
ABSTRACT
The identification of multilocus imprinting disturbances (MLID) appears fundamental to uncover molecular pathways underlying imprinting disorders (IDs) and to complete clinical diagnosis of patients. However, MLID genetic associated mechanisms remain largely unknown. To characterize MLID in Beckwith-Wiedemann (BWS) and Silver-Russell (SRS) syndromes, we profiled by MassARRAY the methylation of 12 imprinted differentially methylated regions (iDMRs) in 21 BWS and 7 SRS patients with chromosome 11p15.5 epimutations. MLID was identified in 50% of BWS and 29% of SRS patients as a maternal hypomethylation syndrome. By next-generation sequencing, we searched for putative MLID-causative mutations in genes involved in methylation establishment/maintenance and found two novel missense mutations possibly causative of MLID: one in NLRP2, affecting ADP binding and protein activity, and one in ZFP42, likely leading to loss of DNA binding specificity. Both variants were paternally inherited. In silico protein modelling allowed to define the functional effect of these mutations. We found that MLID is very frequent in BWS/SRS. In addition, since MLID-BWS patients in our cohort show a peculiar pattern of BWS-associated clinical signs, MLID test could be important for a comprehensive clinical assessment. Finally, we highlighted the possible involvement of ZFP42 variants in MLID development and confirmed NLRP2 as causative locus in BWS-MLID.
Introduction
Beckwith-Wiedemann (BWS, OMIM #130650) and Silver-Russell (SRS, OMIM #180860) syndromes are paired imprinting disorders (IDs) caused by opposite molecular defects at the 11p15.5 chromosomal region, which harbors two imprinted domains, IGF2/H19 and CDKN1C/KCNQ1OT1, regulated by different imprinting control regions, ICR1 and ICR2, respectively [Citation1,Citation2]. Loss of methylation (LOM) at ICR1 accounts for over 50% of SRS cases, while gain of methylation (GOM) at ICR1 and LOM at ICR2 trigger BWS in approximately 10% and 60% of cases, respectively. In addition, maternal uniparental disomy of chromosome 7 (mUPD7) and paternal UPD of 11p15.5 are present in 7–10% of SRS and 20% of BWS, respectively [Citation3–Citation5].
The two syndromes exhibit opposite phenotypes. BWS is an overgrowth condition characterized by macrosomia, macroglossia, hemihyperplasia, visceromegaly with consequent abdominal wall defects, and increased risk of embryonal tumors [Citation1]. Conversely, growth restriction, relative macrocephaly, peculiar small and triangular face, the fifth finger clinodactyly, and hemihypoplasia are characteristic of SRS [Citation2].
The phenotypic heterogeneity of BWS/SRS and the presence of clinical features overlapping with other IDs [Citation6–Citation8], have prompted the investigation of methylation defects at additional imprinted genes in patients with epimutations at the disease-specific locus, leading to the identification of a subset of patients with aberrant methylation affecting other imprinted differentially methylated regions (iDMRs), a condition described as multi-locus imprinting disturbance (MLID). MLID has been identified in BWS patients at frequencies of up to 30% in patients with LOM at ICR2 [Citation6–Citation14], while it is very rare among cases with GOM at ICR1 [Citation15]. Some BWS patients with MLID show LOM or GOM at both maternal and paternal DMRs [Citation8,Citation15], while others have a hypomethylation syndrome restricted to maternally imprinted genes [Citation16–Citation18]. MLID has less frequently been described in patients with SRS (~ 15%) [Citation8].
The phenotypic consequences of MLID in BWS and SRS patients are disputed, since not all patients with MLID show peculiar phenotypic features, probably as a consequence of the (epi)dominance of one locus above the others [Citation19] or as a result of mosaicism. However, some exceptions have been reported. Specifically, birth weight is usually lower in patients with MLID than in those with mono-locus BWS, and macrosomia is less frequent; patients with MLID can present features not related to BWS (speech retardation, apnea, and feeding difficulties) [Citation7]. SRS patients with MLID more frequently exhibit growth delay and additional congenital abnormalities [Citation6]. MLID-associated clinical signs may also only manifest as patients grow up; therefore, MLID analysis, after molecular confirmation of a specific ID, may guide a patient-tailored follow-up to monitor for subclinical signs before their manifestation [Citation17,Citation20].
The etiology of MLID remains to be elucidated, as do the mechanisms underlying the co-regulation of imprinting marks across the genome. Despite the identification of MLID causative mutations in members of the NLRP and zinc-finger protein families in few BWS/SRS patients [Citation10,Citation21,Citation22], the role of these proteins in the imprinting process and the pattern of inheritance remain to be fully elucidated. A role for NLRP mutations in MLID was suggested based on the observation of shared hypomethylated loci between MLID and hydatiform mole [Citation23]. In particular, NLRP7 mutations have been associated with the most severe form of MLID, the biparental hydatiform mole [Citation24]. In addition, maternally-derived mutations of NLRP2 and NLRP5 genes, already associated to molar pregnancies, have been identified in BWS/SRS patients with MLID; however, the type of epimutations (hypo/hypermethylation), the extent of the methylation defect, and the number/type of affected iDMRs in MLID suggest a different mechanism of pathogenicity compared to molar pregnancies [Citation21,Citation22]. Indeed, in NLRP mutated MLID cases, the affected iDMRs appear to vary, and intermediate levels of LOM suggest a post-zygotic effect. In addition, both homozygous and heterozygous causative mutations in the NLRP genes have been reported in BWS patients, suggesting a variable pattern of inheritance [Citation8,Citation21,Citation22,Citation25]. Furthermore, incomplete penetrance may account for the variability observed among patients with the same pathogenic mutations [Citation21].
