
ABSTRACT
Temple syndrome (TS14) is a rare imprinting disorder caused by genetic and epigenetic alterations on chromosome 14q32. A subset of these patients shows an imprinting defect (ID) where the paternal allele harbors a maternal epigenotype thus silencing the paternally expressed genes and leading to an increased expression of the maternally expressed genes. We investigated the grandparental origin of the incorrectly imprinted chromosome 14 in a cohort of 13 TS14 ID patients and their families. In seven families grandmaternal and, in six families, grandpaternal inheritance was observed. These results indicate that the ID occurred after imprint erasure in the paternal germ line. While the complete lack of methylation as observed in the majority of TS14 ID patients may be due to an imprint establishment error in the paternal germ line, cases with methylation mosaicism suggest that in general many IDs (TS14, AS, BWS, and SRS) are in fact of somatic origin in the early or late embryo.
Introduction
Temple syndrome (TS14, OMIM 616222) is a rare imprinting disorder characterized by low birth weight and height, muscular hypotonia and feeding difficulties in the infant period, early puberty and short stature with small hands and feet and often truncal obesity [Citation1,Citation2]. It is caused by genetic and epigenetic alterations on chromosome 14q32. These alterations include paternal deletions, maternal uniparental disomy of chromosome 14, and imprinting defects, all of which lead to an erroneous expression of the imprinted genes in the region (). The maternally expressed genes, MEG3, RTL1as and MEG8, as well as the paternally expressed genes, DLK1 and RTL1, are regulated by two differentially methylated regions in 14q32: the germline-derived, primary MEG3/DLK1-intergenic (IG)-DMR (IG-DMR) and the post-fertilization-derived, secondary MEG3:TSS-DMR (MEG3-DMR; [Citation3,Citation4], for nomenclature see [Citation5]). They are methylated on the paternal and unmethylated on the maternal chromosome. Both DMRs act as imprinting control centers, whereby the MEG3/DLK1:IG-DMR functions as an upstream regulator of the MEG3:TSS-DMR [Citation3,Citation4]. A third post-fertilization-derived, secondary DMR located in intron 2 of the MEG8 gene (MEG8:Int2-DMR) is unmethylated on the paternal and methylated on the maternal chromosome [Citation6]. So far, its function and regulation are unknown.
Figure 1. Overview of the chromosomal region 14q32.
The figure gives an overview of the imprinted gene cluster on chromosome 14q32. Maternally expressed genes are indicated in red, paternally expressed genes in blue. The region harbors three differentially methylated regions (DMRs): the MEG3/DLK1:IG-DMR and the MEG3:TSS-DMR that are methylated on the paternal allele (pat) and the MEG8:Int2-DMR which is methylated on the maternal allele (mat). The methylation status is indicated by filled (methylated) and empty (unmethylated) lollipops. Expression of the genes is indicated by arrows.
The situation in TS14 patients is given below. The MEG3/DLK1:IG-DMR and the MEG3:TSS-DMR are unmethylated on both alleles while the MEG8:Int2-DMR is methylated on both alleles. This leads to an increased expression of the maternally expressed genes (indicated by bold gene names) while the paternally expressed genes are silenced (indicated by grey gene names).
Modified from Beygo et al. 2017 [Citation8].
![Figure 1. Overview of the chromosomal region 14q32.The figure gives an overview of the imprinted gene cluster on chromosome 14q32. Maternally expressed genes are indicated in red, paternally expressed genes in blue. The region harbors three differentially methylated regions (DMRs): the MEG3/DLK1:IG-DMR and the MEG3:TSS-DMR that are methylated on the paternal allele (pat) and the MEG8:Int2-DMR which is methylated on the maternal allele (mat). The methylation status is indicated by filled (methylated) and empty (unmethylated) lollipops. Expression of the genes is indicated by arrows.The situation in TS14 patients is given below. The MEG3/DLK1:IG-DMR and the MEG3:TSS-DMR are unmethylated on both alleles while the MEG8:Int2-DMR is methylated on both alleles. This leads to an increased expression of the maternally expressed genes (indicated by bold gene names) while the paternally expressed genes are silenced (indicated by grey gene names).Modified from Beygo et al. 2017 [Citation8].](/cms/asset/41dbab83-2e33-4616-a7b4-1f32e7344c39/kepi_a_1514233_f0001_c.jpg)
Patients with TS14 and an imprinting defect show biparental inheritance of chromosomes 14 but the paternal allele is hypomethylated at the MEG3/DLK1:IG-DMR and the MEG3:TSS-DMR and hypermethylated at the MEG8:Int2-DMR (; [Citation7,Citation8]).
