
ABSTRACT
Saliva is a non-invasive, easily accessible tissue, which is regularly collected in large epidemiological studies to examine genetic questions. Recently, it is becoming more common to use saliva to assess DNA methylation. However, DNA extracted from saliva is a mixture of both bacterial and human DNA derived from epithelial and immune cells in the mouth. Thus, there are unique challenges to using salivary DNA in methylation studies that can influence data quality. This study assesses: (1) quantification of human DNA after extraction; (2) delineation of human and bacterial DNA; (3) bisulfite conversion (BSC); (4) quantification of BSC DNA; (5) PCR amplification of BSC DNA from saliva and; (6) quantitation of DNA methylation with a targeted assay. The framework proposed will allow saliva samples to be more widely used in targeted epigenetic studies.
Introduction
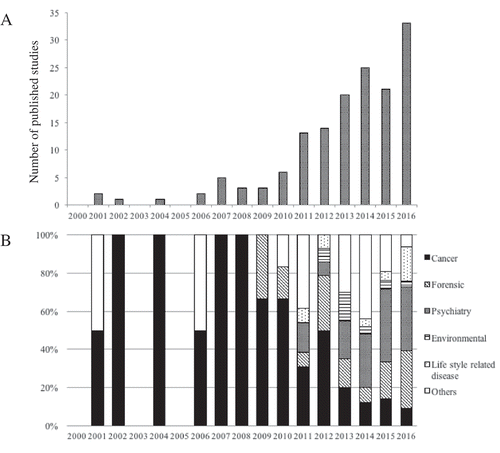
DNA methylation is an epigenetic modification that occurs via addition of a methyl (CH3) group to the cytosine of a CpG dinucleotide, which can affect gene expression. Interindividual differences in DNA methylation patterns have been implicated in the development or progression of a growing number of human pathologies [1–4] and are thought to capture environmental states that moderate genetic risk, serving as an important pathway for the interaction between genes and environment. However, DNA methylation studies in epidemiologic cohorts are complicated by the tissue-specific nature of epigenetic patterns. Growing evidence from studies that evaluate DNA methylation from peripheral tissues, such as blood and saliva, support the utility of peripheral tissues as proxy tissues when an ideal tissue, such as brain, is not available [Citation5–Citation9]. Indeed, there has been an increase in the number of published DNA methylation studies that focus on saliva as a proxy tissue ()). Before 2010, published reports using methylation in saliva mainly focused on studies of cancer or forensics; however, in recent years, the use of saliva in studies of methylation has become more common ()), in part, because of technical improvements in saliva sampling and storage.
Figure 1. Number of published studies that focus on salivary DNA methylation in PubMed search from 2000 to 2016. (A) Total number of published studies. (B) Published studies by discipline.
Saliva sampling also has technical advantages over blood [Citation10]. It is non-invasive, does not require specialized equipment or training and can be stored at room temperature for more than a year, making it feasible to sample outside of a laboratory. When compared to a blood draw, saliva is also more easily collected from young children [Citation11–Citation14]. Finally, it is convenient for repetitive collection from the same participant, making saliva a useful proxy tissue for epidemiological studies such as birth cohorts [Citation15,Citation16].
DNA extracted from saliva is a mixture of human and bacterial DNA. Previous studies have reported a large amount of bacterial DNA in saliva samples, which can result in higher failure rates for genetic and epigenetic assays [Citation17,Citation18]. For example, Feigelson and colleagues found that the median percentage of bacterial DNA in mouthwash samples was 66% of the total DNA [Citation19]. Similarly, Garcia-Closas and colleagues found that the median percentage of bacterial DNA was 50.5% in mouthwash samples and 88.5% in cytobrush samples [Citation20]. Standard quantification methods, including spectrophotometry and spectrofluorimetry, cannot discriminate between bacterial and human DNA. Though bacterial DNA should not compete with human DNA in genome-wide and targeted methods that use human-specific oligonucleotides, bacterial DNA contamination may lead to insufficient template being used in such applications. More recent collection methods have a lower median bacterial DNA content (~11.8%) [Citation21] and may enable more reproducible DNA methylation studies in saliva samples.