Mutations in genes encoding members of the zinc-finger protein family have been considered good candidates as causative defects of MLID, given the well-established role of ZFP57 mutation in patients with transient neonatal diabetes mellitus and MLID [Citation26,Citation27]. However, no mutations have been found to date in other members of the protein family in MLID patients.
Here, we report the methylation profiling of 12 iDMRs investigated simultaneously by MALDI-TOF Mass Spectrometry (MassARRAY) in 21 BWS and 7 SRS patients presenting with chromosome 11p15.5 epimutations. This approach allowed the identification of MLID in approximately 50% and 29% of patients with BWS and SRS, respectively. Targeted next-generation sequencing (NGS) of a panel of genes involved in methylation establishment/maintenance highlighted two novel missense mutations possibly associated with MLID.
Results
Multi-locus methylation disturbances in patients with BWS and SRS
The MassARRAY methylation assays were optimized using 50 blood age-matched controls and 20 amniotic fluids (AF) to define the physiological methylation ranges at each iDMR ( and Supplementary Figure S1). Methylation analysis of the 12 iDMRs allowed the identification of MLID in 10 of 21 cases with BWS (50%) and 2 of 7 with SRS (29%) (). Notably, among the BWS patients, MLID was detected only in those with ICR2 LOM (10 of 17 cases, 59%).
Figure 1. Methylation analyses of 12 imprinted iDMRs by MassARRAY in patients with BWS and SRS with known epimutations at ICR1 or ICR2. Cases with MLID are underlined. Aberrant methylation was defined as methylation values deviating +/- two standard deviations from the mean value observed in amniotic fluid (AF) or blood control samples. Different shades of yellow indicate 5% intervals of LOM (with light yellow corresponding to slight LOM and dark yellow corresponding to heavy LOM), while different shades of blue represent 5% intervals of GOM (from light blue corresponding to slight GOM to dark blue indicating heavy GOM). The last two columns contain ICR1/ICR2 methylation values determined by pyrosequencing, according to protocols previously described [Citation41]. *t-test ICR1 MassARRAY vs. ICR1 pyrosequencing: P = 0.2302. §t-test ICR2 MassARRAY vs. ICR2 pyrosequencing: P = 0.4391.
![Figure 1. Methylation analyses of 12 imprinted iDMRs by MassARRAY in patients with BWS and SRS with known epimutations at ICR1 or ICR2. Cases with MLID are underlined. Aberrant methylation was defined as methylation values deviating +/- two standard deviations from the mean value observed in amniotic fluid (AF) or blood control samples. Different shades of yellow indicate 5% intervals of LOM (with light yellow corresponding to slight LOM and dark yellow corresponding to heavy LOM), while different shades of blue represent 5% intervals of GOM (from light blue corresponding to slight GOM to dark blue indicating heavy GOM). The last two columns contain ICR1/ICR2 methylation values determined by pyrosequencing, according to protocols previously described [Citation41]. *t-test ICR1 MassARRAY vs. ICR1 pyrosequencing: P = 0.2302. §t-test ICR2 MassARRAY vs. ICR2 pyrosequencing: P = 0.4391.](/cms/asset/0f5daa57-89aa-4b7d-aa57-e76cb5a29de7/kepi_a_1514230_f0001_oc.jpg)
MLID involved only maternally imprinted loci in patients with both BWS and SRS, although the affected iDMRs and the degree of the methylation defect varied among cases (). One BWS case (BWS-3) showed hypomethylation at only one additional iDMR, while all other cases exhibited LOM at two or more iDMRs (). Case BWS-7 exhibited the most extensive MLID, with methylation defects at five additional iDMRs.
DIRAS3-CG1, FAM50B, MEST, and GNASXL iDMRs were the most frequently involved loci: DIRAS3-CG1 LOM was observed in seven cases (25%), while FAM50B, MEST, and GNASXL LOM were detected in five samples (18%). Among the analyzed iDMRs, only PLAGL1 and MEG3 did not show aberrant methylation in any BWS/SRS case. In addition, both cases with SRS and MLID exhibited severe hypomethylation at MEST (7q32), involved in maternal UPD(7q) in SRS patients [Citation28].
Finally, ICR1 (P = 0.2302) and ICR2 (P = 0.4391) methylation results obtained by MassARRAY overlapped those obtained by Pyrosequencing for diagnostic purposes ().
