
ABSTRACT
Most tissue samples available for cancer research are archived as formalin-fixed paraffin-embedded (FFPE) samples. However, the fixation process and the long storage duration lead to DNA fragmentation and hinder epigenome analysis. The use of droplet digital PCR (ddPCR) to detect DNA methylation has recently emerged. In this study, we compare an optimized ddPCR assay with a conventional qPCR assay by targeting a dilution series of control DNA. In addition, we compare the ddPCR technology with results from Infinium arrays targeting two separate CpG sites on a set of colon adenoma FFPE samples. Our data demonstrate that qPCR and ddPCR assess methylation status equally well on dilution controls with a high DNA input. However, the methylation detection on low-input samples is more accurate using ddPCR. The proposed primer design (methylation-independent primers with amplification of solely the converted DNA target) will allow for methylation detection, independent of bisulfite conversion efficiency. Those data show that ddPCR can be used for methylation analysis on FFPE samples with a wide range of DNA input and that the precision of the assay depends largely on the total amount of amplifiable DNA fragments. Due to accessibility of the ddPCR technology and its accuracy on high- as well as low-DNA input samples, we propose the use of this approach for studies involving degraded FFPE samples.
Introduction
Epigenetic control mechanisms play a central role in a wide variety of natural and disease processes [Citation1]. The best known epigenetic mechanisms include DNA methylation, histone alterations, and noncoding RNA-associated gene silencing. Aberrant DNA methylation is described in many diseases, including cancer, vascular diseases, and neurodegenerative disorders [Citation2–Citation4]. Epigenetic factors may also contribute to the interpersonal variations in drug response [Citation5]. The ability to detect these altered methylation patterns is key in understanding the complex biology of diseases. To date, several methods for methylation analysis are available, each with their (dis)advantages [Citation6,Citation7], and the selection of the appropriate technology should be based on the nature of the clinical samples [blood sample, fresh-frozen tissue, formalin-fixed paraffin-embedded (FFPE) samples], the amount and quality of the DNA, the required sensitivity and precision, and the availability of the equipment. Whole-genome methylation profiling methods include (but are not limited to) array hybridization, bisulfite (next-gen) sequencing, immunoprecipitation-based methods, and chromatography methods [Citation6,Citation8,Citation9]. Recently, bisulfite-free sequencing methods (Nanopore sequencing and Single-Molecule, Real-Time (SMRT) sequencing with the PacBio platform) have emerged to assess the methylation status [Citation10–Citation13]. PCR-based methods (including Methylight qPCR, pyrosequencing, methylation-sensitive restriction endonuclease methods) are widely used for the confirmation of the findings from initial screenings performed with one of the whole-genome profiling methods [Citation7,Citation14].
In recent years, the droplet digital PCR (ddPCR) system has gained interest for the detection of rare mutations, copy number variations, gene rearrangements, and pathogen detection [Citation15]. ddPCR is based on sample partitioning, by randomly distributing a single sample into small droplets, such that some droplets have no template molecules and others have one or more. Each droplet undergoes PCR amplification and analysis separately. The droplets are then individually assessed and scored as positive or negative for fluorescence. The Poisson limit theorem allows for the estimation of the per-droplet distribution of the number of target copies. This then allows for the absolute quantitation of the target sequence [Citation16]. The use of ddPCR for DNA methylation detection has recently emerged, mainly for use with liquid biopsies [Citation17,Citation18]; a limited number of studies have examined tissue samples or other body fluids [Citation18–Citation21]. Most tissue samples available for cancer research are archived as Formalin-Fixed Paraffin-Embedded (FFPE) samples for many years. Although those retrospective sample repositories are valuable for biomarker research, the fixation process and the long storage duration have a major impact on the DNA quality [Citation22–Citation24]. Bisulfite treatment, which involves the deamination of unmodified cytosine residues to uracil, will cause further DNA degradation. In this study, we compare an optimized ddPCR assay with a conventional qPCR assay by targeting a dilution series of control DNA. In addition, we compare the ddPCR technology with results from Infinium arrays targeting two separate CpG sites on a set of colon adenoma FFPE samples.