The majority of both genome-wide [Citation22,Citation23] and targeted [Citation24–Citation28] methods to assess DNA methylation rely on bisulfite conversion (BSC) of genomic DNA, which requires accurate quantification before and after BSC. Targeted analyses also require PCR amplification, which is complicated because the PCR assay must be specific for the methylated and unmethylated templates that are generated by BSC. Successful PCR is critical for subsequent quantification of methylation levels. The goal of this study is to assess: (1) quantification of human DNA after extraction; (2) delineation of human and bacterial DNA; (3) BSC; (4) quantification of BSC DNA; (5) PCR amplification of BSC DNA from saliva and; (6) quantitation of DNA methylation with a targeted assay.
Results
Quantification of DNA extracted from saliva
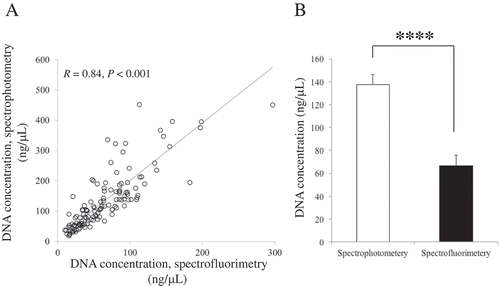
Concentrations of saliva DNA collected from 128 subjects under experimental conditions were measured by spectrophotometry and spectrofluorimetry (). DNA concentrations from both quantification methods are highly correlated (R = 0.84, P <0.001; )), but concentrations obtained from spectrophotometry were approximately two-fold higher than concentrations obtained by spectrofluorimetry (); P <0.001). The discrepancy between these two methods can lead to overestimation of the concentrations of salivary DNA and underestimation of the starting template for PCR.
Figure 2. Comparison of quantification methods for DNA extracted from saliva. (A) Scatterplots showing the significant positive relationship between spectrophotometry and spectrofluorimetry measurements. (B) Concentration differs by the method of measurement. n = 128. Bar represents standard error (SE). **** represents P < 0.001.
Delineating human from bacterial DNA
We quantified the relative proportions of human and bacterial DNA for a reference sample using quantitative PCR assays specific to a single-copy human gene (36B4) and to the 16s rRNA region of the bacterial genome. We noted that a small proportion (<3%) of the reference sample was contributed by bacterial DNA. To evaluate the specificity of this approach, we ran a series of positive and negative controls for each quantitative PCR assay. We noted no bacterial contamination and robust amplification of the human-specific assay in DNA from 3 human cell lines (CEPH, K562 and TCL1301). Similarly, in a non-human fecal sample, we observed robust amplification of the bacteria-specific assay and no contamination with human DNA.
Quantification of BSC DNA
As shown in Supplemental , the reference sample showed some degradation before BSC but was still of high molecular weight. After BSC, the reference sample was clearly fragmented difficult to resolve by electrophoresis. BSC DNA is almost entirely single-stranded, preventing intercalation of ethidium bromide.
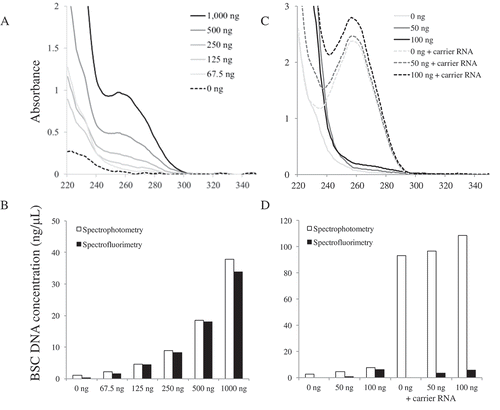
We measured DNA concentrations after BSC from the reference sample at varying initial concentrations using spectrophotometry and spectrofluorimetry. As expected, the concentration of BSC DNA appears proportional to the initial concentration used in the BSC (). Yield is consistent across different concentrations of DNA used in the conversion reaction. The amount of input DNA is important for obtaining adequate resolution of the UV spectrum for accurate quantification (,b)), although samples with low concentration following BSC can be of sufficient quality for PCR amplification (Supplemental ). For example, the use of as little of 2 ng of BSC DNA was difficult to quantify by spectrofluorimetry but amplified in PCR. Minimizing the elution volume after DNA extraction and BSC will increase sample concentrations and increase the chance of successful downstream applications. Multiple elutions may be performed, but rarely resulted in quantifiable DNA (data not shown). BSC kits that have smaller elution requirements should be considered when a higher concentration is needed. In general, 96-well plate format BSC kits often require larger elution volumes and may not be ideal for all applications.