MLID putative causative mutations identified by NGS
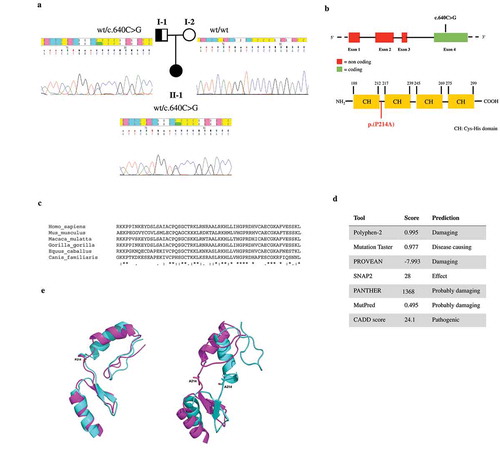
Targeted NGS analysis was performed for all BWS and SRS cases with molecular evidence of MLID. Genes included in the NGS panel (Supplementary Table S1) were chosen among those involved in the establishment and maintenance of parental allele-specific DNA methylation during embryonic development [Citation29]. The use of a targeted gene panel can be considered as a proof of concept for the research of pathogenic variants of MLID. By NGS analysis, we identified two likely pathogenetic mutations. The first was a novel missense mutation in the NLRP2 gene, c.656A> C (p.(K219T); ENST00000339757.11), in patient BWS-15 ()). The mutation was heterozygous, inherited from the father and absent in a subsequent pregnancy of the family (healthy at birth) ()). The mutation was absent in 100 controls analyzed. The observed mutation falls in NLRP2 exon 6, encoding the NOD nucleotide binding domain crucial for protein activation by oligomerization ()) [Citation30]. Moreover, the affected amino acid is phylogenetically conserved, suggesting a role in protein activity ()) and the mutation was predicted to be pathogenic using in silico tools ()). The second variant identified was a heterozygous SNP in the ZFP42 gene (alias REX1, REX-1) in patient SRS-4, c.640C>G (p.(P214A); ENST00000326866.4, rs61748605) ()), which was present in both the Broad Institute ExAC Browser and the 1000 Genomes database, with minor allele frequencies (MAF) of 0.004 and 0.005, respectively. This variant was inherited from the father ()) and was not identified in controls. It lies in the only coding exon and affects the first linker domain between consecutive C2H2-domains involved in DNA binding ()). The affected amino acid residue is phylogenetically conserved ()) and was predicted to be damaging by in silico tools ()). Interestingly, the carrier fathers of both mutated patients did not show multi-locus defects nor phenotypic alterations (data not shown).
Figure 2. In silico analysis of the NLRP2 mutation. (a) Pedigree of the index case identified as heterozygous for a novel c.656A> C mutation in the NLRP2 gene, as shown in electropherograms. The observed mutation was inherited from the father. (b) Schematic representation of the NLRP2 gene and its encoded protein, indicating the position of the identified mutation. (c) Phylogenetic conservation of the amino acid affected by the observed mutation. (d) In silico pathogenic prediction. (e) wt- and mutNLRP2 modelling. In wtNLRP2 ADP is bound to the NBD1 subdomain of NOD: R169 atoms form hydrogen bonds with the N1 and N6 atoms of the adenine base, the T221 atoms form three hydrogen bonds with the α-phosphate group of ADP and the T220 G216 and K219 atoms form three hydrogen bonds with the b-phosphate groups of ADP. ADP also binds to the WHD subdomain of NOD by forming three hydrogen bonds between the H503 residue and the α-phosphate group. In mutNLRP2, ADP makes hydrogen bonds only with residues of the NBD1 subdomain: R169 atoms form hydrogen bonds with the N1 and N6 atoms of the adenine base, the G218, T221 and T220 atoms form four hydrogen bonds with the α-phosphate group, while the E136 atoms make four hydrogen bonds with the ribose hydroxyl groups.
Symbols: * (asterisk) indicates a fully conserved residue; colon (:) indicates conservation between groups of strongly similar amino acids; dot (.) indicates conservation between groups of weakly similar amino acids.
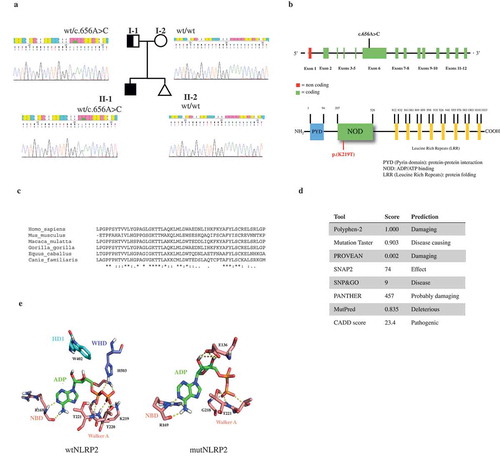
Figure 3. In silico analysis of the ZFP42 mutation. (a) Pedigree of the index case identified as heterozygous for the reported c.640C>G variant in the ZFP42 gene, as shown in electropherograms. The observed mutation was inherited from the father. (b) Schematic representation of the ZFP42 gene and the encoded protein indicating the position of the identified mutation. (c) Phylogenetic conservation of the amino acid affected by the observed mutation. (d) In silico prediction of pathogenicity. (e) wt and mutZFP42 modelling. Representative conformations of zinc finger motif conformations of the two most populated clusters (obtained from clustering molecular dynamics trajectories) are shown in magenta and cyan cartoons. The side chains of the P214 and A214 residues are represented by sticks.