Results
Accurate methylation detection with droplet digital PCR
We have evaluated the performance of conventional qPCR and ddPCR for methylation detection of the CpG sites cg07164631 and cg25249613 on two series of mixtures of methylated and unmethylated DNA controls. Primer pairs targeting only converted cytosine residues were designed. This allowed for the amplification of only the bisulfite-converted DNA sequence, but not of the non-converted DNA template. To enforce that the primers efficiencies are methylation-independent, no CpG sites were included in the primer pairs. Two probes per primer pair were designed with one probe, labeled with FAM, targeting the methylated sequence and the second probe, labeled with HEX, targeting the unmethylated sequence (). Identical primer/probe combinations were used for both technologies and optimal PCR conditions were determined by varying the annealing temperatures.
Table 1. Primer and probe sequences.
A linear detection over the whole methylation range is observed for both qPCR and ddPCR (expected vs. measured percentage methylation: R² > 0.99 for all assays). The mean 95% confidence intervals (CI) for the methylation status were small and comparable between qPCR and ddPCR [cg07164631: 4.67% (qPCR) vs. 5.73% (ddPCR) and cg25249613: 3.51% (qPCR) vs. 4.29% (ddPCR)]. A higher accuracy is observed in ddPCR (; ), as demonstrated with a 4- to 5-fold lower median difference between the expected and measured methylation status for the ddPCR assay compared to the qPCR assay (cg07164631: 1.04% vs. 5.52% and cg25249613: 4.71% vs. 1.16%; ).
Figure 1. Bland-Altman plots for assessment of accuracy of the two CpG sites cg07164631 (A) and cg25249613 (B) using ddPCR and qPCR. The difference in measured and expected percentage methylation (y-axis) is plotted against the expected percentage methylation of the dilution series (x-axis). Two series of mixtures with methylated and unmethylated DNA controls were analyzed. The upper figures (control series 1) have a 5-fold higher DNA input. Dashed line = median; dotted lines = 2.5th and 97.5th percentile; curved blue solid line = Loess; dashed blue delimit 95% confidence band around Loess.
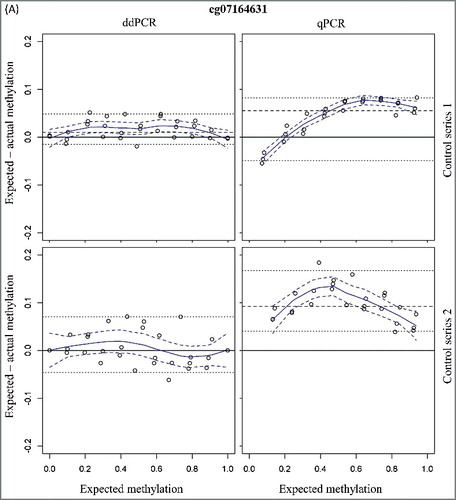
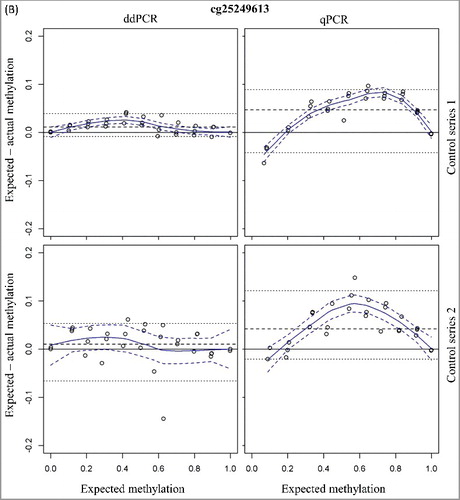
Table 2. Summary of the median differences between expected and measured methylation status. Numbers between brackets represent the 2.5th and 97.5th percentiles.
To simulate the methylation detection on low-input samples, a second series of mixtures with a 5-fold lower DNA input was analyzed. The 95% CI for the methylation detection were higher as compared to the first series of methylation mixtures [cg07164631: 8.62% (qPCR) and 11.60% (ddPCR) and cg25249613: 7.18% (qPCR) and 10.22% (ddPCR)]. In analogy with the dilution series with higher total DNA input, the accuracy for ddPCR is also higher compared to the qPCR assays with the low-input control series (; ). In addition, the accuracy of the low- and high-input control series is comparable for both ddPCR assays (). This is not the case for the cg07164631 qPCR assay where a 2-fold decrease in accuracy is observed with the low-input samples ().
These data have demonstrated the similar performances of qPCR and ddPCR (for those two analyzed CpG sites) for the methylation detection on high-input samples. However, the methylation detection on low-input samples is more accurate using ddPCR.
