
ABSTRACT
Histone posttranslational modifications control the organization and function of chromatin. In particular, methylation of lysine 36 in histone H3 (H3K36me) has been shown to mediate gene transcription, DNA repair, cell cycle regulation, and pre-mRNA splicing. Notably, mutations at or near this residue have been causally linked to the development of several human cancers. These observations have helped to illuminate the role of histones themselves in disease and to clarify the mechanisms by which they acquire oncogenic properties. This perspective focuses on recent advances in discovery and characterization of histone H3 mutations that impact H3K36 methylation. We also highlight findings that the common cancer-related substitution of H3K36 to methionine (H3K36M) disturbs functions of not only H3K36me-writing enzymes but also H3K36me-specific readers. The latter case suggests that the oncogenic effects could also be linked to the inability of readers to engage H3K36M.
KEYWORDS:
Introduction
Histone proteins are main components of the nucleosome, the fundamental building block of chromatin in eukaryotic cells. In addition to playing a critical role in chromatin structure and dynamics, histones undergo posttranslational modifications (PTMs) that provide mechanisms for mediating diverse cellular processes [Citation1–Citation3]. PTMs or epigenetic marks are deposited, removed and recognized by writers, erasers and readers, respectively, which individually or in combination initiate, halt, or propagate biological signals in the nucleus [Citation4,Citation5,Citation6] (). Disruption of the intricate balance between activities of writers, erasers, and readers is linked to a host of human diseases, including cancer and autoimmune, developmental, and neurodegenerative disorders [Citation7].
Figure 1. Histone lysine PTMs and oncogenic mutations. (a) Schematic representation of writers, readers and erasers targeting lysine residues in histone tails of the nucleosome. (b) Residues of the H3 tail and mutations that have been linked to cancer are colored green and red, respectively.
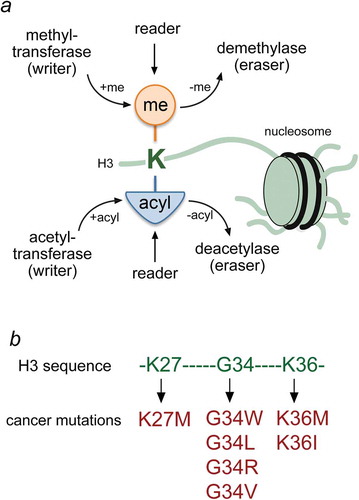
One such histone modification that has intersected at the cross-roads between normal and diseased states is methylation of lysine 36 of histone H3 (H3K36me). This chromatin mark is conserved from yeast to humans and has been shown to have a variety of functions that range from the control of gene transcription and DNA repair, to cell cycle regulation and nutrient stress response [Citation8]. H3K36 methylated domains are associated with active transcription, and aberrant regulation of this chromatin mark has been linked to several cancers. Particularly, histone mutations that affect H3K36 methylation were identified as likely drivers of some types of pediatric cancers [Citation9,Citation10,Citation11,Citation12,Citation13], ()). Genetic studies found that about one fifth of high-grade astrocytomas (HGA) arising in the cerebral hemispheres of adolescents contain aberrations affecting K36, including H3 mutations at G34 or loss of H3K36-specific writer, the methyltransferase SETD2. Two bone cancers, such as chondroblastoma and giant cell tumors (GCT) of the bone that affect adolescents and younger children, carry H3 mutations at K36 and G34, respectively. Importantly, these mutations are the sole genetic alterations identified in these tumors, and the tumor type and pattern suggest that the histone mutations themselves likely drive tumor formation in these neoplasms. Furthermore, the specificity of the histone gene affected in relation to patient age and tumor type or location further points toward a distinctive cell of origin for these tumors and, subsequently, histones are now referred to as ‘oncohistones’, since it is clear that these mutations act as classical cancer drivers.