Symbols: * (asterisk) indicates a fully conserved residue; colon (:) indicates conservation between groups of strongly similar amino acids; dot (.) indicates conservation between groups of weakly similar amino acids.
NLRP2 and ZFP42 mutations affect protein conformation and activity
The NLRP2 variant (c.656A>C) identified in patient BWS-15 affects the NOD central domain, comprising the nucleotide-binding domain (NBD), the helical domain (HD) and the winged-helix domain (WHD). These subdomains contain characteristic motifs (Walker A and B, sensor 1 and 2) involved in ADP binding domain and ATP hydrolysis. The K219 mutated residue is localized in the Walker A motif. No differences in the protein secondary structures were predicted in silico (data not shown). Comparison between wild type (wt) and mutated (mut) NLRP2 molecular dynamics highlighted a significantly different conformation of the NOD domain. In the wtNLRP2, ADP makes hydrogen bonds with specific residues of the NBD1 and WHD subdomains of NOD ()). These interactions allow the subdomains of NOD to be packed in a closed conformation. In the mutNLRP2, ADP makes hydrogen bonds only with residues of the NBD1 domain ()). According to literature data, the conformation of the mutNLRP2, would facilitate conformational changes in the WHD, leading to reduced ADP binding [Citation30].
Significant differences in binding free energies of ADP between wt and mutNLRP2 confirmed the effect of the K219T mutation on ADP binding. The mutNLRP2 showed a consistent decrease in the binding energy compared to the wtNLRP2/ADP complex (−103.5710 Kcal/mol for wtNLRP2 vs. −52.6973 Kcal/mol for mutNLRP2 – data not shown). To obtain a detailed profile of the binding process, the total binding free energy was further decomposed to each residue (Supplementary Figure S2). In the wtNLRP2, the K219 residue appeared to significantly contribute to the binding energy (with value of −60 kcal/mol), thus supporting its key role in ADP binding and subsequent protein activation.
The ZFP42 variant (c.640C>G) is localized in the linker region between the first and second zinc finger of the ZFP42 protein and is not predicted to impact protein secondary structure (data not shown). Molecular dynamics highlighted a greater flexibility of the linker in the mutZFP42 compared to the wtZFP42 ()). Proline imposes a conformational restriction on linker, that is lost when it is substituted with alanine. This amino acid change introduces conformational freedom in the linker, decreasing the probability of binding to the target DNA.
Epigenotype-phenotype correlations
In our cohort of BWS cases, MLID was more frequent in females (P = 0.03 Fisher’s exact test), with a male-to-female ratio of 1:4 ( and Supplementary Figure S3); this finding is not ascribable to sex bias in the original cohort, composed by 10 males and 11 females.
Figure 4. Clinical features of pre- and postnatal BWS cases (Weksberg criteria). Cases with MLID are underlined. Empty circles indicate prenatal signs. §No other phenotypic data available. a, Polyhydramnios; b, Myopia, lombo-sacral cutaneous spots; c, Frontal angioma; d, Renal asymmetry, lypoma right leg. Abbreviations: TA, therapeutic abortion; B, born; IUFD, intrauterine fetal death; d, days; m, months; y, years.
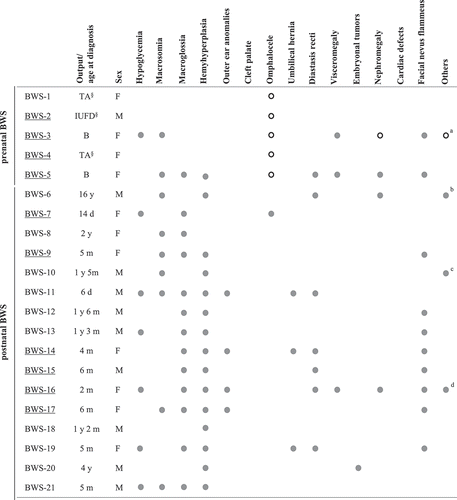
No significant differences between the clinical features of patients with mono-locus and multi-locus BWS were observed, as previously reported [Citation19]. Nevertheless, it is intriguing that a number of clinical features appeared to be more frequent in patients with MLID (), such as facial nevus flammeus (5/6 MLID vs. 3/10 non-MLID), and, in contrast with previous reports [Citation7], macroglossia (6/6 MLID vs. 6/10 non-MLID) and outer ears anomalies (3/6 MLID vs. 1/10 non-MLID). Also, abdominal wall defects were more frequent in both pre- and postnatal BWS-MLID patients (8/10 MLID vs. 4/11 non-MLID).