No impact of low bisulfite conversion efficiency on the methylation detection
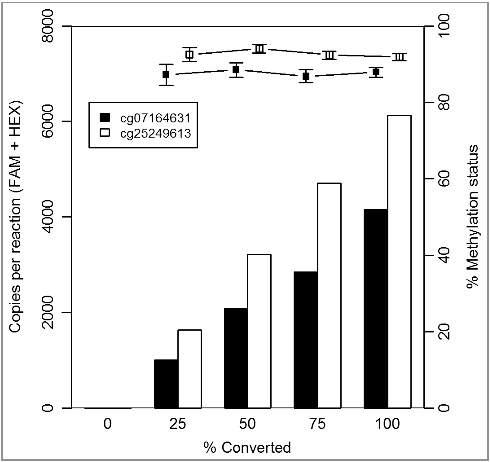
The first step in the methylation detection of clinical samples is an optimal DNA extraction and bisulfite conversion method. To avoid any effect of an incomplete bisulfite conversion on the accurate methylation detection, the primers we have designed target only converted cytosine residues. Consequently, only the bisulfite converted DNA will be amplified and not the non-converted DNA. We have experimentally evaluated the performance of the two ddPCR assays (cg07164631 and cg25249613) on varying degrees of bisulfite converted control DNA (0%, 25%, 50%, 75%, and 100%) in a background of non-converted control DNA. As shown in , an increase in the absolute amount of detected copies is observed by an increasing percentage of converted DNA in the samples. However, the methylation status is stable [mean methylation status ± 95% CI of 87.6 ± 3.8% (cg07164631) and 92.7 ± 2.5% (cg25249613); ]. These data confirmed that an optimal design of the primers/probe can avoid any potential impact of an incomplete bisulfite conversion on the accurate methylation detection.
Figure 2. Optimal design of the primers/probe can avoid any potential impact of an incomplete bisulfite conversion method on the accurate methylation detection. The total copies/reaction (left y-axis; column) and percentage methylation (right y-axis; bullets) are plotted against percentage of converted DNA in the dilutions series (x-axis).
Droplet digital PCR can assess methylation statuses on FFPE samples
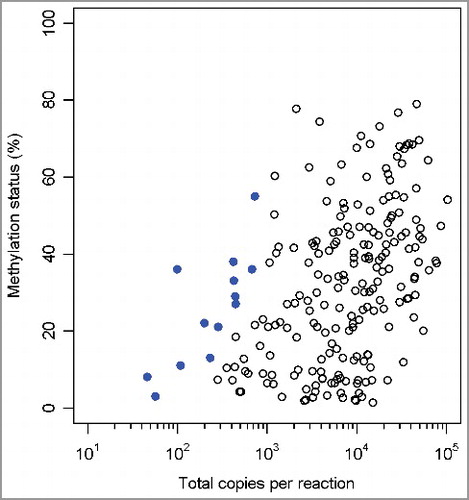
The methylation statuses of the two CpGs (cg07164631 and cg25249613) were tested on FFPE tissue samples (n = 109) using ddPCR. To assess the success of the bisulfite conversion, an additional assay was designed using primers targeting cytosine-free sequences (adapted from [Citation25]). This allowed for the amplification of both the converted and non-converted sequences. Two probes, one labeled with FAM and one with HEX, were designed to bind converted and non-converted DNA sequence, respectively. A mean bisulfite conversion efficiency of 99.57 ± 2.03% is detected on those clinical samples. One sample was excluded from further analysis due to low amount (<8000) of generated droplets in the ddPCR. As shown in , a wide range of both the total copies per reaction and the percentage methylation in those 108 clinical samples was detected. The mean 95% CI were small [3.09% (cg07164631) and 4.35% (cg25249613)], and the magnitude was comparable to the experiments with the control DNA mixtures. Only 13 (6%) out of the 216 generated data points have an associated 95% CI greater than 10% and all these have a low amount (<750) of total copies per reaction. The five data points with a 95% CI >15% have an even smaller amount (<250) of positive droplets in the reaction. Those data have demonstrated that droplet digital PCR can be used for methylation analysis on FFPE samples with a wide range of DNA input and that precision of the assay depends largely on total copies per reaction, i.e., total amount of amplifiable DNA fragments.
Figure 3. Methylation status of cg07164631 and cg25249613 across 108 clinical FFPE samples by ddPCR. Scatterplot with percentage methylation (y-axis) vs. total copies per reaction (x-axis). The data points with a 95% CI >10% methylation status units are indicated in bold.
