
ABSTRACT
Epigenetic mechanisms integrate both genetic variability and environmental exposures. However, comprehensive epigenome-wide analysis has not been performed across major childhood allergic phenotypes. We examined the association of epigenome-wide DNA methylation in mid-childhood peripheral blood (Illumina HumanMethyl450K) with mid-childhood atopic sensitization, environmental/inhalant and food allergen sensitization in 739 children in two birth cohorts (Project Viva–Boston, and the Generation R Study–Rotterdam). We performed covariate-adjusted epigenome-wide association meta-analysis and employed pathway and regional analyses of results. Seven-hundred and five methylation sites (505 genes) were significantly cross-sectionally associated with mid-childhood atopic sensitization, 1411 (905 genes) for environmental and 45 (36 genes) for food allergen sensitization (FDR<0.05). We observed differential methylation across multiple genes for all three phenotypes, including genes implicated previously in innate immunity (DICER1), eosinophilic esophagitis and sinusitis (SIGLEC8), the atopic march (AP5B1) and asthma (EPX, IL4, IL5RA, PRG2, SIGLEC8, CLU). In addition, most of the associated methylation marks for all three phenotypes occur in putative transcription factor binding motifs. Pathway analysis identified multiple methylation sites associated with atopic sensitization and environmental allergen sensitization located in/near genes involved in asthma, mTOR signaling, and inositol phosphate metabolism. We identified multiple differentially methylated regions associated with atopic sensitization (8 regions) and environmental allergen sensitization (26 regions). A number of nominally significant methylation sites in the cord blood analysis were epigenome-wide significant in the mid-childhood analysis, and we observed significant methylation – time interactions among a subset of sites examined. Our findings provide insights into epigenetic regulatory pathways as markers of childhood allergic sensitization.
Introduction
The increase in prevalence of allergic diseases has continued on a global scale [Citation1]. In the United States, allergy accounts for more than $18 billion of the annual healthcare cost, with more than 50 million people being affected each year [Citation2]. Children suffer disproportionally from this increasing trend, and allergic conditions have become the third most common chronic disease of childhood [Citation2].
Childhood allergy is a complex disease phenotype determined by both genetic predisposition and environmental exposures. The strong familial aggregation of allergy suggests a genetic basis and a number of genetic polymorphisms have been implicated [Citation3–Citation8]. In addition, prenatal and early life environmental exposures, such as tobacco smoking [Citation9–Citation12], air pollution [Citation13–Citation16] and dietary factors [Citation17–Citation21] – alone or through interactions with genetic predisposition – also alter the risk of childhood allergy. However, the molecular underpinnings of allergic disease susceptibility and severity are still not well-understood.
DNA methylation – through the addition of a methyl group to cytosine residues in the cytosine-phosphate-guanine (CpG) dinucleotide sequence – is as an epigenetic regulatory mechanism influenced by both genetic variability and environmental exposures, and has been postulated to be a biomarker of allergy. Previous epigenome-wide association studies (EWAS) have linked the blood DNA methylome with atopy [Citation22], food allergy [Citation23,Citation24], serum IgE measured as a continuous trait [Citation22,Citation25,Citation26] and asthma [Citation27]. Further, the nasal [Citation28,Citation29] and bronchial mucosal tissue methylomes [Citation30] have also been associated with atopy-related phenotypes. However, no comprehensive epigenome-wide analysis has been performed across major allergic phenotypes in the same subjects from birth to mid-childhood.
In this study, we sought to evaluate the association between epigenome-wide DNA methylation measured at two time points – birth and mid-childhood – with allergy-related phenotypes (atopic sensitization, and two intermediate phenotypes: environmental/inhalant and food allergen sensitization) measured during mid-childhood. We performed the cord blood and mid-childhood epigenome-wide analyses separately. For the cord blood analysis, we performed a meta-analysis of a total of 1071 newborns from two independent birth cohort – Project Viva (Boston, USA) (N = 232) and the Generation R Study (Rotterdam, the Netherlands) (N = 839); the mid-childhood analysis was conducted in 739 child blood samples from Project Viva (N = 396) and the Generation R Study (N = 343). We hypothesized that childhood allergen sensitization maybe associated with differential DNA methylation in cord blood and mid-childhood peripheral blood. We anticipated finding associations for the mid-childhood analysis given the priming role of childhood environmental exposures, but considered that methylation patterns established during fetal development and measured at birth may set the epigenetic trajectories and mark the susceptibility for allergic sensitization in childhood.