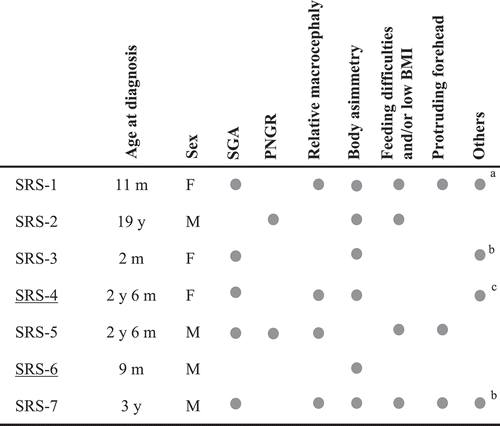
Differently from BWS cases, in which the diagnosis was clinically suspected, in both SRS-MLID patients the clinical diagnosis was misleading because of the presence of uncommon clinical features (i.e., nevus flammeus, hypoglycemia and abdominal wall defects, ). In these cases, the molecular evaluation was resolving, showing ICR1 hypomethylation in both patients, thus classified as SRS.
Figure 5. Clinical features of postnatal SRS patients (Netchine-Harbison criteria). Cases with MLID are underlined. a, Diastasis recti; b, Fifth finger clinodactyly; c, Neonatal hypoglycemia, diastasis recti, facial nevus flammeus, neuromotor delay. Abbreviations: SGA, small for gestational age; PNGR, postnatal growth retardation; m, months; y, years.
Discussion
Molecular characterization of MLID is emerging to be fundamental not only to better define the clinical diagnosis of IDs but also to evidence common functional networks at the basis of the genome-wide deregulation of imprinting. The frequency of MLID appears to vary depending on the sensitivity of the technology used for analyses and the iDMRs investigated [Citation6–Citation8,Citation15,Citation19]. In this study, we set up a panel of 12 iDMRs by MassARRAY technology and applied it to a cohort of patients with BWS, presenting with primary GOM at ICR1 or LOM at ICR2, and with SRS, showing LOM at ICR1. We found a higher frequency of MLID in both BWS (50%) and SRS (29%) patients, compared with previous reports [Citation6–Citation8,Citation15,Citation19], probably because we selected the iDMRs most frequently associated with BWS/SRS. Our data confirm that the iDMRs involved in both BWS- and SRS-MLID can vary among patients, supporting that the defect is non-recurrent. Nevertheless, we found that DIRAS3-CG1, FAM50B, MEST, and GNASXL iDMRs were the most frequently affected loci.
In our cohort, we only identified MLID in BWS patients with ICR2 LOM, and hypomethylation was found only at maternally iDMRs. This phenomenon, known as multiple maternal hypomethylation syndrome, has previously been described for BWS [Citation7,Citation16,Citation17]. Only two studies reported on MLID as GOM at paternally methylated iDMRs in BWS patients [Citation8,Citation15]; however, also in these cases, MLID was supposed to arise as a consequence of a methylation defect of the maternal allele.
Considering the parental derivation and the variable degree of LOM of the affected allele, suggestive of mosaicism, in patients with BWS MLID is likely due to post-zygotic loss of maintenance of methylation at maternally imprinted alleles. This hypothesis is also supported by the following evidences: i) both BWS epimutations (ICR1 GOM or ICR2 LOM) result from defective methylation of the maternal allele, and ii) no [Citation7,Citation19], or very few [Citation15] cases with MLID have been identified among ICR1 hypermethylated patients.
Regarding SRS, although our study included a limited number of cases, the results indicate that SRS-MLID is less frequent and involves fewer iDMRs than BWS-MLID. Similar to BWS, SRS-MLID can be defined as a hypomethylation syndrome; however, SRS-MLID patients did not exhibit any correspondence between the affected allele of the causative epimutation (paternal ICR1) and the parental origin of the other iDMRs involved (all maternal). This may be because of different mechanisms underlying the MLID phenomenon in patients with SRS. Also, in SRS, the methylation impairment appears to be post-zygotic.
Interestingly, the only two SRS patients with MLID shared severe hypomethylation at the MEST gene. MEST hypomethylation in SRS-MLID has also been reported by other authors [Citation8,Citation19,Citation28,Citation31] as a distinct mechanism compared to mUPD(7), leading to GOM at this iDMR [Citation32]. In our cases, LOM at MEST in SRS-MLID patients further sustains a key role of this iDMR in SRS. Since both pathogenic mutations and isolated epimutations at this iDMR have never been reported [Citation33], it is conceivable a different mechanism of pathogenicity, possibly mediated by a cross-talk between MEST and ICR1. According, recent advances sustain an interplay among ICR1 and other iDMRs (our unpublished data) and chromatin interactions between H19 and portions of chromosome 7 [Citation34]. MEST methylation may also be part of an imprinting gene network (IGN), involving the trans-acting functions of H19 lncRNA [Citation35,Citation36]. Further investigations are requested to better elucidate this finding.
