
ABSTRACT
Genomic imprinting is the process of epigenetic modification whereby genes are expressed in a parent-of-origin dependent manner; it plays an important role in normal growth and development. Parthenogenetic embryos contain only the maternal genome. Parthenogenetic embryonic stem cells could be useful for studying imprinted genes. In humans, mature cystic ovarian teratomas originate from parthenogenetic activation of oocytes; they are composed of highly differentiated mature tissues containing all three germ layers. To establish human parthenogenetic induced pluripotent stem cell lines (PgHiPSCs), we generated parthenogenetic fibroblasts from ovarian teratoma tissues. We compared global DNA methylation status of PgHiPSCs with that of biparental human induced pluripotent stem cells by using Illumina Infinium HumanMethylation450 BeadChip array. This analysis identified novel single imprinted CpG sites. We further tested DNA methylation patterns of two of these sites using bisulfite sequencing and described novel candidate imprinted CpG sites. These results confirm that PgHiPSCs are a powerful tool for identifying imprinted genes and investigating their roles in human development and diseases.
Introduction
Genomic imprinting is an epigenetic phenomenon whereby the expression of a gene depends on the parental origin of its chromosome, genomic imprinting plays an important role in normal growth and development [Citation1–4]. Approximately 100 imprinted genes have been described in human and mouse [Citation5]. Regulation of imprinted genes associated with DNA methylation is specific for tissue, cell type, and developmental stage [Citation6–8]. Given the fact that disruption of imprinting can lead to severe developmental defects and disease [Citation3,Citation9,Citation10], identification of novel imprinted genes is important [Citation11].
Parthenogenesis occurs spontaneously in some species without male genetic contribution. Only maternally derived chromosomes are present in parthenogenetic embryos. In mouse, parthenogenetic embryonic stem cells (ESCs) have been established following artificial activation of unfertilized eggs [Citation12–14]. These uniparental embryos or cells are valuable for screening the parent-of-origin-specific DNA methylation profiles to find novel imprinted CpG sites. However, research on human embryos is limited due to destruction of parthenogenetic embryos derived from in vitro activation of unfertilized human eggs. Interestingly, in humans, natural parthenogenetic activation of oocytes is observed in vivo and results in formation of mature cystic ovarian teratomas (MCT) [Citation15]. MCT contain only the maternal genome (maternally expressed and paternally imprinted) and have been used to identify novel imprinted differentially methylated regions [Citation16]. MCT are composed of differentiated tissues from the three germ layers (endoderm, mesoderm, and ectoderm) [Citation17,Citation18]. The presence of various types of tissues makes it difficult to identify novel imprinted genes because DNA methylation patterns are distinct among different tissue types. Therefore, it is desirable to use a homogenous cell population to compare genome-wide DNA methylation for screening novel imprinted genes.
In this study, we aimed to perform a comprehensive analysis of global DNA methylation status in biparental and parthenogenetic human cells for screening imprinted CpG sites. To obtain a homogenous cell population, we first established parthenogenetic fibroblasts (PgFibs) from MCT and generated human parthenogenetic induced pluripotent stem cell lines (PgHiPSCs). Biparental induced pluripotent stem cells (HiPSCs) were obtained from human foreskin fibroblast cells (BJ cells). We compared the methylation status between PgHiPSCs and HiPSCs by using the Illumina Infinium HumanMethylation450 BeadChip (450K) array to identify novel imprinted CpG sites.
Results
Establishment and characterization of human induced pluripotent stem cells from uniparental and biparental fibroblasts
We first derived uniparental PgFibs from three independent MCT tissues. To generate PgHiPSC lines, we reprogrammed three different parthenogenetic fibroblast cell lines (PgFib-1, PgFib-2, and PgFib-3) into PgHiPSC-1, PgHiPSC-2, and PgHiPSC-3 with retroviruses encoding OCT4, SOX2, KLF4, and C-MYC, respectively. We also generated biparental HiPSCs from BJ cells (WtFibs).