In this perspective, we highlight the latest reports underlining the impact of oncohistone mutations on writing and reading the H3K36me mark. We summarize the biological significance of H3K36 methylation for normal cell processes and discuss recently proposed oncohistone-driven tumorigenic mechanisms.
H3K36 methylation and chromatin regulation
In all mammals, H3K36 is methylated by a range of lysine-specific enzymes, including SETD2, the homolog of yeast Set2, NSD1, NSD2/MMSET/WHSC1, NSD3, SETMAR, SMYD2, and ASH1L [Citation14,Citation15–Citation19]. While the NSD1-3 enzymes mediate the bulk of H3K36me1 and H3K36me2 throughout the genome, SETD2 specially trimethylates H3K36 in a co-transcriptional manner through this enzyme’s ability to interact with the transcribing RNA polymerase II (RNAPII) [Citation20,Citation21]. Consistent with the co-transcriptional role of SETD2, H3K36me3 is found almost exclusively in transcribed regions in yeast and mammals. In contrast to SETD2, the NSD enzymes are not co-transcriptional and are known to methylate large stretches of intergenic regions across the genome that serve, at least in part, as domain boundaries that act to prevent the spread of polycomb-mediated H3 lysine 27 (H3K27) methylation [Citation22].
Studies into the function of H3K36me reveal a wide range of activities, which include the suppressing histone exchange and intergenic transcription, promoting DNA repair, cell cycle progression and maintaining proper pre-mRNA splicing [Citation8]. These functions, in large part, are carried out by various reader complexes that associate with this mark. Initially characterized in yeast, H3K36 methylation was shown to recruit and/or activate a number of chromatin-associated enzymes, including the Rpd3S deacetylase complex containing the Eaf3 chromodomain, ISW1b ATP-dependent remodeling complex and the NuA3b acetyltransferase complex [Citation23–Citation27]. Although the function of NuA3b is less clear, the Rpd3S and ISW1b complexes act to maintain a deacetylated chromatin state within RNAPII transcribed regions that serves to enforce the suppression of histone exchange and inappropriate transcription from occurring within these regions [Citation28–Citation32]. From a cellular perspective, this process has been linked to life-span control, DNA double-stand break repair, cell cycle progression, nutrient stress response and, more recently, pre-mRNA splicing in budding yeast [Citation33–Citation42].
Like Set2 in yeast, H3K36me3 mediated by mammalian SETD2 is also known to recruit an Rpd3S-related HDAC complex that associates via the Eaf3 homolog MRG15; MRG15 also maintains reduced acetylation levels and functions in alternative mRNA splicing [Citation43,Citation44]. Beyond HDACs, SETD2 and the other H3K36 methyltransferases have been shown to recruit a variety of other chromatin-associated protein complexes that have functions in transcription elongation, heterochromatin formation, mRNA splicing, and DNA repair [Citation8]. For example, ZMYND11 was shown to bind H3.3K36me3 through its PWWP domain and to regulate pre-mRNA splicing and transcription elongation [Citation45,Citation46]. In addition, SETD2-mediated H3K36me3 regulates the recruitment of the DNA methyltransferase DNMT3b to transcribed regions also through a PWWP domain that controls gene body DNA methylation [Citation47–Citation49], which has remained somewhat elusive in function but may contribute to suppression of intergenic transcription. Outside of transcriptional control, H3K36 methylation functions in homologous recombination and mismatch repair through the recruitment of LEDGF and MHS6, respectively [Citation40,Citation50,Citation51]. Intriguingly, H3K36 methylation also associates with the Tudor domains of PHF1/PHF19 of the polycomb repressive complex 2 (PRC2) [Citation52–Citation59], which appears to function as a boundary element to isolate or restrict the spread of polycomb H3K27me domains. With so many biological activities and readers, H3K36 methylation has emerged to be a critical mark for genome function. As discussed below, a critical component for how H3K36 mutation likely contributes to cancer progression is the inability of these aforementioned readers to bind to H3K36 methylation.