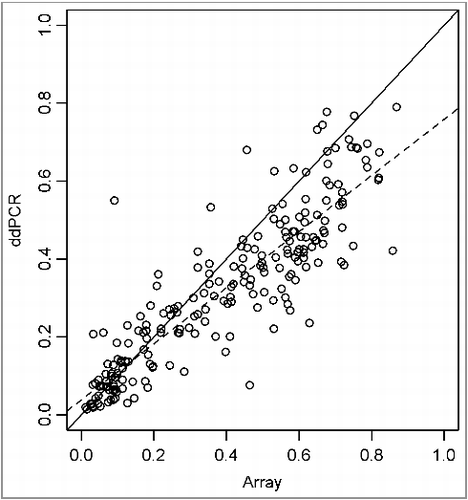
In addition, we have compared the methylation data for those 2 CpG sites from the Infinium arrays and the ddPCR assays. The correlation between those two technologies is demonstrated in (slope = 1.2 ± 0.03; r² = 0.78). The ddPCR technology could therefore be valuable for confirmation of genome-wide methylation screenings.
Figure 4. Scatterplot with linear regression of β-values from Infinium arrays (y-axis) vs. percentage methylation of ddPCR (x-axis).
Discussion
A limited amount of reports regarding methylation detection using ddPCR are available in the literature, most of them with a focus on the sensitivity of this technology [Citation19]. In most cases, methylation-specific PCR primers, which do contain CpG sites and specifically amplify the methylated DNA sequence, and cytosine-less primers (to control for DNA input) were used [Citation19]. In this study, we have used a different approach for the primer/probe design. Since no CpG sites were included in the primer pairs, the primers were methylation-independent, which would allow for a proportional amplification of methylated and unmethylated bisulfite converted templates and minimize any PCR bias. Optimal bisulfite conversion should allow for the sufficient conversion of cytosine residues while leading to only limited inappropriate conversion of methylated cytosine residues and limited DNA degradation. With the experiments in this study, we have demonstrated that the proposed primer design [i.e., methylation-independent primers with amplification of solely the converted DNA target, in combination with (un)methylation-specific probes] will allow for an accurate and reliable methylation detection, independent of bisulfite conversion efficiency.
Droplet digital PCR technology is based on sample partitioning by randomly diluting a single sample into many droplets, such that some droplets have no template molecules and others have one or more. This endpoint PCR method does not require standard curves for absolute quantification (unlike conventional qPCR), can be easily optimized and is therefore rapidly emerging in many applications [Citation15]. Since each droplet in the ddPCR assays independently undergoes PCR amplification, there is less impact of PCR inhibition. By using series of mixtures of methylated and unmethylated DNA controls, we have demonstrated the similar performances of qPCR and ddPCR (for the two primer pairs used) for the methylation detection on high-input samples. Both methods have demonstrated a linear and accurate detection over the complete methylation range. Moreover, the high accuracy of the methylation detection obtained with ddPCR was also obtained with low-DNA input samples, which was opposite to qPCR. Although it should be noted that the excellent amplification achieved may not be generalizable to all primer pairs, the primers used allow us to explore the characteristics of both methods. Due to the high accuracy on both high- and low-input samples, the convenient optimization and readily access to the ddPCR technology, ddPCR has many advantages for studies using highly-degraded FFPE samples. In this study, a precise methylation detection was obtained for most clinical samples (which contain a wide range of amplifiable DNA fragments). Since the fixation process and the long storage duration of such samples have a major impact on the DNA quality, care should be given to those parameters and for certain sample sets the DNA could be too degraded to obtain reliable methylation data. This precision could be further enhanced by running multiple replicates in separate wells and merging those data for the analysis (a well-known feature of the QuantaSoft software).
In conclusion, our data have demonstrated that droplet digital PCR can be a valuable tool for the precise and accurate methylation detection on clinical samples and can be used for confirmation of genome-wide screenings.
Materials and methods
Samples
Colon adenoma samples of patients undergoing surveillance colonoscopy were acquired from Avaden Biosciences. The median storage time of the samples was 12 years.