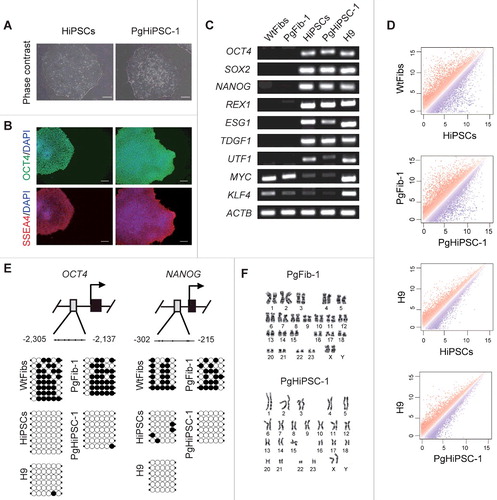
HiPSCs and PgHiPSC-1 expressed the pluripotency markers OCT4, SSEA4, SOX2, NANOG, TRA-1-60, and TRA-1-81 as revealed by immunocytochemistry analysis and were positive for alkaline phosphatase (A, B and Figure S1A). As shown in C, the expression levels of the OCT4, SOX2, NANOG, REX1, ESG1, TDGF1, UTF1, MYC, and KLF4 genes in HiPSCs and PgHiPSC-1 were similar to those of H9 (a human embryonic stem cell line used; as a positive control) as revealed by RT-PCR; WtFibs and PgFib-1 samples were used as negative controls and showed no expression of any of these genes except for MYC and KLF4. We compared the global gene expression profiles of HiPSCs, PgHiPSC-1, and H9 by using DNA microarrays and confirmed that both iPSC lines had expression patterns similar to that of H9 (D). To measure the methylation status of the OCT4 and NANOG promoters in PgHiPSC-1 and their parental fibroblasts, we conducted bisulfite sequencing analysis. OCT4 and NANOG were unmethylated in HiPSCs, PgHiPSC-1, and H9, but methylated in their parental fibroblasts (E). All these cell lines have a normal karyotype (46, XX) (F and Figure S1B). Additionally, we also confirmed that pluripotency status of two additional PgHiPSC lines (PgHiPSC-2 and PgHiPSC-3) was similar to those of H9 (Figure S2).
Figure 1. Characterization of parthenogenetic HiPSC lines. (A) Morphology of HiPSCs and parthenogenetic (Pg) HiPSC-1. (B) Immunocytochemistry for pluripotency markers (OCT4 and SSEA4) in biparental HiPSCs and parthenogenetic HiPSC-1. Scale bar = 100 μm. (C) RT-PCR analysis of pluripotency-specific gene expression in WtFibs, PgFib-1, HiPSCs, PgHiPSC-1, and H9. (D) Scatter plots comparing the global gene expression patterns between WtFibs and HiPSCs, PgFib-1 and PgHiPSC-1, H9 and HiPSCs, and H9 and PgHiPSC-1 analyzed by oligonucleotide microarrays. (E) Bisulfite sequencing analysis of OCT4 and NANOG promoter regions in WtFibs HiPSCs, PgFib-1, PgHiPSC-1, and H9. (F) Confirmation of a normal 46, XX karyotype of PgFib-1 and PgHiPSC-1.
To determine the in vitro differentiation ability of PgHiPSC-1, we cultured embryoid bodies (EB) and directed the differentiation of EBs by overexpressing the genes specific for the endoderm (AFP, ALB, GATA4, SOX17, and TDO2), mesoderm (MSX and BRACHYURY), and ectoderm (PAX6 and MASH1) (Figure S3A). We also confirmed the expression of the endoderm (AFP), mesoderm (α-SMA), and ectoderm (TUBB3) markers in the differentiated cells by immunocytochemistry analysis (Figure S3B). To investigate the in vivo differentiation ability of PgHiPSC-1, we transplanted them subcutaneously into NOD/SCID mice. Twelve weeks after injection, we observed formation of a teratoma containing various tissues (Figure S3C). These results indicate that PgHiPSC-1 can be generated from uniparental somatic cells through reprogramming.
To confirm the homozygosity of PgHiPSC-1, we performed genome-wide SNP analysis using the Affymetrix Human SNP 6.0 Array in PgHiPSC-1 and HiPSCs. Graphs showed that HiPSCs were heterozygous at a random SNP marker along each chromosome and PgHiPSC-1 homozygous (Figure S4). These results indicate that PgHiPSC-1 were truly parthenogenetic.