Table 1. Representative concentrations of BSC DNA measured by two different methods. DNA from a reference sample was used to limit bias due to variations in DNA quality.
Figure 3. Comparison of the quantification methods after BSC. (A) UV spectra of different amounts of DNA used to BSC. (B) The concentrations of different amounts of DNA used to BSC. (C) UV spectra of 0 ng, 50 ng and 100 ng DNA used to BSC with or without using carrier RNA. (D) The concentrations of 0 ng, 50 ng, and 100 ng DNA used to BSC with or without using carrier RNA.
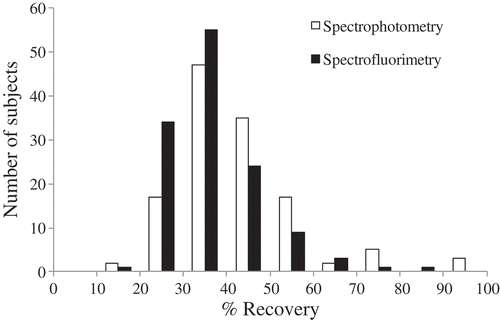
The concentration measured by spectrofluorimetry was proportionately lower than that of spectrophotometry. For the reference sample, spectrofluorimetry estimated the amount of BSC DNA at 365 ng (range 187–806 ng, 36.5% recovery). Using spectrophotometry, the same sample was estimated at 420 ng (range 104–1035 ng, 42.0% recovery). In the experimentally collected samples, the concentration measured by spectrofluorimetry (18.2 ± 0.5 ng/μL; ranged 9.4–40.3 ng/μL) was significantly lower than that of spectrophotometry (21.0 ± 0.7 ng/μL; ranged 5.2–51.8 ng/μL) (P <0.001), but the concentrations were correlated (R = 0.58, P <0.001). As shown in , estimation of the recovery proportion following BSC also varies based on the quantification method used. These results suggest that spectrophotometry overestimates BSC DNA concentration compared to methods designed for ssDNA. Thus, spectrofluorimetry may be preferable when precise quantification is required.
Figure 4. Distribution of percent yield of BSC DNA/dsDNA. X-axis indicates the distribution categories of percent yield and Y-axis indicates the number of subjects in each category.
Carrier RNA
Some protocols [Citation29,Citation30] recommend using carrier RNA during the cleanup process to increase the yield when the DNA input is less than 100 ng. In this study, we found % recovery increased when 50 ng was used for BSC with the addition of carrier RNA, but not with 100 ng used in BSC as measured by spectrofluorimetry (). However, carrier RNA was detectable in spectrophotometric measurements when no DNA was included in the BSC ( and ), suggesting that spectrophotometric measurement cannot discriminate between the carrier RNA and BSC DNA. This may be particularly problematic for samples at lower concentrations following BSC (), which may be attributable to residual reaction material detectable at wavelengths below 260 nm. In contrast, spectrofluorimetry did not appear to be influenced by the addition of carrier RNA or the BSC reaction. Thus, carrier RNA should be used with caution particularly if it is to be quantified with spectrophotometry, which can overestimate the actual concentration of BSC samples complicating downstream PCR performance and optimization.
Table 2. Comparison of the total DNA yield following BSC based on the addition of carrier RNA. Quantifications vary based on the method used (spectrophotometry or spectrofluorimetry), and the presence of carrier RNA. Note that spectrophotometer was blanked with elution buffer from the BSC kit.
PCR and interrogation of BSC DNA
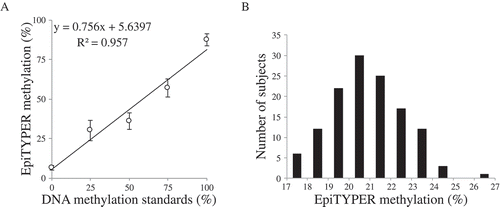
Amplification of OXTR was evaluated in positive and negative controls as well as for all concentrations of the reference saliva sample (Supplemental ). Of note, OXTR was amplified from a minimal concentration of BSC DNA, and there was no evidence of PCR interference when carrier RNA was used (Supplemental , lane 6 and 7). There was a linear relationship between OXTR methylation levels across the standard curve () and Supplemental ). Experimentally collected samples were parametrically distributed within the standard curve () and Supplemental ).