Oncogenic H3K36M mutation blocks H3K36 methylation
Ground-breaking discoveries of H3 variant mutations in pediatric cancers represent ‘game changers’ in our understanding of cancer epigenetics [Citation9–Citation13]. Several genes encoding H3 were found to contain lysine-to-methionine (K-to-M) mutations with a high frequency in certain tumor types. Specifically, H3K27M is found in 85% of diffuse intrinsic potine gliomas (DIPG) and in a majority of mid-line high-grade astrocytomas, whereas H3K36M is found in 95% of chondroblastomas and also at lower frequencies in head and neck squamous cell carcinomas and undifferentiated sarcomas.
Expression of K-to-M mutant histones in cells has been shown to lead to a global loss of lysine methylation on the cognate histone residues in vivo [Citation60–Citation62]. Multiple lines of biochemical and structural evidence converge on the idea that methionine substitution at particular lysines in H3 transform histones from serving as substrates into potent orthosteric inhibitors of methyltransferases [Citation60,Citation62–Citation68]. For example, H3K36M peptides and H3K36M-containing nucleosomes inhibit catalytic activities of H3K36-specific methyltransferases in vitro, and expression of H3K36M results in a global loss of H3K36 methylation in vivo [Citation22,Citation62,Citation69,Citation70].
The oncogenic capacity of the H3K36M mutant histone has recently been demonstrated in studies with animal models. Introduction of the H3K36M mutation into mesenchymal progenitor cells causes a profound impairment of mesenchymal differentiation. Furthermore, the H3K36M-expressing mesenchymal progenitor cells form tumors resembling undifferentiated sarcomas in mice xenograft studies. In addition to reduced H3K36me2/3 levels, H3K36M-expressing tumors exhibit increased H3K27me3 levels [Citation22]. The loss of H3K36me2/3 and the gain of H3K27me3 are linked and result from the availability of new nucleosome substrate for the PRC2 complex, responsible for H3K27 methylation. It is likely that PRC2 ‘senses’ the methylation status on H3K36 through a cis-acting mechanism and fails to act catalytically on histones containing K36me2/3 [Citation71,Citation72]. Genome-wide profiling of H3K36M-expressing cells showed a dramatic loss of H3K36me2 in intergenic regions, which was now replaced by H3K27me3. The increase of intergenic H3K27me3 led to a redistribution of PRC1 and de-repression of its target genes known to block mesenchymal differentiation. Paradoxically, H3K36M cells contain more H3K27me3 but exhibit selective loss of Polycomb-repressed genes. As we noted earlier, H3K36 methylation is linked to a transcriptional elongation, RNA splicing, and DNA damage repair, and misregulation of these processes may also contribute to H3K36M-mediated tumorigenesis.
H3K36M entraps the H3K36me3-specific writer
The atomic-resolution structure of the catalytic domain (CD) of SETD2 that writes H3K36me3, bound to H3K36M peptide sheds light on the mechanism by which this oncogenic mutation blocks H3K36 methylation [Citation70,Citation73], (). The structure of the complex was determined in the presence of S-adenosylhomocystein (SAH), a reaction product formed after the methyl group is transferred from the methyl-donor S-adenosyl methionine (SAM). In the complex, the C-terminal part of CD, comprising the last loop of the SET region and a long loop of the post-SET region, covers bound H3K36M peptide from the top, completely burying the histone sequence from G33 to M36 (, pre-SET is shown in pink). Interestingly, the C-terminal part undergoes large conformational changes upon binding of SAH and H3K36M. While in the apo-state the last loop of the SET region intramolecularly inserts arginine in the active center resulting in a closed conformation and catalytically inactive protein, binding of SAH, and presumably SAM, flips arginine away from the active center relieving autoinhibition and opening the catalytic site for binding of substrate. However, the arginine-inserting pocket of the CD apo-state is occupied by M36 in the structure of the H3K36M-bound state of CD. The extended side chain of M36 points directly toward bound SAH and is locked in a narrow channel lined with hydrophobic residues (colored yellow in ) and topped with a large cap formed by the CD loops. An additional restraint for M36 arises from the formation of a pseudo-β-sheet in which the G33-K36 sequence of the H3K36M peptide adopts a β-strand conformation and inserts between two β-strands of CD. The structure of the H3K36M-CD-SAH complex clearly demonstrates how the H3K36M tail, securely locked in the substrate binding site of the SETD2 catalytic domain, sequesters SETD2. In cells, such sequestration can prevent spreading of H3K36 methylation, accounting for the reduction of global H3K36 methylation levels.