Genomic imprints run through a so-called life cycle where epigenetic marks like DNA methylation, histone modifications and chromatin structure, are erased in the primordial germ cells before they are established again during germ cell development and afterwards maintained in the zygote and through all following cell divisions [Citation9–Citation11]. Therefore, the allele affected by an imprinting defect indicates the mechanism involved in the development of an ID. In case of TS14, the absence of methylation at the MEG3/DLK1:IG-DMR on the paternal allele suggests a failure in the establishment of the methylation imprint in the male germline or its maintenance after fertilization (). To investigate this in detail we determined the grandparental origin of the affected chromosome 14 in a cohort of 13 TS14 ID families by studying the parent-of-origin specific methylation of the three DMRs in 14q32 in combination with informative single nucleotide variants (SNPs).
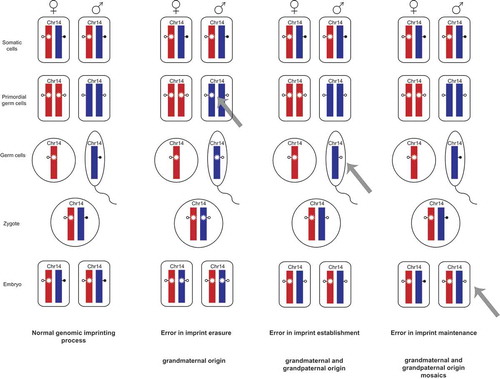
Figure 2. Genomic imprinting and the occurrence of imprint errors.
This scheme shows the so called imprint life cycle exemplarily for the germline-derived MEG3/DLK1:IG-DMR on chromosome 14q32. The normal process of genomic imprinting is depicted in the column on the left-hand side. The methylation imprints of the DMRs are indicated by lollipops (filled – methylated status of the DMR, empty – unmethylated status of the DMR) and shown for the maternal (red) and paternal (blue) alleles.
Somatic cells with a normal methylation imprint are shown at the top. These imprints are then erased in the primordial germ cells. The asterisk on the maternal allele indicates an epigenetic mark that prevents DNA methylation on the normally unmethylated maternal allele e.g., H3K4me2 or H3K4me3. The methylation imprints are then newly established in the male and female germ cells, respectively and are subsequently maintained in the zygote and through all following cell divisions.
The other three columns display errors in imprint erasure, imprint establishment and imprint maintenance and their outcome in regard to chromosome 14.
A failure to erase the maternal imprint in the paternal primordial germ cells would lead to a maternal epigenotype on the paternal allele. In these cases, a TS14 ID patient would have inherited the allele harboring the ID exclusively from the paternal grandmother. (Other marks may apply for mature sperm because the genome is largely devoid of histones.)
In case the imprint erasure (second column) works correct but the imprint is not established in the male germ cells. Here, the ID allele could be inherited either from the paternal grandmother or the grandfather. The same holds true for errors in imprint maintenance. Additionally, mosaic IDs occur when the failure took place after fertilization in the early embryo so that only a subset of cells harbor an ID while the other cells have a normal epigenotype.
Results and discussion
We investigated 13 TS14 patients with an ID and their families to determine the underlying mechanism leading to the ID. For this we searched for informative SNPs within or adjacent to the three DMRs on chromosome 14q32. In informative families, the methylation status in the father was then investigated by next generation bisulfite sequencing. Seven families were informative for the germline-derived, primary MEG3/DLK1:IG-DMR (). In two families, the paternal allele harboring the ID was methylated in the fathers’ blood DNA indicating that it is of grandpaternal origin. In the other five cases, the incorrectly imprinted allele was unmethylated in the fathers’ blood DNA and therefore of grandmaternal origin (Supplementary Figure 1(a)). Similar results could be obtained for the MEG3:TSS-DMR in three informative families. For two families, we could show that the allele carrying the ID was inherited from the paternal grandfather, whereas in one family it was inherited from the paternal grandmother (Supplementary Figure 1(b)). For the MEG8:Int2-DMR, three families were informative as well. Here, the alleles inherited from the father were unmethylated in two families and therefore of grandpaternal origin as this DMR is – in contrast to the other two – methylated on the maternal allele. In the third family, the allele was methylated and of grandmaternal origin (Supplementary Figure 1(c)).