Materials and methods
Study populations
This study included mother-child pairs from Project Viva (Boston, USA) and mother-child pairs from the Generation R Study (Rotterdam, the Netherland) – both of which are population-based prospective longitudinal pre-birth cohorts. Project Viva enrolled 2128 pregnant women during 1999–2002 at their initial obstetrical visit at Atrius Harvard Vanguard Medical Associates in Boston Massachusetts; the Generation R Study enrolled 8,879 pregnant women with a delivery date between April 2002 and January 2006 from midwifery practices, hospitals and child health centers located in Rotterdam, the Netherlands. Detailed protocols have been published previously for both cohorts [Citation31–Citation33]. Seven hundred and thirty-nine mother-child pairs (Project Viva N = 396; the Generation R Study N = 343) had complete information on peripheral blood DNA methylation and allergy – both measured at mid-childhood. Two-hundred and thirty-two mother-child pairs in Project Viva had complete information on cord blood DNA methylation and allergy measures, and 839 mother-child pairs in the Generation R Study had complete information on cord blood DNA methylation and allergy measures. In our previous work, we showed that the subset of participants with DNA methylation and serum IgE measures had similar personal characteristics compared to the overall population in Project Viva [Citation34]. Written informed consent was obtained for all children in both studies. All study protocols were reviewed and approved by the Institutional Review Boards of the participating institutions.
DNA methylation measurements
In Project Viva, we extracted cord blood and peripheral blood DNA using the Qiagen Puregene Kit (Valencia, CA) and bisulfite converted using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA). We randomized samples by chips and plates and generated DNA methylation data using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA). In the sample quality control step, we removed samples that were technical replicates, samples with low quality, and samples with genotype or sex mismatches. We removed low quality probes (based on detection p-values >0.05), probes on sex chromosomes, SNP probes, probes within 10bp of a known SNP with minor allele frequency ≥ 1%, non-CpG probes, as well as non-specific and cross-reactive probes. We applied the normal-exponential out-of-band (‘noob’) method for background correction and dye bias adjustment, and we normalized our data using Beta Mixture Quantile Dilation (BMIQ) – a model based intra-array normalization strategy to reduce bias of type 2 probes, while controlling type 1 probe enrichment bias [Citation35].
In the Generation R Study, DNA was extracted from blood samples taken at birth (cord blood) and the 9-year follow-up visit. Five hundred nanograms of DNA per sample underwent bisulfite conversion using the EZ-96 DNA Methylation kit (Shallow) (Zymo Research Corporation, Irvine, USA). Samples were plated randomly onto 96-well plates. Samples were processed with the Illumina Infinium HumanMethylation450 BeadChip (Illumina Inc., San Diego, USA). Preparation and normalization of the HumanMethylation450 BeadChip array data were performed according to the Control Probe Adjustment and reduction of global CORrelation (CPACOR) workflow [Citation36] using the software package R [Citation37]. Quality control of analyzed samples was performed using standardized criteria. Probes that had a detection p-value ≥ 1E-16 were set to missing per array. Next, the intensity values were stratified by autosomal and non-autosomal probes and quantile normalized for each of the six probe type categories separately: type II red/green, type I methylated red/green and type I unmethylated red/green. Arrays with observed technical problems such as failed bisulfite conversion, hybridization or extension, as well as arrays with a mismatch between sex of the proband and sex determined by the chromosome X and Y probe intensities were removed from subsequent analyses. Additionally, only arrays with a call rate > 95% per sample were processed further.
For both cohorts, we reported methylation as beta values, which were expressed as the percentage of methylated (M) cytosine residuals over the sum of methylated (M) and unmethylated (U) cytosine residues at the 5 carbon position (i.e. beta value = [M/(M + U)]*100%).