Figure 2. Structural basis for entrapping H3K36M in the SETD2 active site. (a) The crystal structure of the SETD2 catalytic domain (SET in white and Post-SET in pink) in complex with the H3K36M peptide (orange) and SAH (blue). SETD2 CD is shown in a surface model with the K36M-binding pocket residues and the G34-binding site residues colored yellow and light green, respectively. (b) A close view of the ribbon diagram of the H3K36M-CD-SAH complex structure. PDB ID code: 5JJY [Citation70].
![Figure 2. Structural basis for entrapping H3K36M in the SETD2 active site. (a) The crystal structure of the SETD2 catalytic domain (SET in white and Post-SET in pink) in complex with the H3K36M peptide (orange) and SAH (blue). SETD2 CD is shown in a surface model with the K36M-binding pocket residues and the G34-binding site residues colored yellow and light green, respectively. (b) A close view of the ribbon diagram of the H3K36M-CD-SAH complex structure. PDB ID code: 5JJY [Citation70].](/cms/asset/fb06afb5-d6e5-461d-97b1-cf94b9d10658/kepi_a_1503491_f0002_oc.jpg)
Oncogenic H3G34 mutations may act in cis
In addition to the K-to-M mutations, missense mutations of glycine 34 (G34R/V/W/L) are found in 20% patients with pediatric supratentorial high-grade astrocytomas and 92% patients with giant cell tumor of the bone [Citation10,Citation11,Citation12]. Strikingly, these mutations are present exclusively in genes encoding histone H3.3 variant but not other H3 variants, including H3.1. The histone H3.3 variant is localized to particular genomic regions, such as promoters, enhancers, actively transcribed genes, and pericentric and telomeric repeats [Citation74]. The direct correlation between G34 mutations and H3.3 genes is likely not coincidental and suggests that deposition of the mutant H3.3 at specific genomic locations plays a role in oncogenic properties of G34 mutants.
Unlike the CD sequestration by the H3K36M mutation, the mechanism underlying aberrant nature of G34R/V/W/L mutations appears to be less clear. Much like M36, the histone residue G34 in the H3K36M-bound CD of SETD2 is fully buried and occupies a hydrophobic pocket formed primarily by aromatic phenylalanine and tyrosine residues of CD [Citation70,Citation73] (colored green in ). Analysis of the structure suggests that the small size of the G34-binding pocket of CD would preclude a bulkier residue to occupy this site. It is conceivable that steric hindrance in cis that prevents priming of CD to the histone tail carrying mutations at G34 is the reason for the reduction in the catalytic activity of SETD2.
Studies have indicated that G34R/V/W/L mutations lead to a local loss of H3K36me3, although delineation of ‘local’ and ‘global’ loss of H3K36 methylation remains ambiguous [Citation62]. Additionally, if the local loss of K36 methylation is linked to oncogenic properties of H3.3G34 mutants, it’s unclear as to why mutations that directly affect K36 methylation (e.g., K36E and K36R) have not been reported in pediatric HGAs and GCTs. It is intriguing to speculate that the H3.3G34 mutant histones found in pediatric HGAs and GCTs create a unique local chromatin environment that cannot be readily achieved by other somatic mutations.