Characterization of known imprinted genes in PgHiPSCs
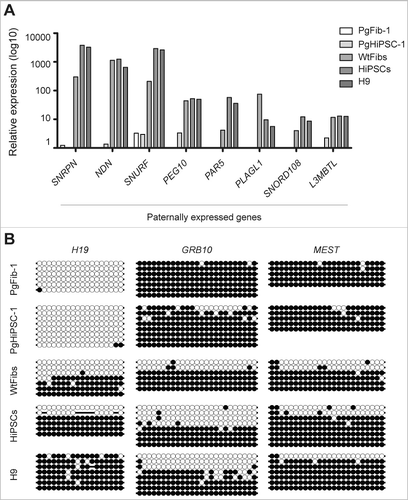
We performed genome-wide microarray analysis to examine the expression of parthenogenesis-specific imprinted genes in PgHiPSCs. As a control, we analyzed the global gene expression in WtFibs, HiPSCs, and H9. Known paternally expressed genes (SNRPN, NDN, SNURF, PEG10, PAR5, PLAGL1, SNORD108, and L3MBTL) were downregulated in parthenogenetic cells compared to biparental cells (A). To determine the parthenogenetic imprinting status of PgHiPSCs, we compared the methylation status in the differentially methylated regions (DMR) of known imprinted genes between HiPSCs and three PgHiPSC lines (PgHiPSC-1, PgHiPSC-2, and PgHiPSC-3) using bisulfite sequencing analysis. Because parthenogenetic cells lack the paternal genome, the paternally imprinted gene H19 are unmethylated, whereas the maternally imprinted genes GRB10 and MEST are methylated. In parthenogenetic cells in our study, H19 was not methylated, whereas GRB10 and MEST were methylated (B and Figure S5). As expected, all three genes had somatic imprinting patterns in WtFibs, HiPSCs, and H9. Alternation of the imprinted genes H19 in human ESCs (hESCs) was previously reported by Rugg-Gunn et al. [Citation19,Citation20], who demonstrated gene-specific differences in the instability of methylation. Thus, the results indicate the parthenogenetic identity of PgHiPSCs was confirmed by the status in known imprinted genes.
Figure 2. Analysis of expression and DNA methylation of known imprinted genes. (A) Relative mRNA expression levels of known paternally expressed genes in WtFibs, HiPSCs, PgFib-1, PgHiPSC-1, and H9. The data shown are averages of gene expression. (B) DNA methylation status of known paternally and maternally imprinted genes (H19, GRB10, and MEST) in PgFib-1, PgHiPSC-1, WtFibs, HiPSCs, and H9 was analyzed by bisulfite sequencing. Each line represents a separate clone. Black and white circles represent methylated and unmethylated CpGs, respectively.
Screening for novel imprinted single CpG sites by microarray analysis
We performed genome-wide DNA methylation analysis using the 450K array. This array contains 485,512 CpG sites at single-nucleotide resolution, including 90% of the sites of the Human Methylation27 (27K) array [Citation21–23]. We confirmed the methylation status of known imprinted genes (e.g., NAT15, ZNF597, GNAS, HM13, HYMAI, KCNQ1OT1, L3MBTL, MAGEL2, MEST, NDN, PEG10, PLAGL1, SGCE, and SNRPN) in parthenogenetic and biparental samples (Figure S6). To screen for differential methylation of CpG sites between parthenogenetic and biparental samples, we filtered the CpG sites using methylation levels β≤0.1 or 0.9≤β in PgFib-1 and PgHiPSC-1, and 0.3≤β≤0.7 in WtFibs, HiPSCs, and H9. It was calculated from the average β-value for each CpG site in PgHiPSC-1 and HiPSCs; the β-value reflects the methylation level (0, unmethylated; 1, methylated) of each CpG site. Using this approach, we selected imprinted single CpG sites. The full list of candidate imprinted CpG sites is shown in the .
Table 1. A survey for candidate imprinted CpG sites
Validation of DNA methylation status at the novel imprinted single CpG sites by bisulfite sequencing
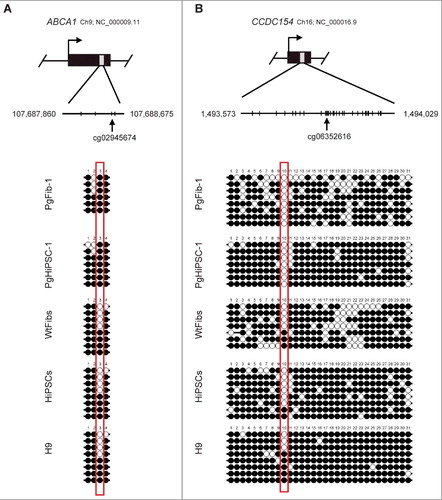
Among the candidates found by microarray analysis, we selected cg02945674 (ABCA1) and cg06352616 (CCDC154) because they were significantly correlated with global sperm methylation status (GSE72444). We conducted bisulfite sequencing analysis to validate the DNA methylation of these CpG sites and to determine the genomic imprinting status in PgFib-1, PgHiPSC-1, WtFibs, HiPSCs, and H9. We found parthenogenetic patterns in the DNA methylation status of the novel imprinted CpG sites: ABCA1 was methylated, whereas CCDC154 was unmethylated in PgFib-1 and PgHiPSC-1 (). We also confirmed parthenogenetic patterns in the DNA methylation status of the novel imprinted single CpG site in parthenogenetic cells (PgFib-2, PgHiPSC-2, PgFib-3, and PgHiPSC-3) (Figure S7). The methylation status data from our 450K array were highly correlated with those from bisulfite sequencing.