Table 1. Summary of the results.
The fact that both grandpaternal and grandmaternal alleles are affected by the ID in TS14 points strongly to a failure in either imprint establishment in the paternal germline or postzygotic imprint maintenance, while an error in imprint erasure is highly unlikely. In that case only grandmaternal inheritance would have been observed. Epigenetic marks are erased in primordial germ cells, which is best investigated for DNA methylation imprints [Citation12]. However, other studies indicated that some kind of mark is still in place distinguishing the two parental alleles. It has been shown for the murine H19 locus – which is also germline-derived and paternally imprinted – that the acquisition of the methylation imprint is asynchronous [Citation13]. Establishment of the methylation imprint takes place earlier on the originally methylated paternal than on the originally unmethylated maternal allele, indicating that the different parental alleles still harbor distinguishing marks even after imprint erasure in the (male) primordial germ cells [Citation13]. Lee and colleagues detected an allelic bias of H3K4me2 at the H19 locus which delays the de novo methylation on the maternal allele [Citation14]. The same allelic bias has been observed for the Meg3/Dlk1:IG-DMR in mice [Citation15]. Here, the paternally derived allele acquired methylation prior to the maternally derived allele, too. Likewise, a difference in methylation of H3K4me3 has been detected at the two parental alleles in the male germ line, its presence on the maternal allele delaying the methylation establishment [Citation16]. Since we detected grandmaternal but also grandpaternal origin of the affected chromosome 14, we conclude that the erasure of methylated H3K4 and other involved factors is functional in TS14 patients.
One would expect that a postzygotic error in imprint maintenance must result in a mosaic state of the ID otherwise all cells would be affected. And, indeed, of the 29 TS ID patients that have been described in the literature up till now six show an ID in a mosaic state [Citation2,Citation8,Citation17–Citation19]. So far, the number of methylation mosaics seems to be rare in TS14, but it is possible that in patients with a high degree of normally methylated cells the phenotype is very mild and that these patients therefore do not come to clinical attention and escape diagnosis. In the cohort investigated in this study, two TS14 patients with a mosaic ID were included, too. At least in these cases, the erroneous methylation imprint is most probably due to a failure in imprint maintenance after fertilization (), while in the non-mosaic TS14 ID patients, the mechanism is most likely an error in imprint establishment during spermatogenesis.
Figure 3. Methylation analysis and model of imprint maintenance error for a mosaic imprinting defect.
Methylation results (upper line) obtained by deep bisulfite sequencing for the three DMRs on 14q32 are depicted for patient 12 who has a mosaic imprinting defect. The paternally methylated MEG3/DLK1:IG-DMR and MEG3:TSS-DMR show a hypomethylation with methylation levels of 38.2% (CpGs6-8 28.7%) and 23.5%, respectively. In accordance with these results is the hypermethylation of the maternally methylated MEG8:Int2-DMR, which exhibits a methylation level of 79.6%.
The scheme below shows the theoretical emergence of a mosaic imprinting defect due to a postzygotic error in imprint maintenance. At first, the methylation imprint is lost at the MEG3/DLK1:IG-DMR. As this DMR governs the MEG3:TSS-DMR in a hierarchical fashion it can be assumed that the methylation is lost at the MEG3:TSS-DMR subsequently. By this, transcription of a hypothesized long transcript starting at the MEG3 promoter and running through the MEG8:Int2-DMR can occur from the paternal allele [Citation8]. This transcription leads to the establishment of methylation at the paternal MEG8:Int2-DMR and its hypermethylation.