Effects of oncogenic mutations on H3K36me3 readers
While the crystal structure of the SETD2 catalytic domain bound to H3K36M peptide helps to explain the effect of oncohistone mutations on the writer’s function, less information is available on how these mutations influence binding activities of H3K36me-specific readers. To determine the effect of oncohistone mutations, we synthesized histone H3 peptides, H3H36me3, H3K36M and H3G34WK36me3 (residues 28–40 of H3.3) and examined their interactions with two established readers of the H3K36me3 mark, the Tudor domain of PHF1 and the PWWP domain of BRPF1 [Citation52,Citation75,Citation76] by NMR (). Large chemical shift perturbations (CSPs) in intermediate exchange regime on the NMR time scale were observed upon titration of the H3K36me3 peptide to the 15N-labeled PHF1 Tudor domain, corroborating previously reported robust binding [Citation52], ()). However, no CSPs were detected upon titration of H3K36M, indicating that the Tudor domain is incapable of binding to this oncohistone peptide. Conversely, addition of H3G34WK36me3 peptide induced CSPs similar in direction and magnitude to CSPs observed upon addition of the H3K36me3 peptide, suggesting that the G34W mutation does not affect the ability of Tudor to recognize H3K36me3. In contrast, either mutation affected binding of the PWWP domain of BRPF1. While the interaction of the 15N-labeled PWWP domain with H3K36M was abolished, binding to H3G34WK36me3 was considerably decreased ()). Together, these results reveal that the oncohistone binding activity of H3K36me3-specific readers is context dependent. While K36M but not G34W abrogates binding of the PHF1 Tudor domain, both mutations impede binding of the PWWP domain.
Figure 3. Oncohistone mutations impact binding of the H3K36me3 readers. (a, b) Superimposed 1H,15N HSQC spectra of PHF1 Tudor (a) and BRPF1 PWWP (b) recorded while H3H36me3, H3K36M and H3G34WK36me3 (residues 28–40 of H3.3) peptides were added stepwise. The spectra are color coded according to protein:peptide molar ratio. The structures of the H3K36me3-bound PHF1 Tudor domain and H3K36me3-bound BRPF1 PWWP domain are shown as surface models with the K36M-binding pocket residues and the G34-binding site residues colored yellow and light green, respectively. PDB ID codes: 4HCZ and 2X4X.
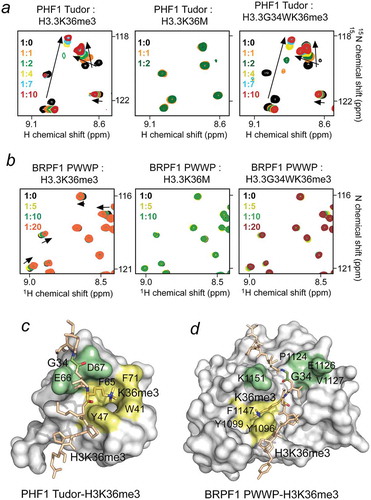
Structural studies reveal that both these readers recognize H3K36me3 via aromatic cages and that cation-π interactions between the trimethylammonium group of H3K36me3 and the aromatic side chains are essential. The inability of Tudor and PWWP to engage with H3K36M reinforces the critical role of these interactions. However, G34 in the Tudor-H3K36me3 complex is solvent exposed and occupies a shallow cavity that can accommodate a much larger residue such as tryptophan without apparent steric clashes. In contrast, priming of H3G34WK36me3 in the binding groove of PWWP is compromised likely because of steric hindrance.