Figure 3. Confirmation of DNA methylation status at novel imprinted CpG sites. Bisulfite sequencing showed imprinted DNA methylation patterns of ABCA1 and CCDC154. Each line represents a separate clone. Black and white circles represent methylated and unmethylated CpGs, respectively.
Discussion
Homozygous hESCs generated from parthenogenetic embryos have potential advantages for studying genomic imprinting, but ethical issues need to be considered [Citation24–26]. An alternative way to obtain parthenogenetic stem cells without ethical controversy is to use ovarian teratomas. Since the ovarian teratomas used in this study were non-cancerous tumors, the possibility of DNA methylation loss in imprinted sites, which is frequently observed in cancer cells [Citation27,Citation28], can be ruled out. However, there are several limitations to screening for new imprinted genes using primary cells obtained from ovarian teratomas. First, it is possible that primary cells from MCT containing three germ layers are heterogeneous [Citation18], which may hinder global DNA methylation analysis and screening for novel imprinted genes. Second, although primary cells have similar biological properties in vivo and can be used for a variety of studies, it is difficult to cultivate homogeneous cell populations. In most cases, the possibility to repeat experiments with primary cells is limited because of cellular senescence that occurs as the cells are passaged [Citation29]. To enable continuous cultures, we first established PgFibs from MCT and then used them to generate a homogeneous population of PgHiPSCs by transduction of four defined transcription factors.
PgFibs attained pluripotency through reprogramming despite having only uniparental imprinting because they had only the maternal genome. Microarray analysis has been used to compare gene expression between parthenogenetic and biparental samples in human and mouse [Citation30–32]. Our results were reasonable because paternally expressed genes are downregulated in parthenogenetic samples. Using bisulfite sequencing analysis, we found that the DNA methylation status of three imprinted genes (H19, GRB10, and MEST) was maintained even after reprogramming. DNA methylation status of imprinted genes may be maintained or altered during the generation of pluripotent stem cells [Citation33]. We previously reported that the androgenetic imprinting status of H19 and Mest is maintained after reprogramming of mouse spermatogonial stem cells into germline-derived pluripotent stem cells [Citation34]. In the present study, we confirmed that our PgHiPSCs are similar to parthenogenetic HiPSCs derived from ovarian teratoma primary fibroblasts (GM01304 and GM01306) [Citation31].
Screening for imprinted genes is important because it can be used to study genomic imprinting disorders and the database catalogue of imprinted genes is still incomplete [Citation5,Citation35,Citation36]. Ovarian teratoma tissues have been useful in screening for imprinted genes but some limitations are indicated because these tissues may contain cells of three germ layers [Citation37]. To overcome these limitations, we used the 450K array to perform genome-wide analysis of DNA methylation in homogeneous PgHiPSC-1. The methylation status of known imprinted genes was accurately measured to ascertain the parthenogenetic characteristics of our PgHiPSC-1. Because the 450K array is an advanced version of the 27K array used in the Stelzer et al.'s study [Citation31] and has a wide range of methylated CpG sites, we were able to build a comprehensive DNA methylation profile. Stelzer et al. screened for somatic and parthenogenetic methylation patterns according to β-value of 0.4≤β≤0.6 in HiPSCs and hESCs and 0.75≤β in PgHiPSC-1 [Citation31]. They identified the known paternally expressed genes (L3MBTL, PEG10, NDN, NAP1L5, SGCE, and SNRPN) and newly found paternally expressed gene (FBLN7, SIGLEC6, and C1orf118). As we set the methylation range same to Stelzer et al. [Citation31] and additionally set the methylation range (β≤0.3 in PgFib-1 and PgHiPSC-1) for screening maternally expressed candidate genes, 108 known paternally imprinted CpG sites, 10 known maternally imprinted CpG sites, 25 novel paternally imprinted CpG sites, and 19 novel maternally imprinted CpG sties were identified (Table S1 and Table S2). In the case of known paternally expressed genes (L3MBTL, PEG10, NDN, NAP1L5, SGCE, and SNRPN) mentioned in Stelzer et al. [Citation31], we were able to also find them in our analysis. It is interesting that although we found SIGLEC6 among the three genes (FBLN7, SIGLEC6, and C1orf118) identified as paternally imprinted genes in our samples, it was not selected by our methylation range (β≤0.1 or 0.9≤β in PgFib-1 and PgHiPSC-1, and 0.3≤β≤0.7 in WtFibs, HiPSCs, and H9).