Allergy measures
We studied three allergy-related outcomes in mid-childhood: atopic sensitization; environmental and food allergen sensitization. In Project Viva, atopic sensitization was defined as any serum specific IgE level ≥ 0.35 IU/ml to common indoor allergens (dust mite, cat dander, dog dander, cockroach), mold (Aspergillus, Alternaria tenuis), outdoor allergens (rye grass, ragweed) or food allergens (egg white, milk, wheat, peanut, soybean), or total serum IgE greater than or equal to 100 IU/ml; environmental allergen sensitization was defined as any serum specific IgE level ≥ 0.35 IU/ml to common indoor allergens (dust mite, cat dander, dog dander, cockroach), mold (Aspergillus, Alternaria tenuis) or grass pollen (ryegrass, ragweed) [Citation17]; and food allergen sensitization was defined as any serum specific IgE level ≥ 0.35 IU/ml to one of the food allergens listed (egg white, milk, wheat, peanut, soybean) [Citation18]. Twenty-two children were sensitized to all three phenotypes; 56 children were sensitized to environmental allergen only and 24 children were sensitized to food allergen only.
In the Generation R Study, allergic sensitization was measured by skin prick tests on the volar surface of the left forearm with single-use sterile 1 mm-tip metal lancets (ALK-Abelló A/S, Hørsholm, Denmark). Histamine dihydrochloride (10 mg/mL) and a saline solution (NaCl 0.9%) served as 2 positive controls and 1 negative control, respectively. Environmental allergens comprised house dust mite (Dermatophagoides pteronyssinus), 5-grass mixture (Dactylis glomerata, Festuca pratensis, Lolium perenne, Phleum pratense, Poa pratensis), birch (Betula verrucosa), cat (Felis catus) and dog (Canis familiaris) (ALK-Abelló B.V., Almere, The Netherlands). Food allergens comprised hazelnut, cashew nut, peanut and peach extracts. For all food allergens, aliquots were stored at −20°C and were defrosted for 1 h prior to the skin prick test. Skin responses were recorded 15 min after applying allergens to the skin by measuring the area of the wheal (in mm2) using the Precise Automated Area Measurement of Skin Test (PAAMOST) software and considered positive if the area of the wheal was ≥40% of the histamine response (i.e. histamine equivalent prick index area ≥0.40) [Citation38]. A skin prick test was not performed if children could not omit antihistamine intake or corticosteroid ointment ≤72 h and ≤48 h prior to the test, respectively, used oral prednisone ≥10 mg daily or had eczema on the volar surface of the left forearm. Data from 343 participants contributed to the mid-childhood analysis, 107 were not sensitized to environmental or food allergen, 105 participants were sensitized to environmental allergen only, and 18 participants were sensitized to food allergen only.
Statistical analysis
Peripheral blood analysis
We performed cohort-specific cross-sectional analyses of peripheral blood DNA methylation and allergic phenotypes – both measured at mid-childhood. We fit covariate-adjusted logistic regression models for each methylation site, and adjusted for maternal covariates [age at enrollment (continuous), smoking status (Project Viva: never, former, or during pregnancy; the Generation R Study: never, stopped in early pregnancy or continued throughout pregnancy), education (college/graduate level or not college/graduate level), atopy history (asthma, eczema or hay fever)], child covariates [sex (female or male), race/ethnicity (white, black or others), gestational age (continuous), age at mid-childhood (questionnaire based)(continuous)], and cell type proxies estimated for peripheral blood (Houseman estimation) (percentages of monocytes, CD8T cells, CD4T cells, NK cells, B cells) [Citation39]. We also performed a sensitivity analyses adjusting for childhood asthma status. We meta-analyzed results from Project Viva and the Generation R Study using fixed-effect meta-analysis weighted by the inverse of the variance using METAL [Citation40]. We reviewed Q-Q plots and reported genomic inflation factor (lambda) to examine whether our associations were subject to systematic bias/inflation. We accounted for multiple comparisons using the false discovery rate (FDR) method of Benjamini and Hochberg [Citation41]. We further compared the overlaps among FDR-significant methylation sites (FDR<0.05) identified for each phenotype (i.e. atopic sensitization, environmental allergen and food allergen sensitization). Using the Database for Annotation, Visualization and Integrated Discovery (DAVID 6.7) pathway analysis [Citation42], we searched for functional enrichment of biological pathways of statistically significant methylation sites identified. Specifically, we selected methylation sites that showed FDR<0.05 based on the meta-analysis results, mapped each methylation site to its corresponding UCSC reference gene name and chose Kyoto Encyclopedia of Genes and Genomes (KEGG) database to perform pathway analysis with default settings, with a minimum of four genes in an annotated group.