Taken together, these findings show that onco-histone mutants that impact H3K36 methylation do not lead to continued association of its readers. In contract to the writer (the SETD2 catalytic domain), the H3K36me3-specific readers do not sequester their host proteins. Rather, these readers are unable to promote down-stream biological effects because their binding to the oncogenic H3K36M tail is impaired. It is still unclear which reader(s) contribute to the onco-histone phenotype; however, we speculate that the compromised histone binding activity could lead to the redistribution of reader-containing proteins, including ZMYND11, MRG15 and DNMT3b, to new genomic locations where they contribute to aberrant transcription elongation, histone deacetylation, and/or DNA methylation. Additionally, the redistribution of the reader-containing proteins and complexes may result in a loss of boundaries that restrict spreading of other marks, such as the repressive H3K27me3 mark, triggering uncontrolled silencing of tumor suppressor genes. Alternately, the loss of H3K36me3-binding activity of readers can decrease or eliminate catalytic functions of writers, as has been demonstrated for the Rpd3S complex in yeast [Citation77]. The reduced catalytic activities in the genomic regions enriched in oncogenic H3 mutations could derail transcriptional and DNA repair programs that contribute to the cancer phenotype. Future work will be needed to better define how H3K36M and related onco-histone mutations lead to cancer.
Concluding remarks
Recent studies have unveiled a novel pathway to cancer implicating mutations of H3K36 or nearby residues. While significant insights have been gained regarding how H3K36 mutation acts to inhibit the writers and readers of this mark, as well as influences the epigenome in ways that favor cancer progression, much remains to be learned. For example, why certain pediatric or adult cancers favor H3K36 mutation in a particular histone H3 variant is still poorly understood, along with how H3K36me-affecting mutants such as H3.3G34 impact cancer progression. It is also unclear whether these mutations act to promote cancer through dysregulation of the epigenome (e.g., cross-talk between H3K36me and H3K27me) over the loss of critical functions endowed in H3K36 (i.e., DNA repair). The next few years will undoubtedly be exciting as these questions become answered. Long-term, these insights stand to open up new targets for therapeutic intervention.
Materials and methods
Peptide synthesis
Peptides were synthesized by UNC peptide synthesis core.
DNA cloning, expression and protein purification
The Tudor domain of human PHF1 (aa 14–87) and the BRPF1 PWWP domain (aa 1064–1214) were expressed and purified as described [Citation52,Citation78]. Briefly, the BRPF1 PWWP domain and the PHF1 Tudor domain were expressed as uniformly 15N-labeled GST-fusion proteins in BL21(DE3) RIL cells grown in minimal media supplemented with 15NH4Cl (Sigma). The bacteria were harvested, and cells were lysed by sonication. The GST-fusion proteins were purified using glutathione agarose resin, and the GST tag was cleaved with either Thrombin (PWWP) or PreScission (Tudor) protease.
NMR spectroscopy
NMR experiments were performed on a Varian INOVA 600 MHz spectrometer at the University of Colorado School of Medicine NMR Core facility. CSPs experiments were carried out at 298K using uniformly 15N-labeled PWWP domain of BRPF1 and Tudor domain of PHF1. 1H,15N heteronuclear single quantum coherence (HSQC) spectra were collected in the presence of increasing concentrations of either H3H36me3, H3K36M and H3G34WK36me3 peptides in buffers: PBS buffer pH 6.8 supplemented with 3 mM DTT for PWWP, and 25 mM Tris-HCl buffer pH 6.8 supplemented with 150 mM NaCl and 3 mM DTT for Tudor.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839:627–643.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395.
- Andrews FH, Strahl BD, Kutateladze TG. Insights into newly discovered marks and readers of epigenetic information. Nat Chem Biol. 2016;12:662–668.
- Taverna SD, Li H, Ruthenburg AJ, et al. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040.
- Musselman CA, Lalonde ME, Cote J, et al. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227.
- Patel DJ, Wang Z. Readout of epigenetic modifications. Annu Rev Biochem. 2013;82:81–118.
- Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. 2018;378:1323–1334.
- McDaniel SL, Strahl BD. Shaping the cellular landscape with Set2/SETD2 methylation. Cell Mol Life Sci. 2017;74:3317–3334.