To screen for novel candidate methylation markers, we set a higher stringency range (methylation levels in range of β≤0.1 or 0.9≤β in PgFib-1 and PgHiPSC-1). We detected novel candidate methylation markers, which were methylation sites in parthenogenetic samples vs. biparental samples. Sperm samples contain paternal genomes and represent androgenetic imprinting status. Association studies based on genome-wide profiling of DNA methylation in sperm have been reported [Citation38,Citation39]. Interestingly, two markers (cg02945674 and cg06352616) among the candidate methylation markers show androgenetic imprinting status in sperm samples from public databases such as the GEO. The DNA methylation status in the DMR of imprinted genes is associated with regulation of paternal- and maternal-specific expression and normal development of the embryo. We performed bisulfite sequencing to confirm the presence of imprinted DMR in the two markers. Contrary to expectations, we found no imprinting status in DMR of these markers. Nonetheless, previous epigenetic studies have reported that a single or few CpG sites are useful as surrogate methylation markers in various fields [Citation40,Citation41].
To investigate the significance of DNA methylation at cg02945674 and cg06352616 CpG sites, we performed real time PCR analysis to examine the expression of two corresponding genes. The expression level of ABCA1 appears not to correlate with the methylation at the CpG site (cg02945674) in either non-pluripotent or pluripotent cells. The expression level of CCDC154 in parthenogenetic fibroblast was higher than that in biparental fibroblast, whereas no difference was found between PgHiPSC-1 and HiPSCs (Figure S8). These results suggest that methylation of CpG site (cg06352616) correlates with expression in non-pluripotent cells but not in pluripotent stem cells. Interestingly, among the known imprinted genes, we found that methylation of the imprinted genes (PEG3 and HYMAI) was not correlated with their expression in pluripotent stem cells while their expression was indeed correlated with methylation status in non-pluripotent cells, which is similar to the CCDC154 gene (data not shown). This notion is also supported by Stelzer et al., who found that the paternally expressed genes, L3MBTL, NAP1L5, and SGCE, were not expressed in pluripotent stem cells [Citation31]. In fact, it has been reported that methylation of a single CpG site was enough to regulate promoter and transcription factor binding in hepatocellular carcinoma [Citation42] and various tissues [Citation43]. As such, it may not rule out that gene expression is regulated by the imprinted single CpG site. Whether the methylation status of a single CpG site can influence a specific transcription-related function is still an open question [Citation41]. There are no precedents of the regulation of transcriptional repression being regulated by methylation of a single CpG site in the promoter region [Citation40]. We suggest that our candidate CpG site approach can be used to identify imprinting DNA markers. Our data demonstrated that PgHiPSCs could be a potentially important research tool for the investigation of maternal and paternal genomes.
Materials and Methods
Derivation of human parthenogenetic fibroblast cells and culture of biparental fibroblast cells
Human mature cystic ovarian teratoma tissues were obtained from Konkuk University Medical Center, Seoul, Korea. Fibroblasts were isolated by collagenase digestion of tissues followed by culture in fibroblast medium [DMEM (Welgene) with 10% FBS (Hyclone), 1% non-essential amino acids (Invitrogen) and 1% penicillin-streptomycin (Welgene)]. The BJ fibroblast cell line was purchased from the American Type Culture Collection and cultured in fibroblast medium.
Generation of human parthenogenetic iPSCs
To prepare the retroviruses, Plat-A cells purchased from the American Type Culture Collection were transfected with 15 μg of pMXs plasmids containing human OCT4, SOX2, KLF4, and c-MYC by using the Infect transfection reagent (iNtRON) according to the manufacturer's instructions. Supernatants were collected 48 h after transfection, filtered through a 0.45 μm pore-size filter and concentrated in a Retro-X™ Concentrator (Clontech) in a 4°C refrigerator overnight. The retrovirus-containing supernatants were cleared by centrifugation (1,500 g, 40 min). Human fibroblasts were plated at 1 × 104 cells per well in a 6-well plate and infected with the retroviruses encoding the four factors supplemented with 4 μg/ml polybrene (Sigma). The retrovirus supernatants were replaced with the second supernatants 24 h after transduction. Four days after transduction, fibroblasts were transferred to hESC medium [Citation44] [Knockout DMEM (Invitrogen) with 20% Knockout serum replacement (Invitrogen), 2 mM L-glutamine (Invitrogen), 1% non-essential amino acids (Invitrogen), 1% penicillin-streptomycin (Welgene), 50 μM β-mercaptoethanol (Invitrogen), 10 ng/ml human basic FGF (Peprotech)], 0.5 mM sodium butyrate (Sigma), 2 μM SB431542 (Tocris), and 0.5 μM PD0325901 (Sigma) on mitomycin C-treated mouse embryonic fibroblasts as feeder cells. hESC-like colonies were mechanically picked up and transferred on a Matrigel (Corning)-coated plate containing mTeSR™ 1 medium (StemCell Technologies). iPSC lines were cultured in mTeSR™ 1 medium on a Matrigel-coated plate.