Figure 5. PCR amplification of OXTR MT2. (A) Linear association between unmethylated (0%), 25%, 50%, 75%, and 100% methylated samples across the target region in the EpiTYPER assay, 2 CpG sites were excluded due to technical issues. Each standard was run in triplicate. The error bars are indicative of standard error (SE). (B) Histogram for mean values across the target region in the EpiTYPER assay of normal adult samples (n = 128) .
Discussion
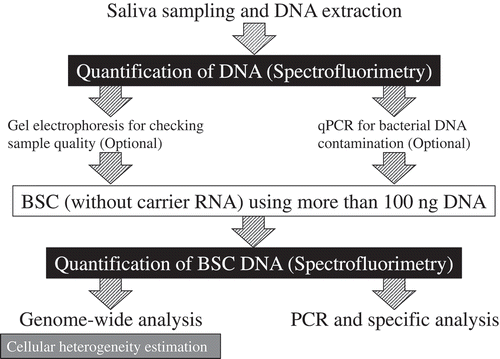
This study evaluated technical considerations for (1) quantification of salivary DNA after extraction; (2) delineation of human and bacterial DNA; (3) BSC; (4) quantification of BSC DNA; (5) PCR amplification of BSC DNA from saliva and; (6) quantitation of DNA methylation with a targeted assay. summarizes these steps as well as the key recommendation based on this study: the discrepancy between quantifications by spectrophotometry and spectrofluorimetry can lead to overestimation of the concentrations of salivary DNA following its initial extraction and again following bisulfite conversion. This can introduce underestimation of the template used in the steps of the subsequent workflow. This study recommends using at least 100 ng of starting template for BSC to avoid the necessity for carrier RNA, which limits the interpretability of spectrophotometric quantification. Spectrofluorimetry is recommended for the quantification of BSC DNA even though the results are correlated to some degree with that obtained from spectrophotometry.
Figure 6. Summary of the recommendations for salivary DNA methylation analysis.
There are other inherent limitations to using DNA extracted from saliva for methylation-based analyses. First, there is cellular heterogeneity in salivary DNA, and methods to estimate cell composition from a mixed sample are only available for genome-wide approaches. There are no comparable methods that can be applied to targeted methylation analyses. Future studies would benefit from a minimal reference panel for estimating the relative proportions of epithelial and leukocyte cells in saliva. The availability of a minimal reference panel that can be included with targeted methylation assays, similar to the bioinformatics approach available for genome-wide studies [Citation23], will enable the power of salivary DNA to be fully harnessed for epidemiological investigations. Second, unlike DNA derived from blood, saliva samples can include bacterial DNA [Citation21] that can interfere with downstream applications, such as PCR, to varying degrees from sample to sample. This study provides protocols for two qPCR assays that can be used to quantify either human or bacterial DNA that can be used to measure and normalize salivary DNA samples. Third, saliva samples are clearly fragmented difficult to resolve by electrophoresis after BSC. This fragmentation renders the template insufficient for specific applications such as long-range PCR or long-read sequencing. However, newer technologies, such as nanopore sequencing, are capable of interrogating DNA without BSC [Citation31].
Saliva samples, compared to blood or other tissues, are easy to handle, can be stored at room temperature, and will help expand the scope of epidemiological studies including repeat sampling for longitudinal studies within the same participants. Though few studies have yet evaluated temporal changes of DNA methylation in saliva, cross-sectional studies that examine the association between salivary DNA methylation and various phenotypes, such as aging [Citation32,Citation33], sex differences [Citation34], pubertal development [Citation35], obesity [Citation36,Citation37], smoking [Citation38], environmental exposures [Citation39,Citation40], stress [Citation28,Citation41], abuse [Citation42] and psychiatric disorders [Citation43–Citation45], are becoming more common. Unlike examining genomic sequences, DNA methylation is dynamic; therefore, repeat sampling can be meaningful to find unknown mechanisms underlying developmental processes or temporal changes. A recent study by, Lévesque and colleagues [Citation46] identified 47 temporally unstable genes across the genome using salivary DNA in monozygotic twin adolescents. They suggested that the DNA methylome is highly responsive in adolescence to experiences and may reveal extraneous signals that occurred during development. Another study investigating temporal changes of DNA methylation in adult blood found dynamic changes of DNA methylation of stress-associated genes within 30 minutes of acute psychosocial stress [Citation47]. More recently, there has been growing interest in identifying dynamic changes in DNA methylation that occur early in life [Citation11–Citation13,Citation48]. Longitudinal studies of such developmental processes and temporal changes in DNA methylation will be required to identify novel epigenetic mechanisms of human traits and behaviors, and salivary DNA is poised to provide new opportunities for biomarker development.