After identification of overlapping methylation marks from the meta-analyses for all phenotypes, we assessed whether the 36 overlapping CpG associations for all phenotypes occur preferentially in transcription factor (TF) binding motifs. The DNA-binding domains (DBD) of the TFs recognize these short consensus sequence motifs (8–20 bp), represented as position weight matrices (PWM). We have downloaded human transcription factor motifs from the Catalogue of Inferred Sequence Binding Preferences (CIS-BP) [Citation43], selected a subset that represent a non-redundant set of transcription factors [Citation44], which were then mapped to the reference human genome (hg19) using Find Individual Motif Occurrence (FIMO) [Citation45], retaining only significant matches (P < 1x10 −5). We then used the intersect feature of BEDTools [Citation46] to search all motifs occurrences within 20 base pairs of the selected CpG sites.
We also performed comb-p, a regional analysis method that combines meta-analysis p-values in sliding windows, while taking into account spatial correlation across the genome [Citation47]. For comb-p analyses, we chose an initial seed of 10−7, a step size of 800 bases and a p value cut-off of 0.10.
Cord blood analysis
For the cord blood associations, we first performed cohort-specific analysis using covariate-adjusted logistic regressions for each methylation site as the predictor. We adjusted for the following covariates selected a priori: maternal covariates [age at enrollment (continuous), smoking status (Project Viva: never, former, or during pregnancy; the Generation R Study: never, stopped in early pregnancy or continued throughout pregnancy), education (college/graduate level or not college/graduate level), atopy history (asthma, eczema or hay fever)], child covariates [sex (female or male), race/ethnicity (white, black or others), gestational age (continuous)], and cell type proxies estimated for cord blood (Balkuski estimation) (percentages of monocytes, CD8T cells, CD4T cells, NK cells, B cells and nucleated red blood cells (nRBC)) [Citation48]. We meta-analyzed results from the two cohorts using fixed-effect meta-analysis weighted by the inverse of the variance [Citation40]. We evaluated Q-Q plots, computed the genomic inflation factor (lambda) and accounted for multiple comparisons using the false discovery rate (FDR) method of Benjamini and Hochberg [Citation41].
Longitudinal trend in methylation trajectories
We reported the number of methylation sites that showed nominal significance (p < 0.05) in the cord blood meta-analysis, and became epigenome-wide significance (FDR<0.05) in the mid-childhood cross-sectional meta-analysis, and plotted trajectory plots of a subset of these sites. We also calculated the difference in methylation values between mid-childhood and cord blood measurements (delta change for 211 subjects with two time points of methylation data), and evaluated the association between methylation change from cord till mid-childhood and allergic outcomes in mid-childhood in Project Viva for sites that showed nominal significance in the cord blood meta-analysis, and became epigenome wide significance in the mid-childhood meta-analysis. We adjusted for covariates reported in the section above; we additionally adjusted for cord blood (baseline) DNA methylation of the same sites. For selected methylation sites, we performed mixed effects models with repeated statement for binary outcomes. We introduced a methylation – time interaction to examine whether there is any time trend among 211 children Project Viva with both cord blood and mid-childhood DNA methylation measurements.
Sensitivity analysis
To evaluate whether the significant associations for the peripheral blood analysis primarily reflect childhood environmental exposures, we evaluated additional models adjusted for cord blood DNA methylation of the same CpG site when examining the association between peripheral blood DNA methylation and childhood allergic sensitization in Project Viva (211 children with methylation measures at both time points).
Using a reference methylation Quantitative Trait Loci (QTL) library, we evaluated the genetic control of methylation for the top 30 associated CpG sites identified from the mid-childhood meta-analysis for atopic sensitization, environmental and food allergen sensitization. We leveraged genetic-epigenetic information from the ARIES large-scale genome-mQTL analysis, which included 1,000 mother-child pairs (http://www.mqtldb.org/) [Citation49]. The genetic influences on DNA methylation were studied across 5 different time points in blood in this reference library: maternal [pregnancy, middle-age]; child [at birth, childhood, adolescence]. We focused our mQTL comparisons in the cis position (i.e. a genetic variant within ±500Kb of a methylation locus) measured from peripheral blood in childhood, and with MAF ≥5%.