RNA isolation and RT-PCR
Total RNA was isolated by using a miRNeasy Mini Kit (Qiagen) and treated with RNase-Free DNase Set (Qiagen) to remove genomic DNA. Total RNA (1 μg) was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed with gene-specific primers (Table S3) as follows: initial denaturation at 94°C (5 min), 35 cycles (94°C for 30 s, annealing for 30 s, 72°C for 30 s), and a final extension at 72°C for 7 min. The PCR products were examined by 1% agarose gel electrophoresis.
Alkaline phosphatase staining and immunocytochemistry
Alkaline phosphatase staining was performed using an Alkaline Phosphatase Detection Kit (Millipore). For immunocytochemistry, cells were fixed with PBS (Hyclone) containing 4% paraformaldehyde for 10 min at room temperature, washed three times with PBS, permeabilized for 10 min with PBS containing 0.5% Triton X-100 (Sigma), and blocked for 1 h with 1% BSA (Sigma) in PBS, followed by incubation with primary antibodies (Pluripotency Antibody Kit, Cell Signaling), AFP (R&D Systems), a-SMA (Abcam), and TUBB3 (Millipore) overnight at 4°C. After washing in PBS, cells were incubated with secondary antibodies (against rabbit IgG, mouse IgG, or mouse IgM; all from Cell Signaling) for 1 h at room temperature and were stained with DAPI (Invitrogen) to detect the nucleus.
Global gene expression and microarray analysis
The microarray study was carried out using Human HT-12 v 4.0 Expression BeadChips (Illumina). Data was quantile normalized using Illumina's Genome Studio data analysis software. Normalized data was analyzed using a scatter plot (log2 scale) to assess the linear correlation between two samples.
In vitro differentiation
For EB formation, human iPSCs were harvested using ReLeSR (StemCell Technologies), and cell clumps were resuspended in hESC medium without human basic FGF. After 14 days, RNA was isolated from EBs. For monolayer differentiation, human iPSCs were cultured in the three germ layer differentiation medium for 3 weeks, as previously described [Citation45]. The cells were fixed with PBS containing 4% paraformaldehyde (Sigma). Immunocytochemistry was performed according to the instructions supplied by the antibody manufacturers.
Teratoma formation
PgHiPSCs were harvested using ReLeSR and then the pellets were suspended in mTeSR™ 1 medium. The cells were injected subcutaneously into NOD/SCID mouse. Twelve weeks after the injection, teratomas were dissected and fixed overnight in Bouin's solution and embedded in paraffin. The paraffin sections were stained with hematoxylin and eosin.
Karyotype analysis
Karyotype analysis was performed according to standard method at the Seoul Clinical Laboratories.
SNP genotyping
Genome-wide Human SNP 6.0 Array (Affymetrix) analysis for SNP genotyping was performed according to the manufacturer's instructions [Citation46].
DNA methylation analysis using bisulfite sequencing and 450K BeadChips
Genomic DNA was isolated using the Total DNA Extraction Kit (iNtRON). Genomic DNA (1 μg) was bisulfite converted using an EpiTect Bisulfite Kit (Qiagen) according to the manufacturer's protocol and used for PCR amplification. For all markers, PCR was performed with a PyroMark PCR kit (Qiagen) and for 45 cycles with primers listed in Table S3. The PCR products were cloned using the PCR Cloning Kit (Qiagen) according to the manufacturer's protocol. Bisulfite-converted DNA was analyzed using Infinium Human Methylation 450K BeadChips (Illumina) according to the manufacturer's protocol [Citation21]. BeadChips were scanned using an Illumina HiScan system, which has a two-color channel (green and red). Methylation data was processed with Methylation Module of Genome Studio software. DNA methylation measurements with P<0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
The authors indicate no potential conflicts of interest.