Materials and methods
DNA extraction from saliva
All samples were collected using Oragene Discover (DNA Genotek, OGR-500). DNA was extracted using prepIT®•L2P reagent (DNA Genotek, PT-L2P-5) using 500 μL of Oragene samples. Two sample types were used for this study. The first was a reference sample collected in a laboratory setting and extracted immediately. The second was a group of samples from 128 adults, described in a previous study [Citation28], that were collected under experimental conditions in which they were stored for up to 6 months between collection and extraction. Participants provided written informed consent as detailed in the Declaration of Helsinki.
Quantification of saliva DNA after extraction
Quantity of the extracted DNA was assessed by spectrofluorimetry using a Synergy 2 Multi-Mode Plate Reader (BioTek Instruments) with PicoGreen® (ThermoFisher Scientific, Inc., Quant-iT™ PicoGreen® dsDNA Assay Kit, P11496) and ultraviolet (UV) spectrophotometry using NanoDrop 2000 (ThermoFisher Scientific, Inc.). The reference sample was used for quantifying the yield of bisulfite converted (BSC) DNA. Its concentration was 151.6 ng/μL according to spectrophotometry and 86.0 ng/μL according to spectrofluorimetry.
Quantification of the proportion of human DNA versus bacterial DNA
To confirm human DNA in the reference sample, we performed qPCR targeted at 36B4 gene that is known to be a single copy gene in human genome [Citation49]. Twenty nanograms of DNA were used in a 25 μL reaction mixture using PerfecCTa SYBR® Green SuperMix, ROX (Quantabio, 95,055–100) with the following primer pairs: 5ʹ-CAGCAAGTGGGAAGGTGTAATCC-3ʹ (forward) and 5ʹ-CCCATTCTATCATCAACGGGTACAA-3ʹ (reverse). The cycling conditions were: denaturation (95°C for 5 min) followed by 30 cycles of amplification (95°C for 15 s and 60°C for 1 min) (QuantStudio™ 6 Flex System, ThermoFisher Scientific, Inc.). We used DNA from a CEPH cell line (Coriell Institute, NA12340) for the standard curve.
To determine the percentage of bacterial DNA in the reference sample, we performed qPCR using Microbial DNA qPCR Assay Kit (Qiagen, 330,025) which detects bacterial 16S rRNA gene, which is known to be conserved across a wide variety of microorganisms and is not found in eukaryotes [Citation50]. One microliter (5.5 ng) of DNA was used in a 12.5 μL reaction mixture. We used reference sample and the other negative control samples (Coriell Institute, NA12340), genomic DNA from K562 (Sigma-Aldrich, 89,121,407) and TCL1301(Sigma-Aldrich, 01051619) for the qPCR. The cycling conditions were: denaturation (95°C for 10 min) followed by 40 cycles of amplification (95°C for 15 s and 60°C for 2 min) (QuantStudio™ 6 Flex System, ThermoFisher Scientific, Inc.). We used ZymoBIOMICS™ Microbial Community Standard (Zymo, D6300) for the standard curve.
Bisulfite conversion (BSC)
The reference sample was normalized to 0, 67.5, 125, 250, 500, and 1,000 ng to assess BSC performance and yield using the EpiTect Bisulfite Kit (Qiagen, 59,104). We evaluated the effect of addition of carrier RNA on the percent yield increase for low concentration samples. For this experiment, we compared the yield for 0 ng (ddH2O), 50 ng, and 100 ng of the reference sample with or without the addition of carrier RNA in BSC process. For this reference sample, 2 μg saliva DNA was bisulfite converted and evaluated via electrophoresis on a 2% agarose gel. For the experimentally collected samples, 1 μg of salivary DNA was converted according to manufacturer instructions. All samples were eluted using 20 μL of Buffer EB. A second elution was also collected and evaluated separately.
Quantification of BSC DNA
Quantification of BSC DNA was assessed by spectrofluorimetry using a Synergy 2 Multi-Mode Plate Reader (BioTek Instruments) with QuantiFluor® ssDNA System (Promega, E3190), employing a ssDNA-specific dye as well as a NanoDrop 2000 set for RNA quantification as recommended by the manufacturer.
