nov;10(5):1107–1117. doi:10.1016/S1097-2765(02)00741-4.
- Hanson RD, Hess JL, Yu BD, et al. Mammalian Trithorax and polycomb-group homologues are antagonistic regulators of homeotic development. Proc Natl Acad Sci U S A. 1999 Dec 07;96(25):14372–14377. doi:10.1073/pnas.96.25.14372.
- Rocha-Viegas L, Villa R, Gutierrez A, et al. Role of UTX in retinoic acid receptor-mediated gene regulation in leukemia. Mol Cell Biol. 2014 Oct 01;34(19):3765–3775. doi:10.1128/MCB.00839-14.
- Issaeva I, Zonis Y, Rozovskaia T, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007 Mar;27(5):1889–1903. doi:10.1128/MCB.01506-06.
- He LZ, Tribioli C, Rivi R, et al. Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci U S A. 1997 May 13;94(10):5302–5307. doi:10.1073/pnas.94.10.5302.
- Lin RJ, Nagy L, Inoue S, et al. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998 Feb 19;391(6669):811–814. doi:10.1038/35895.
- Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998 Feb 19;391(6669):815–818. doi:10.1038/35901.
- Di Croce L, Raker VA, Corsaro M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002 Feb 08;295(5557):1079–1082. doi:10.1126/science.1065173.
- Giampaolo A, Felli N, Diverio D, et al. Expression pattern of HOXB6 homeobox gene in myelomonocytic differentiation and acute myeloid leukemia. Leukemia. 2002 Jul;16(7):1293–1301. doi:10.1038/sj.leu.2402532.
- Alharbi RA, Pettengell R, Pandha HS, et al. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013 Apr;27(5):1000–1008. doi:10.1038/leu.2012.356.
- Cole CB, Verdoni AM, Ketkar S, et al. PML-RARA requires DNA methyltransferase 3A to initiate acute promyelocytic leukemia. J Clin Invest. 2016 Jan;126(1):85–98. doi:10.1172/JCI82897.
- Ntziachristos P, Tsirigos A, Welstead GG, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014 Oct 23;514(7523):513–517. doi:10.1038/nature13605.
- Lanotte M, Martin-Thouvenin V, Najman S, et al. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood. 1991 Mar 01;77(5):1080–1086.
- Duprez E, Benoit G, Flexor M, et al. A mutated PML/RARA found in the retinoid maturation resistant NB4 subclone, NB4-R2, blocks RARA and wild-type PML/RARA transcriptional activities. Leukemia. 2000 Feb;14(2):255–261. doi:10.1038/sj.leu.2401683.
- Rosenauer A, Raelson JV, Nervi C, et al. Alterations in expression, binding to ligand and DNA, and transcriptional activity of rearranged and wild-type retinoid receptors in retinoid-resistant acute promyelocytic leukemia cell lines. Blood. 1996 Oct 01;88(7):2671–2682.
- Drabkin HA, Parsy C, Ferguson K, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002 Feb;16(2):186–195. doi:10.1038/sj.leu.2402354.
- Starkova J, Zamostna B, Mejstrikova E, et al. HOX gene expression in phenotypic and genotypic subgroups and low HOXA gene expression as an adverse prognostic factor in pediatric ALL. Pediatr Blood Cancer. 2010 Dec 01;55(6):1072–1082. doi:10.1002/pbc.22749.
- Das ND, Jung KH, Choi MR, et al. Gene networking and inflammatory pathway analysis in a JMJD3 knockdown human monocytic cell line. Cell Biochem Funct. 2012 Apr;30(3):224–232. doi:10.1002/cbf.1839.
- Yang J, Fang X. [Expression of DNMT1, DNMT3a, and DNMT3b in eutopic endometrium]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2012 Jan;37(1):94–99.
- Liu J, Mercher T, Scholl C, et al. A functional role for the histone demethylase UTX in normal and malignant hematopoietic cells. Exp Hematol. 2012 Jun;40(6):487–498. doi:10.1016/j.exphem.2012.01.017.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012 Mar 04;9(4):357–359. doi:10.1038/nmeth.1923.
- Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi:10.1186/gb-2008-9-9-r137. PMID:18798982
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010 Mar 15;26(6):841–842. doi:10.1093/bioinformatics/btq033.