![Figure 3. Methylation analysis and model of imprint maintenance error for a mosaic imprinting defect.Methylation results (upper line) obtained by deep bisulfite sequencing for the three DMRs on 14q32 are depicted for patient 12 who has a mosaic imprinting defect. The paternally methylated MEG3/DLK1:IG-DMR and MEG3:TSS-DMR show a hypomethylation with methylation levels of 38.2% (CpGs6-8 28.7%) and 23.5%, respectively. In accordance with these results is the hypermethylation of the maternally methylated MEG8:Int2-DMR, which exhibits a methylation level of 79.6%.The scheme below shows the theoretical emergence of a mosaic imprinting defect due to a postzygotic error in imprint maintenance. At first, the methylation imprint is lost at the MEG3/DLK1:IG-DMR. As this DMR governs the MEG3:TSS-DMR in a hierarchical fashion it can be assumed that the methylation is lost at the MEG3:TSS-DMR subsequently. By this, transcription of a hypothesized long transcript starting at the MEG3 promoter and running through the MEG8:Int2-DMR can occur from the paternal allele [Citation8]. This transcription leads to the establishment of methylation at the paternal MEG8:Int2-DMR and its hypermethylation.](/cms/asset/90fe049d-db94-47fa-829c-e96258939d08/kepi_a_1514233_f0003_c.jpg)
The underlying molecular mechanism of primary epimutations, i.e., the presence of an aberrant methylation pattern in the absence of a DNA sequence change, has been reported in detail for Prader-Willi syndrome (PWS) and Angelman syndrome (AS). Buiting and colleagues investigated the grandparental origin of the allele harboring an imprinting defect in patients with PWS and in patients with AS using a combined methylation/restriction polymorphism analysis for the SNRPN locus [Citation20]. In case of PWS ID, only the allele inherited from the paternal grandmother was found in all 19 informative trios, pointing to a failure in imprint erasure in the paternal germline. In 18 informative AS ID patients and their parents, inheritance from both the maternal grandmother and the maternal grandfather was observed (seven and eleven times, respectively). Thus, indicating a failure in imprint establishment and/or maintenance. Furthermore, a study investigating the grandparental origin in case of BWS due to a hypermethylation of the ICR1 showed that the allele affected by the ID was inherited either from the maternal grandmother or the maternal grandfather (one and five times, respectively) [Citation21]. Therefore, the ID at the ICR1 is most likely due to an error in the establishment or maintenance of the methylation imprint, too.
Interestingly, AS patients with an ID in a mosaic state have been described in approximately 40% of the AS ID cases [Citation22]. In addition, imprinting defects on chromosome 11p15.5 leading to a hypomethylation of the ICR1 (H19-IGF2:IG-DMR) or ICR2 region (KCNQ1OT1:TSS-DMR) and Silver-Russell syndrome (SRS) and Beckwith-Wiedemann syndrome (BWS), respectively, are present in a mosaic state in the majority of cases [Citation23,Citation24].
On the other hand, a mosaic imprinting defect is extremely rare in PWS [Citation20,Citation25]. Here, the paternally unmethylated DMR in the SNRPN promoter/exon 1 region (PWS imprinting center) appears to be resistant to aberrant de novo methylation after fertilization, probably because this region is actively engaged in transcription [Citation26]. In this case, hypermethylation results from the maintenance of the inactive state through incomplete reprogramming during germ cell development (imprint erasure error) before fertilization [Citation20].
It would be interesting to see whether the hypermethylation of the maternal 14q32 allele in patients with Kagami-Ogata syndrome (KOS14), the counterpart of TS14, is due to a failure to erase the (grand)paternal methylation imprint in the maternal germline, similar to the situation in PWS. Although the number of published KOS14 ID cases is low, none were found to have an ID in a mosaic state so far [Citation8,Citation27].
Based on the data available on the origin of primary imprinting defects in TS14, AS, BWS (ICR2) and SRS we can draw the following conclusions: The absence of a methylation imprint appears to be of somatic origin in at least 50% of these patients. These manifest as mosaics. In the other 50%, the complete absence of methylation results from either a failure in imprint establishment in the germ line or from a failure to maintain the methylation imprint in the very early embryo, although a distinction between those two scenarios is not possible. In view of the fact that more than 1,000 regions are methylated during oogenesis and that most of these regions are demethylated shortly after fertilization, maternal methylation imprints may occasionally be lost in the early embryo [Citation28,Citation29]. The same could apply for paternal methylation imprints. Our data suggests three plausible mechanisms for the occurrence of IDs in TS14 patients: i) a stochastic error in imprint establishment in the early spermatogenesis (prospermatogenesis); ii) a failure to maintain the paternal methylation imprint during the global wave of demethylation in the zygote or; iii) during (early) embryonal development in case of methylation mosaics.
Material and methods
All samples were obtained after written informed consent. The study was approved by the local ethics committee (AZ 08–3858).