Assessment of cellular heterogeneity
DNA methylation was interrogated for the reference sample using the Infinium® MethylationEPIC BeadChip (Illumina) and cellular heterogeneity was estimated by the approach described in Smith and colleagues [Citation23,Citation51]. We found that the reference sample had an estimate 55% buccal epithelial cells.
PCR and epityper
To evaluate PCR amplification and interrogation of BSC saliva DNA, we used a well-established assay for the oxytocin receptor (OXTR) (chr3: 8,792,095 to 8,811,300: GRCh37/hg19 build), which has used in studies of psychiatric disorders [Citation52–Citation54] and social behaviors [Citation55,Citation56]. DNA methylation of 27 CpG sites in the promoter region (Supplemental ), which contains the MT2 region, [Citation57] was assessed using the following primer pairs: 5ʹ- aggaagagagGAGGTTTTAGTGAGAGATTTTAGTTTAG-3ʹ (forward) and 5ʹ- cagtaatacgactcactatagggagaaggctTCCCTACTAAAAAAACCCCTACCTC-3ʹ (reverse). The primer set was designed for EpiTYPER using EpiDesigner (Agena Bioscience), and the spectrum characteristics were validated with RSeqMeth [Citation58]. One microliter of BSC DNA (10 ng for standard samples, 0, 2, 5, 8, 18, and 34 ng for reference sample and average 18 ng for group of samples) was used in a 5 μL reaction mixture containing 0.4 U of PCR enzyme, 1 X PCR Buffer with 20 mM MgCl2, 200 μM dNTPs (Agena Bioscience, 11,327) and 2 pmoles of each primer. The cycling conditions were: denaturation (94°C for 15 min) followed by 50 cycles of amplification (94°C for 30 s, 58°C for 60 s, and 72°C for 30 s), and a final extension step (72°C for 10 min) (GeneAmp® PCR System 9700, ThermoFisher Scientific, Inc.). Unmethylated (0%) and methylated (100%) DNA standards (Qiagen, EpiTect control DNA, 59,695) were used to create a standard curve (0%, 25%, 50% 75%, 100% methylation) and served as positive controls. To confirm amplification, each sample was run on a 2% agarose gel. Reproducibility and sensitivity of the product of the OXTR EpiTYPER assay was assessed using the positive controls run in triplicate and measured on a MALDI-TOF mass spectrometer. The mass spectra methylation ratios were generated using EpiTYPER v. 1.2 (Agena Biosciences).
Statistical analysis
The association between DNA quantifications performed with different methods (e.g., spectrofluorimetry vs. spectrophotometry) was assessed using Pearson correlations. Student t-tests were used to compare the mean values between methods. All analyses were conducted using R (v. 3.2.1) via the RStudio platform (v. 0.99.489).
Supplemental Material
Download MS Power Point (1.4 MB)Acknowledgments
The authors would like to acknowledge the participants of the study and the staff for their assistance with participant recruitment and data collection.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun. 2014;38:227–236.
- Adalsteinsson BT, Ferguson-Smith AC. Epigenetic control of the genome-lessons from genomic imprinting. Genes (Basel). 2014;5(3):635–655.
- Almli LM, Stevens JS, Smith AK, et al. A genome-wide identified risk variant for PTSD is a methylation quantitative trait locus and confers decreased cortical activation to fearful faces. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(5):327–336.
- Kato T, Iwamoto K. Comprehensive DNA methylation and hydroxymethylation analysis in the human brain and its implication in mental disorders. Neuropharmacology. 2014;80:133–139.
- Aslibekyan S, Demerath EW, Mendelson M, et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring). 2015;23(7):1493–1501.
- Parets SE, Conneely KN, Kilaru V, et al. DNA methylation provides insight into intergenerational risk for preterm birth in African Americans. Epigenetics. 2015;10(9):784–792.
- Houseman EA, Kim S, Kelsey KT, et al. DNA methylation in whole blood: uses and challenges. Curr Environ Health Rep. 2015;2(2):145–154.
- Fuchikami M, Morinobu S, Segawa M, et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One. 2011;6(8):e23881.
- Córdova-Palomera A, Fatjó-Vilas M, Gastó C, et al. Genome-wide methylation study on depression: differential methylation and variable methylation in monozygotic twins. Transl Psychiatry. 2015;5:e557.
- Nunes AP, Oliveira IO, Santos BR, et al. Quality of DNA extracted from saliva samples collected with the Oragene™ DNA self-collection kit. BMC Med Res Methodol. 2012;12:65.