To explain MLID phenomenon, an underlying genetic mechanism has been suggested, such as a mutation in pleiotropic genes regulating DNA methylation. This hypothesis is intriguing and has been corroborated by the identification of few genes (e.g., NLRP family genes), mutated in MLID patients. Nevertheless, the real role of these genes has to be fully elucidated, because mutated patients reported in literature show clinical and epigenetic heterogeneity. These genes are generally considered to have a ‘maternal effect’ since they have a key role during the early embryonic stages before full zygotic genome activation. Maternally derived mutations of these genes cause impaired or delayed preimplantation development and have been associated to MLID development through oocyte-stored transcripts [Citation37]. Nevertheless, our data do not sustain this hypothesis, since the paternally-derived NLRP2 mutation could impair methylation, suggesting a function of NLRP2 occurring after embryonic-genome activation rather than via oocyte-stored transcripts.
Our hypothesis, based on in silico modeling and literature data of NLR protein family in the inflammasome response [Citation30], is that mutNLRP2 remains in an open conformation, leading to inappropriate LOM at iDMRs. Although intriguing, this suggestion is speculative in relation to NLRP2 embryonic epigenetic function. Alternatively, mutNLRP2 may act as dominant negative through oligomerization/sequestration of wtNLRP2 proteins. Moreover, all other described NLRP2 mutations associated with MLID in BWS/SRS patients [Citation8,Citation21] affect the NOD domain, that delineates as a possible mutational hotspot with a deleterious effect on methylation regulation.
The ZFP42 variant, identified in patient SRS-4, although annotated as a polymorphism, is very rare in the general population and is predicted to be pathogenic. In silico modeling suggested a reduced binding specificity of mutZFP42 on target DNA and consequently a diminished binding competition with the antagonist YY1 [Citation38]. Based on SRS-4 MLID pattern and according to literature data, showing that the ZFP42 knockout causes LOM at several iDMRs not including H19 [Citation39], we speculate that that the loss of binding specificity of the mutZFP42 may lead to LOM at iDMRs not normally controlled by this factor. According to the paternal derivation of the ZFP42 variant, we can assume that the timing of the methylation aberration would be post embryonic genome activation, working in a dominant-negative fashion, and resulting in LOM mosaicism.
Overall, the absence of MLID and phenotypic effect in both carrier fathers may be explained by incomplete penetrance, as already reported for NLRP2 [Citation21,Citation37] and ZFP57 mutations [Citation40]. Defining and proving the pathogenic effect of MLID-associated mutations is, indeed, challenging, given the poorly established mechanism of transmission and lack of information about the roles of analyzed factors in methylation. However, there is strong justification for reporting these variants in order to understand whether they are really pathogenetic or only confer higher susceptibility to single or multiple methylation defects, likely in conjunction with additional yet unidentified variants in methylation factors.
Regarding epigenotype-phenotype correlations in patients with BWS-MLID, we confirmed the absence of MLID-specific or recurrent clinical signs. Given the variability of affected iDMRs, no significant associations between epimutations and phenotypes could be identified. Nevertheless, MLID should be always suspected in female patients with nevus flammeus, macroglossia, outer ears anomalies and abdominal wall defects.
It is intriguing that both patients with MLID-SRS were clinically misdiagnosed as BWS: one SRS patient showed only hemihyperplasia (reclassified as hemihypoplasia after clinical re-evaluation and in light of molecular results), while the other exhibited BWS-associated clinical signs.
Conclusions
Overall, our data indicate that MLID is frequent in BWS/SRS and restricted to maternal iDMRs. While searching for genetic defects associated with MLID, we identified two additional nucleotide variants mapping at functional sites of NLRP2 and ZFP42 genes. By checking for the pattern of inheritance, we found a paternal derivation, never reported for such mutations. This evidence suggests yet unexplored functions of these genes, or different mechanisms of pathogenicity and underline the importance of father’s testing.
BWS-MLID patients do not show peculiar clinical signs, although MLID appears more frequent in females with a specific phenotypic pattern. All this information should be considered for genetic counseling.
Methods
Study population
The study population included 21 patients with BWS and ICR1/ICR2 epimutations: five (BWS 1–5) were referred to our laboratory during pregnancy, after detection of omphalocele by ultrasound imaging, and 16 were postnatal cases (BWS 6–21). Among these latter, 11 were clinically diagnosed according to Weksberg classification [Citation1], while diagnosis was only suspected in the remaining five. We also studied seven cases with SRS (SRS 1–7) and ICR1 LOM: three of these cases achieved the Netchine-Harbison criteria for clinical diagnosis of SRS [Citation2], while diagnosis was only suspected in the remaining four.
All clinical features of BWS and SRS patients are reported in and , respectively.
All cases were collected between 2011 and 2016. Appropriate informed consent was obtained from all parents. The study was approved by the Ethics Committee of Fondazione IRCSS Ca’ Granda Ospedale Maggiore Policlinico (no. 526/2015).
All cases included in the study had a primary methylation defects at ICR1/ICR2, as confirmed by molecular diagnosis by pyrosequencing [Citation41–Citation43]. Genomic imbalances or UPD were excluded by SNP array (Supplementary materials and methods).
The parents of two cases (BWS-15 and SRS-4) were investigated to define the inheritance of variants detected by NGS. The mutation identified in patient BWS-15 was also evaluated in the DNA from amniotic fluid from a subsequent pregnancy of the family.