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447.
- Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231.
- Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253.
- Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–1482.
- Papillon-Cavanagh S, Lu C, Gayden T, et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet. 2017;49:180–185.
- Strahl BD, Grant PA, Briggs SD, et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298–1306.
- Rayasam GV, Wendling O, Angrand PO, et al. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003;22:3153–3163.
- Angrand PO, Apiou F, Stewart AF, et al. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 2001;74:79–88.
- Eram MS, Kuznetsova E, Li F, et al. Kinetic characterization of human histone H3 lysine 36 methyltransferases, ASH1L and SETD2. Biochim Biophys Acta. 2015;1850:1842–1848.
- Yi X, Jiang XJ, Li XY, et al. Histone methyltransferases: novel targets for tumor and developmental defects. Am J Transl Res. 2015;7:2159–2175.
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420.
- Sun XJ, Wei J, Wu XY, et al. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005;280:35261–35271.
- Kizer KO, Phatnani HP, Shibata Y, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316.
- Lu C, Jain SU, Hoelper D, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–849.
- Carrozza MJ, Li B, Florens L, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592.
- Keogh MC, Kurdistani SK, Morris SA, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605.
- Smolle M, Venkatesh S, Gogol MM, et al. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol. 2012;19:884–892.
- Maltby VE, Martin BJ, Schulze JM, et al. Histone H3 lysine 36 methylation targets the Isw1b remodeling complex to chromatin. Mol Cell Biol. 2012;32:3479–3485.
- Gilbert TM, McDaniel SL, Byrum SD, et al. A PWWP domain-containing protein targets the NuA3 acetyltransferase complex via histone H3 lysine 36 trimethylation to coordinate transcriptional elongation at coding regions. Mol Cell Proteomics. 2014;13:2883–2895.
- Lee CH, Wu J, Li B. Chromatin remodelers fine-tune H3K36me-directed deacetylation of neighbor nucleosomes by Rpd3S. Mol Cell. 2013;52:255–263.
- Venkatesh S, Smolle M, Li H, et al. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455.
- Venkatesh S, Li H, Gogol MM, et al. Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat Commun. 2016;7:13610.
- Li B, Gogol M, Carey M, et al. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007;21:1422–1430.
- Li B, Gogol M, Carey M, et al. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007;316:1050–1054.
- Dronamraju R, Jha DK, Eser U, et al. Set2 methyltransferase facilitates cell cycle progression by maintaining transcriptional fidelity. Nucleic Acids Res. 2018;46:1331–1344.
- Jha DK, Strahl BD. An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat Commun. 2014;5:3965.
- McDaniel SL, Hepperla AJ, Huang J, et al. H3K36 methylation regulates nutrient stress response in saccharomyces cerevisiae by enforcing transcriptional fidelity. Cell Rep. 2017;19:2371–2382.
- Sen P, Dang W, Donahue G, et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015;29:1362–1376.
- Sorenson MR, Jha DK, Ucles SA, et al. Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae. RNA Biol. 2016;13:412–426.
- Pai CC, Deegan RS, Subramanian L, et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat Commun. 2014;5:4091.
- Carvalho S, Vitor AC, Sridhara SC, et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. 2014;3:e02482.
- Li F, Mao G, Tong D, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600.
- Pai CC, Kishkevich A, Deegan RS, et al. Set2 methyltransferase facilitates DNA replication and promotes genotoxic stress responses through MBF-dependent transcription. Cell Rep. 2017;20:2693–2705.
- Pfister SX, Ahrabi S, Zalmas LP, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006–2018.
- Kumar GS, Chang W, Xie T, et al. Sequence requirements for combinatorial recognition of histone H3 by the MRG15 and Pf1 subunits of the Rpd3S/Sin3S corepressor complex. J Mol Biol. 2012;422:519–531.