All patients were tested at least at three additional imprinted loci for the presence of further methylation disturbances. Some of the patients have also been tested for methylation abnormalities on chromosome 15q11q13 and chromosome 11p15.5 (see clinical descriptions of the patients in the cited literature) with no abnormalities being detected.
The 13 TS ID patients have been described previously:
TS14 ID patients 1–4 were described in Beygo et al. 2017 [Citation8], TS14 ID patients 5 and 6 were described in Buiting et al. 2008 [Citation30], TS14 patient 7 was described in Mitter et al. 2006 [Citation17] and Buiting et al. 2008 [Citation30], TS14 ID patients 8–13 in Gillessen-Kaesbach et al. 2018 [Citation2].
DNA extraction
DNA extraction was performed using the Flexigene Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
All samples were analyzed by MS-MLPA (methylation-specific multiplex ligation dependent probe amplification, MRC Holland, Amsterdam, The Netherlands, Kit ME032-A1) and subsequent microsatellite analysis for chromosome 14 to determine an ID as cause of the Temple syndrome in the patients.
SNP identification and methylation analyzes
The TS ID patients and their families were investigated for informativity of different SNPs in or adjacent to the three DMRs on chromosome 14q32 (MEG3/DLK1:IG-DMR, MEG3:TSS-DMR and the MEG8:Int2-DMR) by PCR and Sanger sequencing. Primer sequences are listed in Table S1.
Methylation analyzes were conducted as described previously [Citation8]. Briefly, DNA was converted using the EZ DNA Methylation-Gold Kit (Zymo Research Europe, Freiberg, Germany) following the manufacturer’s protocol. Bisulfite amplicon libraries were generated for each sample and specifically tagged then purified, diluted and clonally amplified in an emulsion PCR. The samples were sequenced on the Roche/454 GS junior system (Branford, CT, USA). Data analysis was conducted using the Amplikyzer software [Citation31].
Supplemental Material
Download PDF (1.2 MB)Acknowledgments
We thank the patients and their families for their participation in the study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Ioannides Y, Lokulo-Sodipe K, Mackay DJ, et al. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014 Aug;51(8):495–501. PubMed PMID: 24891339; eng.
- Gillessen-Kaesbach G, Albrecht B, Eggermann T, et al. Molecular and clinical studies in 8 patients with Temple syndrome. Clin Genet. 2018 Jun;93(6):1179–1188. PubMed PMID: 29468661; eng.
- Kagami M, O’Sullivan MJ, Green AJ, et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 2010 Jun;6(6):e1000992. PubMed PMID: 20585555; PubMed Central PMCID: PMC2887472. eng.
- Beygo J, Elbracht M, de Groot K, et al. Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur J Hum Genet. 2015 Feb;23(2):180–188. PubMed PMID: 24801763; PubMed Central PMCID: PMC4297900. eng.
- Monk D, Morales J, den Dunnen JT, et al. Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics. 2016 Dec 2. PubMed PMID: 27911167; eng. DOI:10.1080/15592294.2016.1264561
- Court F, Tayama C, Romanelli V, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014 Apr;24(4):554–569. PubMed PMID: 24402520; PubMed Central PMCID: PMC3975056. eng.
- Bens S, Kolarova J, Gillessen-Kaesbach G, et al. The differentially methylated region of MEG8 is hypermethylated in patients with Temple syndrome. Epigenomics. 2015 Oct;7(7):1089–1097. PubMed PMID: 26541061; eng.
- Beygo J, Kuchler A, Gillessen-Kaesbach G, et al. New insights into the imprinted MEG8-DMR in 14q32 and clinical and molecular description of novel patients with Temple syndrome. Eur J Hum Genet. 2017 Aug;25(8):935–945. PubMed PMID: 28635951; PubMed Central PMCID: PMC5567157. eng.
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001 Jan;2(1):21–32. PubMed PMID: 11253064; eng.
- Li Y, Sasaki H. Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming. Cell Res. 2011 Mar;21(3):466–473. PubMed PMID: 21283132; PubMed Central PMCID: PMC3193417. eng.
- Kaneda M. Genomic imprinting in mammals-epigenetic parental memories. Differentiation. 2011 Sep;82(2):51–56. bPubMed PMID: 21680080; eng.
- Stewart KR, Veselovska L, Kelsey G. Establishment and functions of DNA methylation in the germline. Epigenomics. 2016 Oct;8(10):1399–1413. PubMed PMID: 27659720; PubMed Central PMCID: PMC5066131. eng.