DNA extraction and bisulfite conversion
Paraffin was removed from one FFPE slice (10 µm) with deparaffinization solution (Qiagen) and DNA was extracted using the QiaAmp DNA FFPE Tissue. This was followed by bisulfite conversion using the EpiTect Fast Bisulfite Conversion Kit (Qiagen). Three extractions/bisulfite conversion reactions per sample were pooled into a total elution volume of 60 µl.
Controls
Synthetic DNA fragments (gBlocks Gene Fragments; stock concentration 5 ng/µl; IDT), corresponding to either the fully methylated or unmethylated bisulfite converted DNA sequence surrounding the CpG site of interest, were synthesized. A series of mixtures of methylated and unmethylated DNA controls (gBlock fragments: 10% methylation differences) were prepared fresh and stored in TE (10 mM Tris-Cl, 1 mM EDTA) buffer with 100 ng/µl carrier RNA. Two series were made; dilution series 2 has a 5-fold lower DNA input than dilution series 1. For the first series of methylation mixtures, a mean of 1157 and 2621 total copies per reaction (sum of the positive droplets for methylated and unmethylated target) are observed for the cg07164631 and cg2524961 ddPCR assays, respectively. For the second series of methylation mixtures, a mean of 222 and 537 total copies per reaction are observed for the cg07164631 and cg2524961 ddPCR assays. In addition, dilution series with varying degrees of converted and non-converted DNA (EpiTect PCR Control DNA; Qiagen) were prepared. EpiTect Control DNAs (completely methylated or completely unmethylated bisulfite converted DNAs and untreated, unmethylated genomic DNA) are added to each PCR reaction.
Primer/probe design
Three assays were designed: two for evaluation of the methylation status and the third assay for evaluation of the bisulfite conversion efficiency. Sequences of the primers and probes of the above assays are provided in .
To evaluate the methylation status of two CpG sites (cg07164631 and cg25249613), primer pairs targeting only converted cytosine residues (optimal number ≥4) were designed. This allowed for the amplification of only the bisulfite-converted DNA sequence, but not of the non-converted DNA template. To enforce that the primers efficiencies are methylation-independent, no CpG sites were included in the primer pairs. Two probes, one labeled with FAM and one with HEX were designed (MethPrimer software [Citation26]) so as to bind methylated and non-methylated CpG sites, respectively. Locked nucleic acid bases (LNA™) were incorporated into the HEX and FAM probes to enhance the melting temperature.
To evaluate the bisulfite conversion efficiency, an additional assay was designed using primers targeting cytosine-free sequences (adapted from [Citation25]). This allowed for the amplification of both the converted and non-converted sequences. Two probes, one labeled with FAM and one with HEX, were designed to bind converted and non-converted DNA sequence, respectively.
ddPCR
The ddPCR reaction mixture consisted of the 2 × ddPCR Supermix for probes (no dUTP) (Bio-Rad), forward and reverse primer (900 nM), probe (250 nM) and 2 to 5 µl bisulfite-converted DNA in a total reaction volume of 25 µl. Methylated and non-methylated controls and a no-template control (water) were added in every experiment. A volume of 20 µl of PCR mixture was added to the middle row of wells of the droplet generation cartridge (DG8) followed by addition of 70 µl of droplet generation oil to the left row. After completion of the droplet generation (average 15,000 droplets/well) in the QX200 droplet generator, the contents were transferred to a PCR plate. Reactions with 8000 droplets or lower were excluded from further processing. Thermal cycling conditions were 95°C for 10 min, followed by 40 cycles at 94°C for 30 secs, 56°C for 60/90 secs with a final 10 min at 98°C (). After PCR amplification using the T100™ Thermal Cycler Bio-Rad, the positive droplets (HEX/FAM) were counted with QX200 droplet reader (Bio-Rad) and data was analyzed using the QuantaSoft 1.7.4 software (Bio-Rad). The total number of copies/reaction was calculated and results were reported as the percentage methylation, i.e., the fractional abundance of methylated DNA alleles. Reactions on the control DNA dilutions were performed in triplicate and the mean with 95% CI was calculated. Clinical samples were analyzed in single reactions.
qPCR
The qPCR reaction mixture consists of 2x Epitect Methylight Master Mix (Qiagen), forward and reverse primer (400 nM) and probe (200 nM) and 2 µl bisulfite-converted DNA. Water is added to a total reaction volume of 20 µl. Thermal cycling conditions were 95°C for 5 min, followed by 45 cycles of 95°C for 15 sec, 60°C for 60 sec; The Bio-Rad CFX-96 was used. Ct values were calculated by the Bio-Rad CFX manager software (version 3.1) using the Single Threshold mode. The percentage methylation was calculated as follows: % methylation = 100/(1+2(CtFAM-CtHEX)). Reactions were performed in triplicate and the mean with 95% CI was calculated.