- Parade SH, Novick AM, Parent J, et al. Stress exposure and psychopathology alter methylation of the serotonin receptor 2A (HTR2A) gene in preschoolers. Dev Psychopathol. 2017;29(5):1619–1626.
- Parade SH, Parent J, Rabemananjara K, et al. Change in FK506 binding protein 5 (FKBP5) methylation over time among preschoolers with adversity. Dev Psychopathol. 2017;29(5):1627–1634.
- Parent J, Parade SH, Laumann LE, et al. Dynamic stress-related epigenetic regulation of the glucocorticoid receptor gene promoter during early development: the role of child maltreatment. Dev Psychopathol. 2017;29(5):1635–1648.
- Oelsner KT, Guo Y, To SB, et al. Maternal BMI as a predictor of methylation of obesity-related genes in saliva samples from preschool-age Hispanic children at-risk for obesity. BMC Genomics. 2017;18(1):57.
- Jovanovic T, Vance LA, Cross D, et al. Exposure to violence accelerates epigenetic aging in children. Sci Rep. 2017;7(1):8962.
- Langie SAS, Szarc Vel Szic K, Declerck K, et al. Correction: whole-genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS One. 2017;12(8):e0183088.
- Philibert RA, Zadorozhnyaya O, Beach SR, et al. Comparison of the genotyping results using DNA obtained from blood and saliva. Psychiatr Genet. 2008;18(6):275–281.
- Abraham JE, Maranian MJ, Spiteri I, et al. Saliva samples are a viable alternative to blood samples as a source of DNA for high throughput genotyping. BMC Med Genomics. 2012;5:19.
- Feigelson HS, Rodriguez C, Robertson AS, et al. Determinants of DNA yield and quality from buccal cell samples collected with mouthwash. Cancer Epidemiol Biomarkers Prev. 2001;10(9):1005–1008.
- García-Closas M, Egan KM, Abruzzo J, et al. Collection of genomic DNA from adults in epidemiological studies by buccal cytobrush and mouthwash. Cancer Epidemiol Biomarkers Prev. 2001;10(6):687–696.
- Birnboim HC, Iwasiow RM, James CMP. Human genomic DNA content of saliva samples collected in Oragene®DNA. Ottawa, Ontario, Canada: DNA Genotek, Inc; 2008.
- Song Y, Miyaki K, Suzuki T, et al. Altered DNA methylation status of human brain derived neurotrophis factor gene could be useful as biomarker of depression. Am J Med Genet B Neuropsychiatr Genet. 2014;165B(4):357–364.
- Smith AK, Kilaru V, Klengel T, et al. DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(1):36–44.
- Nohesara S, Ghadirivasfi M, Mostafavi S, et al. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J Psychiatr Res. 2011;45(11):1432–1438.
- Antunes J, Silva DS, Balamurugan K, et al. High-resolution melt analysis of DNA methylation to discriminate semen in biological stains. Anal Biochem. 2016;494:40–45.
- Swift-Scanlan T, Smith CT, Bardowell SA, et al. Comprehensive interrogation of CpG island methylation in the gene encoding COMT, a key estrogen and catecholamine regulator. BMC Med Genomics. 2014;7:5.
- Armstrong DA, Lesseur C, Conradt E, et al. Global and gene-specific DNA methylation across multiple tissues in early infancy: implications for children’s health research. FASEB J. 2014;28(5):2088–2097.
- Haas BW, Filkowski MM, Cochran RN, et al. Epigenetic modification of OXT and human sociability. Proc Natl Acad Sci USA. 2016;113(27):E3816–23.
- Patterson K, Molloy L, Qu W, et al. DNA methylation: bisulphite modification and analysis. J Vis Exp. 2011 Oct 21;56.
- Rusiecki JA, Byrne C, Galdzicki Z, et al. PTSD and DNA methylation in select immune function gene promoter regions: a repeated measures case-control study of U.S. Military service members. Front Psychiatry. 2013;4:56.
- Simpson JT, Workman RE, Zuzarte PC, et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods. 2017;14(4):407–410.
- Soares Bispo Santos Silva D, Antunes J, Balamurugan K, et al. Evaluation of DNA methylation markers and their potential to predict human aging. Electrophoresis. 2015;36(15):1775–1780.
- Bocklandt S, Lin W, Sehl ME, et al. Epigenetic predictor of age. PLoS One. 2011;6(6):e14821.