- Jelinic P, Pellegrino J, David G. A novel mammalian complex containing Sin3B mitigates histone acetylation and RNA polymerase II progression within transcribed loci. Mol Cell Biol. 2011;31:54–62.
- Guo R, Zheng L, Park JW, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell. 2014;56:298–310.
- Wen H, Li Y, Xi Y, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–268.
- Baubec T, Colombo DF, Wirbelauer C, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247.
- Morselli M, Pastor WA, Montanini B, et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife. 2015;4:e06205.
- Neri F, Rapelli S, Krepelova A, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543:72–77.
- Pradeepa MM, Sutherland HG, Ule J, et al. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717.
- Zhu L, Li Q, Wong SH, et al. ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 2016;6:770–783.
- Musselman CA, Avvakumov N, Watanabe R, et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol. 2012;19:1266–1272.
- Brien GL, Gambero G, O’Connell DJ, et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol. 2012;19:1273–1281.
- Cai L, Rothbart SB, Lu R, et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell. 2013;49:571–582.
- Ballare C, Lange M, Lapinaite A, et al. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat Struct Mol Biol. 2012;19:1257–1265.
- Qin S, Guo Y, Xu C, et al. Tudor domains of the PRC2 components PHF1 and PHF19 selectively bind to histone H3K36me3. Biochem Biophys Res Commun. 2013;430:547–553.
- Gibson MD, Gatchalian J, Slater A, et al. PHF1 Tudor and N-terminal domains synergistically target partially unwrapped nucleosomes to increase DNA accessibility. Nucleic Acids Res. 2017;45:3767–3776.
- Gatchalian J, Kingsley MC, Moslet SD, et al. An aromatic cage is required but not sufficient for binding of Tudor domains of the Polycomblike protein family to H3K36me3. Epigenetics. 2015;10:467–473.
- Musselman CA, Gibson MD, Hartwick EW, et al. Binding of PHF1 Tudor to H3K36me3 enhances nucleosome accessibility. Nat Commun. 2013;4:2969.
- Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–672.
- Chan KM, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–990.
- Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861.
- Lewis PW, Allis CD. Poisoning the “histone code” in pediatric gliomagenesis. Cell Cycle. 2013;12:3241–3242.
- Brown ZZ, Muller MM, Jain SU, et al. Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J Am Chem Soc. 2014;136:13498–13501.
- Brown ZZ, Muller MM, Kong HE, et al. Targeted histone peptides: insights into the spatial regulation of the methyltransferase PRC2 by using a Surrogate of heterotypic chromatin. Angew Chem Int Ed Engl. 2015;54:6457–6461.
- Jayaram H, Hoelper D, Jain SU, et al. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc Natl Acad Sci USA. 2016;113:6182–6187.
- Justin N, Zhang Y, Tarricone C, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316.
- Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015;350:aac4383.
- Fang D, Gan H, Lee JH, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 2016;352:1344–1348.
- Yang S, Zheng X, Lu C, et al. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 2016;30:1611–1616.
- Schmitges FW, Prusty AB, Faty M, et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42:330–341.
- Yuan W, Xu M, Huang C, et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286:7983–7989.
- Zhang Y, Shan CM, Wang J, et al. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep. 2017;7:43906.
- Banaszynski LA, Allis CD, Lewis PW. Histone variants in metazoan development. Dev Cell. 2010;19:662–674.
- Vezzoli A, Bonadies N, Allen MD, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–619.
- Klein BJ, Muthurajan UM, Lalonde ME, et al. Bivalent interaction of the PZP domain of BRPF1 with the nucleosome impacts chromatin dynamics and acetylation. Nucleic Acids Res. 2016;44:472–484.
- Drouin S, Laramee L, Jacques PE, et al. DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet. 2010;6:e1001173.
- Andrews FH, Gatchalian J, Krajewski K, et al. Regulation of methyllysine readers through phosphorylation. ACS Chem Biol. 2016;11:547–553.