Eosinophils and neutrophils are central effector immune cells that often associate with allergic outcomes. To further ensure the associations we observed were due to changes in methylation levels, rather than shifts in eosinophil/neutrophil cell proportions, we applied a reference-free method based on principal component analysis (PCA) with low rank approximation (ReFACTor) to control for cell-type heterogeneity [Citation50]. Previous studies have shown that ReFACTor components correlate well with measured eosinophil and neutrophil counts [Citation50].
We also compared the FDR-significant methylation sites identified from a previously published epigenome-wide association study of IgE across various age groups [Citation25].
All analyses were performed in SAS® version 9.4 (SAS Institute, Inc.) and R version 3.4.1.
Results
Population characteristics
This study includes a total of 739 mother-child pairs for the mid-childhood peripheral blood analysis (Project Viva N = 396; the Generation R Study N = 343), and 1071 mother-child pairs for the cord blood analysis (Project Viva N = 232; the Generation R Study N = 839) (). For the mid-childhood subset, mean maternal age was 32.3 (standard deviation (SD) = 5.5) years for Project Viva and 32.3 (SD = 3.9) years for the Generation R Study. The percentage of mothers who smoked actively during pregnancy was comparable in both cohorts; however, a larger proportion of mothers were former smokers in Project Viva (Project Viva: 20.2% vs. the Generation R Study 12.2%). Approximately 39% of mothers in Project Viva and in the Generation R Study had a history of atopy (asthma, eczema and hay fever) reported during their pregnancy interviews. Children from the Generation R Study were slightly older when the allergy tests were conducted (age mean±SD: Project Viva 7.7 ± 0.7; the Generation R Study 9.8 ± 0.3). Sixty-three percent of children were white in the Project Viva subset, while all children were white in the Generation R Study subset. Higher proportions of children had positive allergic tests in Project Viva than in the Generation R Study (Project Viva: 54.3%, 42.2% and 26.3% for atopic sensitization, environmental allergen sensitization and food allergen sensitization, respectively; the Generation R Study: 31.2%, 30.6% and 5.2% for atopic sensitization, environmental allergen sensitization and food allergen sensitization, respectively). Participant characteristics were similar for the cord blood subset compared to the mid-childhood subset ().
Table 1. Characteristics of children and their mothers in project viva and the generation R study.
Mid-childhood peripheral blood analysis
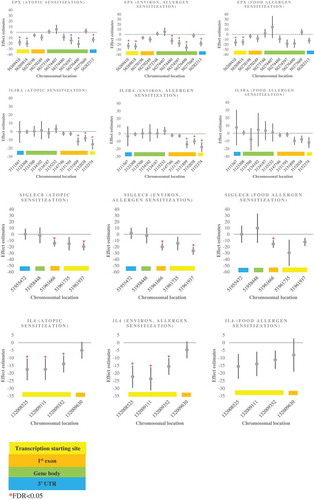
Our epigenome-wide meta-analysis identified 705 (representing 505 annotated genes), 1411 (representing 905 annotated genes), and 45 (representing 36 annotated genes) differentially methylated CpG sites in mid-childhood peripheral blood associated with mid-childhood atopic sensitization, environmental allergen sensitization and food allergen sensitization, respectively (FDR< 0.05) (; Table S1 – full list; ). Genomic inflation factor ranges from 1.16–1.26 were acceptable for the three allergic phenotypes, suggesting our findings were unlikely due to systematic bias (Figure S1). We observed differential methylation associations for all three phenotypes in multiple genes previously associated with asthma (EPX [Citation51,Citation52], IL4 [Citation53], IL5RA [Citation53], PRG2 [Citation54], SIGLEC8 [Citation55–Citation57]) (). Adjusting for current asthma status of the child did not change the results, as we observed similar magnitude of effect estimates and p values in models with and without the adjustment of asthma status (Table S2). For atopic sensitization, the biological processes suggested from the DAVID pathway analysis of the FDR significant methylation sites included mTOR signaling pathway (number of genes contributing to the pathway = 7, p value = 0.0029), lipid metabolism (number of genes contributing to the pathway = 5, p value = 0.015), Fc gamma R-mediated phagocytosis (number of genes contributing to the pathway = 8, p value = 0.016), endocytosis (number of genes contributing to the pathway = 11, p value = 0.031), and asthma (number of genes contributing to the pathway = 4, p value = 0.045). For environmental allergen sensitization, the biological processes implicated in the pathway analysis included inositol phosphate metabolism (number of genes contributing to the pathway = 9, p value = 0.007), lysosome (number of genes contributing to the pathway = 14, p value = 0.0086), N-Glycan biosynthesis (number of genes contributing to the pathway = 8, p value = 0.0099), asthma (number of genes contributing to the pathway = 6, p value = 0.017), mTOR signaling pathway (number of genes contributing to the pathway = 8, p value = 0.019), type II diabetes mellitus (number of genes contributing to the pathway = 7, p value = 0.036), phosphatidylinositol signaling system (number of genes contributing to the pathway = 5, p value = 0.041). . For food allergen sensitization, only the asthma-related pathway appeared to be significant (number of genes contributing to the pathway = 2, p value = 0.039). A list of genes contributing to each pathway is shown in Table S3a.