DNA samples from 100 aged-matched healthy controls were included in the study: i) to set up the MassARRAY methylation panel and define the normal interval of methylation for each locus (50 individuals), and ii) to calculate the frequency of NGS-identified variants in the general population (100 individuals). In addition, 20 DNAs from amniotic fluid of healthy pregnancies were used to define normal methylation ranges of iDMRs included in the MassARRAY platform for the analysis of pre-natal BWS cases.
DNA isolation and bisulfite conversion
Genomic DNA was extracted from the peripheral blood and amniotic fluid of patients and controls using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). DNA samples (500–700 ng) were bisulfite converted using the EZ Direct DNA Methylation Kit (Zymo Research, Irvine, CA). In prenatal cases, maternal DNA contamination was excluded by qfPCR.
Methylation analysis by MassARRAY
Methylation profile of 12 iDMRs frequently involved in MLID in BWS/SRS [Citation7,Citation8,Citation10,Citation15] was investigated by the MassARRAY methylation platform (Agena Bioscience, Hamburg, Germany). Details on the setting up and on protocol are reported in Supplementary materials and methods. The investigated iDMRs, their genomic positions, specific primers, and CpG sites are described in Supplementary Table S2, which also includes primers and CpGs investigated for BWS/SRS molecular diagnosis by pyrosequencing. The MassARRAY ICR1 assay comprises the four CpG sites analyzed by pyrosequencing, while the MassARRAY ICR2 sequence maps downstream of the pyrosequencing assay. Aberrant methylation of iDMR in patients was defined when the observed methylation value fell outside the methylation range defined in controls (mean ± two standard deviations).
Next-generation sequencing (NGS)
A panel of 25 genes (Supplementary Table S1) involved in methylation establishment/maintenance was designed for targeted NGS using the Agilent SureDesign online tool (Agilent Technologies, Santa Clara, CA). NGS was performed on DNA from patients with BWS- and SRS-MLID, using the Agilent HaloPlex Target Enrichment kit (Agilent Technologies) and an Illumina MiSeq sequencer with MiSeq Reagent Kit V2 (Illumina, CA). A detailed description of NGS procedures is reported in Supplementary materials and methods.
Prediction of pathogenicity and computational modeling studies
The pathogenic effects of variants identified by NGS were predicted using online tools reported in Supplementary materials and methods.
The structure of NLRP2 NOD (nucleotide-binding and oligomerization domain) was obtained by homology modeling using as template the crystal structure of the NOD2 in the ADP-bound (PDB: 1irm). The effect of the mutation on the NOD structure and function was assessed by performing 200 ns of molecular dynamics simulations of wt and mutated models.
To gain insight into the contribution spectrum of binding energy for ADP-wtNLRP2 and ADP-mutNLRP2, the enthalpy contributions during the last 15 ns of simulations were computed.
The unbound zinc finger motifs of the ZFP42 protein were modeled by homology using the crystal structure of the transcriptional repressor protein YY1-DNA complex (PDB: 1ubd) as template. The impact of the amino acid substitution on protein folding and on ability to bind DNA was assessed by performing 30 ns of molecular dynamics using the ff03 force filed. To maintain the tetrahedral structures of the zinc-binding site of the C2H2 coordination, the cationic dummy atom approach was used instead of a simple non-bonded model. The conformational change of zinc finger motifs during simulation was evaluated by cluster analysis determining the most representative structures from molecular dynamic simulations.
Statistical analyses
T-tests were used to compare ICR1 and ICR2 methylation values determined by MassARRAY and pyrosequencing. For (epi)genotype-phenotype correlations, Fisher’s exact tests were performed.
Supplemental Material
Download Zip (306.7 KB)Acknowledgments
We would acknowledge patients and families.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14.
- Azzi S, Salem J, Thibaud N, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015;52(7):446–453.
- Gicquel C, Rossignol S, Cabrol S, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37(9):1003–1007.
- Netchine I, Rossignol S, Dufourg MN, et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab. 2007;92(8):3148–3154.
- Ibrahim A, Kirby G, Hardy C, et al. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics. 2014;6(1):11.
- Poole RL, Docherty LE, Al Sayegh A, et al. Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am J Med Genet A. 2013;161A(9):2174–2182.
- Bliek J, Verde G, Callaway J, et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2009;17(5):611–619.
- Court F, Martin-Trujillo A, Romanelli V, et al. Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum Mutat. 2013;34(4):595–602.
- Arima T, Kamikihara T, Hayashida T, et al. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005;33(8):2650–2660.
- Bens S, Kolarova J, Beygo J, et al. Phenotypic spectrum and extent of DNA methylation defects associated with multilocus imprinting disturbances. Epigenomics. 2016;8(6):801–816.
- Docherty LE, Rezwan FI, Poole RL, et al. Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J Med Genet. 2014;51(4):229–238.
- Rossignol S, Steunou V, Chalas C, et al. The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J Med Genet. 2006;43(12):902–907.
- Lim D, Bowdin SC, Tee L, et al. Clinical and molecular genetic features of Beckwith-Wiedemann syndrome associated with assisted reproductive technologies. Hum Reprod. 2009;24(3):741–747.