Choi_et_al_Supplementary_table.docx
Download MS Word (33.1 KB)Choi_et_al_Supplemental_figure.docx
Download MS Word (3.6 MB)Additional information
Funding
Related Research Data
References
- Morison IM, Paton CJ, Cleverley SD. The imprinted gene and parent-of-origin effect database. Nucleic Acids Research. 2001;29:275–6. doi:10.1093/nar/29.1.275. PMID:11125110
- Ishida M, Moore GE. The role of imprinted genes in humans. Molecular Aspects of Medicine. 2013;34:826–40. doi:10.1016/j.mam.2012.06.009. PMID:22771538
- Morison IM, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet. 2005;21:457–65. doi:10.1016/j.tig.2005.06.008. PMID:15990197
- Surani MA, Kothary R, Allen ND, et al. Genome imprinting and development in the mouse. Dev Suppl.1990:89–98.
- Smith RJ, Dean W, Konfortova G, et al. Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res. 2003;13:558–69. doi:10.1101/gr.781503. PMID:12670997
- Paulsen M, Ferguson-Smith AC. DNA methylation in genomic imprinting, development, and disease. J Pathol. 2001;195:97–110. doi:10.1002/path.890. PMID:11568896
- Moore GE, Ishida M, Demetriou C, et al. The role and interaction of imprinted genes in human fetal growth. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140074. doi:10.1098/rstb.2014.0074. PMID:25602077
- Szabo P, Mann JR. Expression and methylation of imprinted genes during in vitro differentiation of mouse parthenogenetic and androgenetic embryonic stem cell lines. Development. 1994;120:1651–60. PMID:8050371
- Glenn CC, Driscoll DJ, Yang TP, et al. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod. 1997;3:321–32. doi:10.1093/molehr/3.4.321. PMID:9237260
- Bressan FF, De Bem TH, Perecin F, Lopes FL, et al. Unearthing the roles of imprinted genes in the placenta. Placenta. 2009;30:823–34. doi:10.1016/j.placenta.2009.07.007. PMID:19679348
- Luedi PP, Dietrich FS, Weidman JR, et al. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723–30. doi:10.1101/gr.6584707. PMID:18055845
- Stevens LC, Varnum DS, Eicher EM. Viable chimaeras produced from normal and parthenogenetic mouse embryos. Nature. 1977;269:515–7. doi:10.1038/269515a0. PMID:909600
- Leeb M, Walker R, Mansfield B, et al. Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development. 2012;139:3301–5. doi:10.1242/dev.083675. PMID:22912412
- Iles SA, McBurney MW, Bramwell SR, et al. Development of parthenogenetic and fertilized mouse embryos in the uterus and in extra-uterine sites. J Embryol Exp Morphol. 1975;34:387–405. PMID:1194837
- Oliveira FG, Dozortsev D, Diamond MP, et al. Evidence of parthenogenetic origin of ovarian teratoma: case report. Hum Reprod. 2004;19:1867–70. doi:10.1093/humrep/deh345. PMID:15192066
- Choufani S, Shapiro JS, Susiarjo M, et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 2011;21:465–76. doi:10.1101/gr.111922.110. PMID:21324877
- Wolff EF, Hughes M, Merino MJ, et al. Expression of benign and malignant thyroid tissue in ovarian teratomas and the importance of multimodal management as illustrated by a BRAF-positive follicular variant of papillary thyroid cancer. Thyroid. 2010;20:981–7. doi:10.1089/thy.2009.0458. PMID:20718682
- Outwater EK, Siegelman ES, Hunt JL. Ovarian teratomas: tumor types and imaging characteristics. Radiographics. 2001;21:475–90. doi:10.1148/radiographics.21.2.g01mr09475. PMID:11259710
- Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA. Status of genomic imprinting in human embryonic stem cells as revealed by a large cohort of independently derived and maintained lines. Hum Mol Genet. 2007;16 Spec No. 2:R243–51. doi:10.1093/hmg/ddm245. PMID:17911167
- Kim KP, Thurston A, Mummery C, et al. Gene-specific vulnerability to imprinting variability in human embryonic stem cell lines. Genome Res. 2007;17:1731–42. doi:10.1101/gr.6609207. PMID:17989250
- Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. doi:10.1016/j.ygeno.2011.07.007. PMID:21839163
- Sandoval J, Heyn H, Moran S, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi:10.4161/epi.6.6.16196. PMID:21593595
- Morris TJ, Beck S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 2015;72:3–8. doi:10.1016/j.ymeth.2014.08.011. PMID:25233806