Table 2. Association between mid-childhood peripheral blood DNA methylation with mid-childhood atopic sensitization, environmental and food allergen sensitization (top 30 methylation sites are presented here sorted by meta-analysis p-values) .
Figure 1. Miami plots. P values based on meta-analysis results of the associations between cord blood or mid-childhood peripheral blood DNA methylation with mid-childhood atopic sensitization, environmental and food allergen sensitization.
Redline – Bonferroni significance (Bonferroni-adjusted p-value<0.05)Green line – FDR significance (FDR-adjusted p-value <0.05)
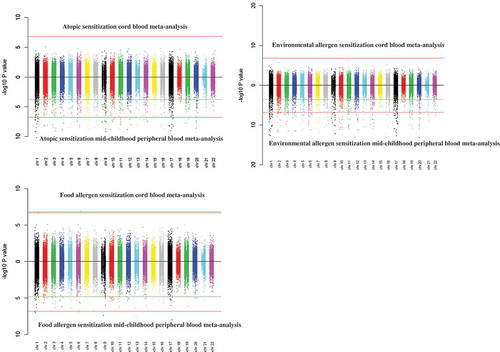
Figure 2. Plots for the associations between EPX, IL5RA, SIGLEC8, IL4 DNA methylation in mid-childhood with mid-childhood atopy (sorted by chromosomal locations) .
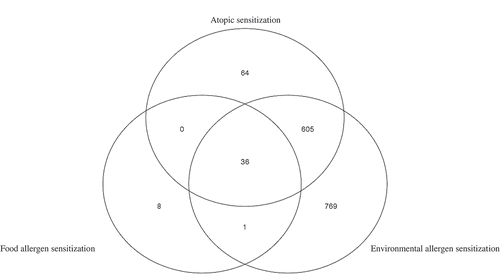
Thirty-six methylation sites with similar magnitude and direction of effect overlapped among the three analyses (atopic sensitization, environmental allergen sensitization and food allergen sensitization) (; ) This list () includes multiple genes that have been implicated in innate immunity (DICER1) [Citation58], the atopic march (AP5B1) [Citation59], eosinophilic esophagitis [Citation60] and chronic sinusitis (SIGLEC8) [Citation61] and asthma (EPX [Citation51,Citation52], PRG2 [Citation54], SIGLEC8 [Citation55–Citation57], and CLU [Citation62]). In addition, all but two of the CpG sites in the overlapping list occur in putative transcription factor binding motifs; motifs from TF families are present for more than one CpG association. For example, five CpG sites overlap putative GATA family transcription sites (GATA2-cg01901579,cg06391412 GATA3-cg08722695, GATA4-cg01901579, cg25854298, GATA6-cg13829849) and three overlap SMAD family motifs (SMAD1-cg26791242, SMAD2-cg25758167, SMAD4-cg25738116) (Supplemental Table S3b) suggesting the common aspects of allergy may include TF-mediated gene regulatory pathways.
Table 3. Overlapping results of all three phenotypes from the mid-childhood meta-analysis (i.e. methylation sites that met FDR<0.05 for all three phenotypes) .