- Davis TL, Trasler JM, Moss SB, et al. Acquisition of the H19 methylation imprint occurs differentially on the parental alleles during spermatogenesis. Genomics. 1999 May 15;58(1):18–28. PubMed PMID: 10331941; eng.
- Lee DH, Singh P, Tsai SY, et al. CTCF-dependent chromatin bias constitutes transient epigenetic memory of the mother at the H19-Igf2 imprinting control region in prospermatogonia. PLoS Genet. 2010 Nov 24;6(11):e1001224. PubMed PMID: 21124827; PubMed Central PMCID: PMC2991272. eng.
- Kato Y, Kaneda M, Hata K, et al. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet. 2007 Oct 01;16(19):2272–2280. PubMed PMID: 17616512; eng.
- Henckel A, Chebli K, Kota SK, et al. Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J. 2012 Feb 1;31(3):606–615. PubMed PMID: 22117218; PubMed Central PMCID: PMC3273379. eng.
- Mitter D, Buiting K, von Eggeling F, et al. Is there a higher incidence of maternal uniparental disomy 14 [upd(14)mat]? Detection of 10 new patients by methylation-specific PCR. Am J Med Genet A. 2006 Oct 1;140(19):2039–2049. PubMed PMID: 16906536; Eng.
- Kagami M, Kurosawa K, Miyazaki O, et al. Comprehensive clinical studies in 34 patients with molecularly defined UPD(14)pat and related conditions (Kagami-Ogata syndrome). Eur J Hum Genet. 2015 Nov;23(11):1488–1498. PubMed PMID: 25689926; PubMed Central PMCID: PMC4613461. eng.
- Lande A, Kroken M, Rabben K, et al. Temple syndrome as a differential diagnosis to Prader-Willi syndrome: identifying three new patients. Am J Med Genet A. 2018 Jan;176(1):175–180. PubMed PMID: 29159982; eng.
- Buiting K, Gross S, Lich C, et al. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003 Mar;72(3):571–577. PubMed PMID: 12545427; PubMed Central PMCID: PMC1180233. eng.
- Cerrato F, Sparago A, Verde G, et al. Different mechanisms cause imprinting defects at the IGF2/H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumour. Hum Mol Genet. 2008 May 15;17(10):1427–1435. PubMed PMID: 18245780; eng.
- Buiting K, Williams C, Horsthemke B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016 Oct;12(10):584–593. PubMed PMID: 27615419; eng.
- Eggermann T, Begemann M, Binder G, et al. Silver-Russell syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis. 2010 Jun 23;5:19. PubMed PMID: 20573229; PubMed Central PMCID: PMC2907323. eng.
- Mussa A, Russo S, Larizza L, et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet. 2016 Apr;89(4):403–415. PubMed PMID: 26138266; eng.
- Wey E, Bartholdi D, Riegel M, et al. Mosaic imprinting defect in a patient with an almost typical expression of the Prader-Willi syndrome. Eur J Hum Genet. 2005 Mar;13(3):273–277. PubMed PMID: 15578038; eng.
- Dittrich B, Buiting K, Korn B, et al. Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat Genet. 1996 Oct;14(2):163–170. PubMed PMID: 8841186; eng.
- Ogata T, Kagami M. Kagami-Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J Hum Genet. 2016 Feb;61(2):87–94. PubMed PMID: 26377239; PubMed Central PMCID: PMC4771937. eng.
- Tomizawa S, Nowacka-Woszuk J, Kelsey G. DNA methylation establishment during oocyte growth: mechanisms and significance. Int J Dev Biol. 2012;56(10–12):867–875. PubMed PMID: 23417409; eng.
- Kelsey G, Feil R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos Trans R Soc Lond B Biol Sci. 2013 Jan 5;368(1609):20110336. PubMed PMID: 23166397; PubMed Central PMCID: PMC3539362. eng.
- Buiting K, Kanber D, Martin-Subero JI, et al. Clinical features of maternal uniparental disomy 14 in patients with an epimutation and a deletion of the imprinted DLK1/GTL2 gene cluster. Hum Mutat. 2008 Sep;29(9):1141–1146. PubMed PMID: 18454453; eng.
- Rahmann S, Beygo J, Kanber D, et al. Amplikyzer: automated methylation analysis of amplicons from bisulfite flowgram sequencing. PeerJ PrePrints. 2013;1:e122v2.