- Lo B, Parham L. Ethical issues in stem cell research. Endocr Rev. 2009;30:204–13. doi:10.1210/er.2008-0031. PMID:19366754
- de Wert G, Mummery C. Human embryonic stem cells: research, ethics and policy. Hum Reprod. 2003;18:672–82. doi:10.1093/humrep/deg143. PMID:12660256
- Hyun I. The bioethics of stem cell research and therapy. J Clin Invest. 2010;120:71–5. doi:10.1172/JCI40435. PMID:20051638
- Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1:299–305. doi:10.1016/S1535-6108(02)00061-2. PMID:12086841
- Holm TM, Jackson-Grusby L, Brambrink T, et al. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell. 2005;8:275–85. doi:10.1016/j.ccr.2005.09.007. PMID:16226703
- Claydon K, Owens L. Attempts at immortalization of crustacean primary cell cultures using human cancer genes. In Vitro Cell Dev Biol Anim. 2008;44:451–7. doi:10.1007/s11626-008-9141-x. PMID:18830775
- Sritanaudomchai H, Ma H, Clepper L, et al. Discovery of a novel imprinted gene by transcriptional analysis of parthenogenetic embryonic stem cells. Hum Reprod. 2010;25:1927–41. doi:10.1093/humrep/deq144. PMID:20522441
- Stelzer Y, Yanuka O, Benvenisty N. Global analysis of parental imprinting in human parthenogenetic induced pluripotent stem cells. Nat Struct Mol Biol. 2011;18:735–41. doi:10.1038/nsmb.2050. PMID:21572443
- Kobayashi H, Yamada K, Morita S, et al. Identification of the mouse paternally expressed imprinted gene Zdbf2 on chromosome 1 and its imprinted human homolog ZDBF2 on chromosome 2. Genomics. 2009;93:461–72. doi:10.1016/j.ygeno.2008.12.012. PMID:19200453
- Kim MJ, Choi HW, Jang HJ, et al. Conversion of genomic imprinting by reprogramming and redifferentiation. J Cell Sci. 2013;126:2516–24. doi:10.1242/jcs.122754. PMID:23525019
- Lee HJ, Choi NY, Lee SW, et al. Epigenetic alteration of imprinted genes during neural differentiation of germline-derived pluripotent stem cells. Epigenetics. 2016;11:177–83. doi:10.1080/15592294.2016.1146852. PMID:26962997
- Henckel A, Arnaud P. Genome-wide identification of new imprinted genes. Brief Funct Genomics. 2010;9:304–14. doi:10.1093/bfgp/elq016. PMID:20591836
- Frost JM, Moore GE. The importance of imprinting in the human placenta. PLoS Genet. 2010;6:e1001015. doi:10.1371/journal.pgen.1001015. PMID:20617174
- Strichman-Almashanu LZ, Lee RS, Onyango PO, et al. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 2002;12:543–54. doi:10.1101/gr.224102. PMID:11932239
- Molaro A, Hodges E, Fang F, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell. 2011;146:1029–41. doi:10.1016/j.cell.2011.08.016. PMID:21925323
- Hammoud SS, Nix DA, Zhang H, et al. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–8. doi:10.1038/nature08162. PMID:19525931
- Pogribny IP, Pogribna M, Christman JK, et al. Single-site methylation within the p53 promoter region reduces gene expression in a reporter gene construct: Possible in vivo relevance during tumorigenesis. Cancer Res. 2000;60:588–94. PMID:10676641
- Medvedeva YA, Khamis AM, Kulakovskiy IV, et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014;15:119. doi:10.1186/1471-2164-15-119. PMID:24669864
- Sohn BH, Park IY, Lee JJ, et al. Functional switching of TGF-beta1 signaling in liver cancer via epigenetic modulation of a single CpG site in TTP promoter. Gastroenterology. 2010;138:1898–908. doi:10.1053/j.gastro.2009.12.044. PMID:20038433
- Furst RW, Kliem H, Meyer HH, et al. A differentially methylated single CpG-site is correlated with estrogen receptor alpha transcription. J Steroid Biochem Mol Biol. 2012;130:96–104. doi:10.1016/j.jsbmb.2012.01.009. PMID:22342840
- Zhang Z, Gao Y, Gordon A, et al. Efficient generation of fully reprogrammed human iPS cells via polycistronic retroviral vector and a new cocktail of chemical compounds. PLoS One. 2011;6:e26592. doi:10.1371/journal.pone.0026592. PMID:22046312
- Kim JB, Greber B, Arauzo-Bravo MJ, et al. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009;461:649–3. doi:10.1038/nature08436. PMID:19718018
- Kim K, Ng K, Rugg-Gunn PJ, et al. Recombination signatures distinguish embryonic stem cells derived by parthenogenesis and somatic cell nuclear transfer. Cell Stem Cell. 2007;1:346–52. doi:10.1016/j.stem.2007.07.001. PMID:18371368