- Hiura H, Okae H, Miyauchi N, et al. Characterization of DNA methylation errors in patients with imprinting disorders conceived by assisted reproduction technologies. Hum Reprod. 2012;27(8):2541–2548.
- Maeda T, Higashimoto K, Jozaki K, et al. Comprehensive and quantitative multilocus methylation analysis reveals the susceptibility of specific imprinted differentially methylated regions to aberrant methylation in Beckwith-Wiedemann syndrome with epimutations. Genet Med. 2014;16(12):903–912.
- Boonen SE, Pörksen S, Mackay DJ, et al. Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings. Eur J Hum Genet. 2008;16(4):453–461.
- Sano S, Matsubara K, Nagasaki K, et al. Beckwith-Wiedemann syndrome and pseudohypoparathyroidism type Ib in a patient with multilocus imprinting disturbance: a female-dominant phenomenon? J Hum Genet. 2016;61(8):765–769.
- Baple EL, Poole RL, Mansour S, et al. An atypical case of hypomethylation at multiple imprinted loci. Eur J Hum Genet. 2011;19(3):360–362.
- Azzi S, Rossignol S, Steunou V, et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet. 2009;18(24):4724–4733.
- Bakker B, Sonneveld LJ, Woltering MC, et al. Beckwith-Wiedemann Syndrome and Pseudohypoparathyroidism Type 1B Due to Multiple Imprinting Defects. J Clin Endocrinol Metab. 2015;100(11):3963–3966.
- Meyer E, Lim D, Pasha S, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet. 2009;5(3):e1000423.
- Docherty LE, Rezwan FI, Poole RL, et al. Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat Commun. 2015;6:8086.
- Li R, Albertini DF. The road to maturation: somatic cell interaction and self-organization of the mammalian oocyte. Nat Rev Mol Cell Biol. 2013 Mar;14(3):141–152. Review. PubMed PMID: 23429793.
- Sanchez-Delgado M, Martin-Trujillo A, Tayama C, et al. Absence of maternal methylation in biparental hydatidiform moles from women with NLRP7 maternal-effect mutations reveals widespread placenta-specific imprinting. PLoS Genet. 2015;11(11):e1005644.
- Caliebe A, Richter J, Ammerpohl O, et al. A familial disorder of altered DNA-methylation. J Med Genet. 2014 Jun;51(6):407–412.
- Mackay DJ, Callaway JL, Marks SM, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(8):949–951.
- Quenneville S, Verde G, Corsinotti A, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44(3):361–372.
- Kagami M, Nagai T, Fukami M, et al. Silver-Russell syndrome in a girl born after in vitro fertilization: partial hypermethylation at the differentially methylated region of PEG1/MEST. J Assist Reprod Genet. 2007;24(4):131–136.
- Kelsey G, Feil R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos Trans R Soc Lond B Biol Sci. 2013;368(1609):20110336.
- Hu Z, Yan C, Liu P, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science. 2013;341:172–175.
- Begemann M, Spengler S, Kanber D, et al. Silver-Russell patients showing a broad range of ICR1 and ICR2 hypomethylation in different tissues. Clin Genet. 2011;80(1):83–88.
- Fuke-Sato T, Yamazawa K, Nakabayashi K, et al. Mosaic upd(7)mat in a patient with Silver-Russell syndrome. Am J Med Genet A. 2012;158A(2):465–468.
- Schöherr N, Jäger S, Ranke MB, et al. No evidence for isolated imprinting mutations in the PEG1/MEST locus in Silver-Russell patients. Eur J Med Genet. 2008;51(4):322–324.
- Zhao Z, Tavoosidana G, Sjölinder M, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38(11):1341–1347.
- Gabory A, Ripoche MA, Le Digarcher A, et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development. 2009;136(20):3413–3421.
- Monnier P, Martinet C, Pontis J, et al. H19 lncRNA controls gene expression of the imprinted gene network by recruiting MBD1. Proc Natl Acad Sci U S A. 2013;110(51):20693–20698.
- Begemann M, Rezwan FI, Beygo J, et al. Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J Med Genet. 2018;pii:jmedgenet-2017–105190.
- Masui S, Ohtsuka S, Yagi R, et al. Rex1/Zfp42 is dispensable for pluripotency in mouse ES cells. BMC Dev Biol. 2008;8:45.
- Kim JD, Kim H, Ekram MB, et al. Rex1/Zfp42 as an epigenetic regulator for genomic imprinting. Hum Mol Genet. 2011;20(7):1353–1362.
- Boonen SE, Mackay DJ, Hahnemann JM, et al. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up. Diabetes Care. 2013 Mar;36(3):505–512.
- Paganini L, Carlessi N, Fontana L, et al. Beckwith-Wiedemann syndrome prenatal diagnosis by methylation analysis in chorionic villi. Epigenetics. 2015;10(7):643–649.
- Tabano S, Colapietro P, Cetin I, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics. 2010;5(4):313–324.
- Calvello M, Tabano S, Colapietro P, et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Epigenetics. 2013;8(10):1053–1060.