Genome-wide methylation screening
DNA was extracted from 2 slices (10 µm) from the FFPE samples (Qiagen), quantified using PicoGreen and checked for quality using the Illumina FFPE QC Kit. The DNA was then subjected to a Bisulfite conversion (EZ DNA methylation Kit, Zymo Research). Next, 250 ng DNA was subjected to Illumina's FFPE restoration protocol, per manufacturer's protocol. Samples were run on Infinium HumanMethylation450 BeadChip (450K). Several selective filters (detection filter, bead-count filter, SNP-filter, cross-reactivity filter, XY-filter) were applied to the probe sets, as described in Nordlund et al [Citation27]. and Dedeurwaerder et al [Citation28]. Within-sample β-values were normalized for probe-design using an adjusted version of the PBC algorithm [Citation28] and corrected for batch-effects using surrogate variable analysis [Citation29].
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
References
- Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. PubMed PMID: 15164071.
- Mikeska T, Bock C, Do H, et al. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Rev Mol Diagn. 2012;12(5):473–487. doi: 10.1586/erm.1245. PubMed PMID: 22702364.
- Zheng J, Cheng J, Zhang Q, et al. Novel insights into DNA methylation and its critical implications in diabetic vascular complications. Biosci Rep. 2017;37(2):1–7. Epub 2017/02/12. doi: 10.1042/bsr20160611. PubMed PMID: 28183874; PubMed Central PMCID: PMCPMC5350598.
- Hwang JY, Aromolaran KA, Zukin RS. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat Rev Neurosci. 2017;18(6):347–361. Epub 2017/05/19. doi: 10.1038/nrn.2017.46. PubMed PMID: 28515491.
- Majchrzak-Celinska A, Baer-Dubowska W. Pharmacoepigenetics: an element of personalized therapy? Expert Opin Drug Metabol Toxicol. 2017;13(4):387–398. Epub 2016/11/20. doi: 10.1080/17425255.2017.1260546. PubMed PMID: 27860490.
- Kurdyukov S, Bullock M. DNA methylation analysis: Choosing the right method. Biology (Basel). 2016;5(1):3–21. doi: 10.3390/biology5010003. PubMed PMID: 26751487; PubMed Central PMCID: PMCPMC4810160.
- Noehammer C, Pulverer W, Hassler MR, et al. Strategies for validation and testing of DNA methylation biomarkers. Epigenomics. 2014;6(6):603–622. doi: 10.2217/epi.14.43. PubMed PMID: 25531255.
- Chowdhury B, Cho IH, Irudayaraj J. Technical advances in global DNA methylation analysis in human cancers. J Biol Eng. 2017;11:10. doi: 10.1186/s13036-017-0052-9. PubMed PMID: 28261325; PubMed Central PMCID: PMCPMC5331624.
- Dirks RA, Stunnenberg HG, Marks H. Genome-wide epigenomic profiling for biomarker discovery. Clin Epigenetics. 2016;8:122. doi: 10.1186/s13148-016-0284-4. PubMed PMID: 27895806; PubMed Central PMCID: PMCPMC5117701.
- Rand AC, Jain M, Eizenga JM, et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Meth. 2017;14(4):411–413. doi: 10.1038/nmeth.4189 http://www.nature.com/nmeth/journal/v14/n4/abs/nmeth.4189.html#supplementary-information.
- Rhoads A, Au KF. PacBio Sequencing and Its Applications. Genomics, Proteomics Bioinform. 2015;13(5):278–289. Epub 2015/11/07. doi: 10.1016/j.gpb.2015.08.002. PubMed PMID: 26542840; PubMed Central PMCID: PMCPMC4678779.
- Schatz MC. Nanopore sequencing meets epigenetics. Nat Methods. 2017;14(4):347–348. Epub 2017/04/01. doi: 10.1038/nmeth.4240. PubMed PMID: 28362434.
- Simpson JT, Workman RE, Zuzarte PC, et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat Meth. 2017;14(4):407–410. doi: 10.1038/nmeth.4184 http://www.nature.com/nmeth/journal/v14/n4/abs/nmeth.4184.html#supplementary-information.