- Liu J, Morgan M, Hutchison K, et al. A study of the influence of sex on genome wide methylation. PLoS One. 2010;5(4):e10028.
- Stueve TR, Wolff MS, Pajak A, et al. CYP19A1 promoter methylation in saliva associated with milestones of pubertal timing in urban girls. BMC Pediatr. 2014;14:78.
- Souren NY, Tierling S, Fryns JP, et al. DNA methylation variability at growth-related imprints does not contribute to overweight in monozygotic twins discordant for BMI. Obesity (Silver Spring). 2011;19(7):1519–1522.
- Gardner KR, Sapienza C, Fisher JO. Genetic and epigenetic associations to obesity-related appetite phenotypes among African-American children. Pediatr Obes. 2015;10(6):476–482.
- Simkin M, Abdalla M, El-Mogy M, et al. Differences in the quantity of DNA found in the urine and saliva of smokers versus nonsmokers: implications for the timing of epigenetic events. Epigenomics. 2012;4(3):343–352.
- Hinhumpatch P, Navasumrit P, Chaisatra K, et al. Oxidative DNA damage and repair in children exposed to low levels of arsenic in utero and during early childhood: application of salivary and urinary biomarkers. Toxicol Appl Pharmacol. 2013;273(3):569–579.
- Kim JH, Rozek LS, Soliman AS, et al. Bisphenol A-associated epigenomic changes in prepubescent girls: a cross-sectional study in Gharbiah, Egypt. Environ Health. 2013;12:33.
- Edelman S, Shalev I, Uzefovsky F, et al. Epigenetic and genetic factors predict women’s salivary cortisol following a threat to the social self. PLoS One. 2012;7(11):e48597.
- Weder N, Zhang H, Jensen K, et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry. 2014;53(4):417–24.e5.
- Abdolmaleky HM, Pajouhanfar S, Faghankhani M, et al. Antipsychotic drugs attenuate aberrant DNA methylation of DTNBP1 (dysbindin) promoter in saliva and post-mortem brain of patients with schizophrenia and Psychotic bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2015;168(8):687–696.
- Andersen AM, Dogan MV, Beach SR, et al. Current and future prospects for epigenetic biomarkers of substance use disorders. Genes (Basel). 2015;6(4):991–1022.
- Kaminsky Z, Wilcox HC, Eaton WW, et al. Epigenetic and genetic variation at SKA2 predict suicidal behavior and post-traumatic stress disorder. Transl Psychiatry. 2015;5:e627.
- Lévesque ML, Casey KF, Szyf M, et al. Genome-wide DNA methylation variability in adolescent monozygotic twins followed since birth. Epigenetics. 2014;9(10):1410–1421.
- Unternaehrer E, Luers P, Mill J, et al. Dynamic changes in DNA methylation of stress-associated genes (OXTR, BDNF) after acute psychosocial stress. Transl Psychiatry. 2012;2:e150.
- Piyasena C, Cartier J, Provencal N, et al. Dynamic changes in DNA methylation occur during the first year of life in preterm infants. Front Endocrinol (Lausanne). 2016;7:158.
- Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37(3):e21.
- Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59(3):695–700.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
- Gregory SG, Connelly JJ, Towers AJ, et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009;7:62.
- Haas BW, Smith AK. Oxytocin, vasopressin, and Williams syndrome: epigenetic effects on abnormal social behavior. Front Genet. 2015;6:28.
- Bell AF, Carter CS, Steer CD, et al. Interaction between oxytocin receptor DNA methylation and genotype is associated with risk of postpartum depression in women without depression in pregnancy. Front Genet. 2015;6:243.
- Dadds MR, Moul C, Cauchi A, et al. Methylation of the oxytocin receptor gene and oxytocin blood levels in the development of psychopathy. Dev Psychopathol. 2014;26(1):33–40.
- Jack A, Connelly JJ, Morris JP. DNA methylation of the oxytocin receptor gene predicts neural response to ambiguous social stimuli. Front Hum Neurosci. 2012;6:280.
- Kusui C, Kimura T, Ogita K, et al. DNA methylation of the human oxytocin receptor gene promoter regulates tissue-specific gene suppression. Biochem Biophys Res Commun. 2001;289(3):681–686.
- Coolen MW, Statham AL, Gardiner-Garden M, et al. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements. Nucleic Acids Res. 2007;35(18):e119.