- Hernandez HG, Tse MY, Pang SC, et al. Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques. 2013;55(4):181–197. doi: 10.2144/000114087. PubMed PMID: 24107250.
- Olmedillas-Lopez S, Garcia-Arranz M, Garcia-Olmo D. Current and emerging applications of droplet digital PCR in oncology. Mol Diagn Ther. 2017;21(5):493–510. Epub 2017/05/10. doi: 10.1007/s40291-017-0278-8. PubMed PMID: 28477149.
- Perkins G, Lu H, Garlan F, et al. Droplet-Based Digital PCR: Application in Cancer Research. Adv Clin Chem. 2017;79:43–91. doi: 10.1016/bs.acc.2016.10.001. PubMed PMID: 28212714.
- Wiencke JK, Bracci PM, Hsuang G, et al. A comparison of DNA methylation specific droplet digital PCR (ddPCR) and real time qPCR with flow cytometry in characterizing human T cells in peripheral blood. Epigenetics. 2014;9(10):1360–1365. doi: 10.4161/15592294.2014.967589. PubMed PMID: 25437051; PubMed Central PMCID: PMCPMC4622657.
- Barault L, Amatu A, Bleeker FE, et al. Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann Oncol. 2015;26(9):1994–1999. doi: 10.1093/annonc/mdv272. PubMed PMID: 26113646.
- Yu M, Carter KT, Makar KW, et al. MethyLight droplet digital PCR for detection and absolute quantification of infrequently methylated alleles. Epigenetics. 2015;10(9):803–809. doi: 10.1080/15592294.2015.1068490. PubMed PMID: 26186366; PubMed Central PMCID: PMCPMC4623055.
- Hayashi M, Guerrero-Preston R, Sidransky D, et al. Paired box 5 methylation detection by droplet digital PCR for ultra-sensitive deep surgical margins analysis of head and neck squamous cell carcinoma. Cancer Prev Res (Phila). 2015;8(11):1017–1026. doi: 10.1158/1940-6207.CAPR-15-0180. PubMed PMID: 26304463; PubMed Central PMCID: PMCPMC4633357.
- Liu Y, Wu H, Zhou Q, et al. Digital quantification of gene methylation in stool DNA by emulsion-PCR coupled with hydrogel immobilized bead-array. Biosens Bioelectron 2017;92:596–601. doi: 10.1016/j.bios.2016.10054. PubMed PMID: 27829567.
- Do H, Dobrovic A. Sequence Artifacts in DNA from Formalin-Fixed Tissues: Causes and Strategies for Minimization. Clin Chem. 2015;61(1):64–71. doi: 10.1373/clinchem.2014.223040. PMID:25421801.
- Einaga N, Yoshida A, Noda H, et al. Assessment of the quality of DNA from various formalin-fixed paraffin-embedded (FFPE) tissues and the use of this DNA for next-generation sequencing (NGS) with no artifactual mutation. PLoS One. 2017;12(5):e0176280. Epub 2017/05/13. doi: 10.1371/journal.pone.0176280. PubMed PMID: 28498833; PubMed Central PMCID: PMCPMC5428915.
- Bak ST, Staunstrup NH, Starnawska A, et al. Evaluating the feasibility of DNA methylation analyses using long-term archived brain formalin-fixed paraffin-embedded samples. Mol Neurobiol. 2018;55(1):668–681. Epub 2016/12/21. doi: 10.1007/s12035-016-0345-x. PubMed PMID: 27995571.
- Holmes EE, Jung M, Meller S, et al. Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One. 2014;9(4):e93933. doi: 10.1371/journal.pone.0093933. PubMed PMID: 24699908; PubMed Central PMCID: PMCPMC3974851.
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. PubMed PMID: 12424112. doi:10.1093/bioinformatics/18.11.1427.
- Nordlund J, Backlin CL, Wahlberg P et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013;14(9):r105. doi: 10.1186/gb-2013-14-9-r105. PubMed PMID: 24063430; PubMed Central PMCID: PMCPMC4014804.
- Dedeurwaerder S, Defrance M, Bizet M, et al. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief Bioinform. 2014;15(6):929–941. doi:10.1093/bib/bbt054. PubMed PMID: 23990268; PubMed Central PMCID: PMCPMC4239800.
- Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. doi: 10.1093/bioinformatics/bts034. PubMed PMID: 22257669; PubMed Central PMCID: PMCPMC3307112.