Figure 3. Venn diagram of overlapping FDR significant methylation sites (mid-childhood cross-sectional meta-analysis) among three phenotypes for the meta-analysis of Project Viva and The Generation R Study.
In addition to the CpG-by-CpG analysis, we also performed regional analysis based on meta-analysis p-values using comb-p. We identified 8 methylation regions associated with mid-childhood atopic sensitization and 26 regions associated with environmental allergen sensitization (with at least two methylation sites) (). All methylation regions identified for atopic sensitization were significantly associated with environmental allergen sensitization (). Those overlapping regions were annotated to VKORC1L1 (7 sites), TLDC2 (5 sites), ACOT7 (4 sites), LOC339524 (3 sites), SIGLEC8 (3 sites), ZFPM1 (3 sites), NHLRC4 (2 sites), and ZNF862 genes (2 sites). There were no significant associations for food allergen sensitization in this regional analyses.
Table 4. Regional analysis (comb-p) of the association between mid-childhood peripheral blood with mid-childhood atopic sensitization and environmental allergen sensitization.
Cord blood analysis
There were nominal p values of at least 10−6 for all three atopic phenotypes (Table S4), but we did not observe statistically significant (FDR<0.05) differentially methylated sites in cord blood DNA CpGs associated with childhood atopic sensitization or environmental allergen sensitization at mid-childhood in the meta-analysis of cord blood methylation. For food allergen sensitization, 2 methylation sites reached epigenome-wide significance level (RPS6KA2 and CHIT1). The significant associations at these sites were predominantly driven by the Generation R Study results (Table S4), and we observed heterogeneous associations for CHIT1 between the two cohorts (pheterogeneity<0.05). The Q-Q plots indicate there is little inflation, and we did not observe systematic bias (Figure S1).
Longitudinal trend
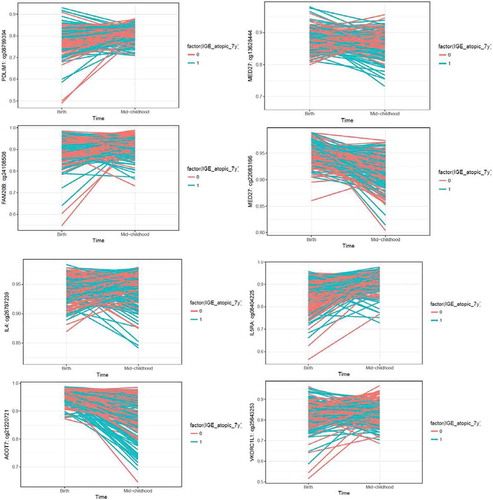
Next, we explored whether the differentially methylated CpG sites (FDR<0.05) in the mid-childhood cross-sectional analysis showed nominal significance (p < 0.05) at birth. We identified 99 methylation sites that showed nominal significance at birth and were epigenome-wide significant at mid-childhood for atopic sensitization; 163 for environmental allergen sensitization and 3 for food allergen sensitization (Table S5). For atopic sensitization and environmental allergen sensitization, multiple sites are in/close to genes related to asthma pathway genes (EPX, IL4, IL5RA). Among the methylation sites that showed nominal significance (p < 0.05) in the cord blood analysis, and were epigenome-wide significance (FDR<0.05) in the mid-childhood analysis, changes in methylation values from cord to mid-childhood were also nominally significantly associated with mid-childhood allergy, with p values in the range of 10−5 to 10−6 (Table S5). In the mixed-effects model analysis, which included methylation – time interactions, we observed significant time trend among 18 out of 19 sites examined, where we selected the top 4 methylation sites (sorted by p-value significance) for the cord blood analysis, 4 methylation sites previously linked to asthma pathways for atopic sensitization and environmental allergen sensitization; and the only 3 methylation sites that met the p-value criteria for food allergen sensitization (Table S6). Trajectory plots are presented in (for atopic sensitization) and Figure S2 (for environmental and food allergen sensitization).
Figure 4. Trajectory plots of methylation changes by atopic status. Methylation sites were selected as they showed nominal significance in the cord blood analysis, and epigenome-wide significance in the mid-childhood analysis. Top 4 plots: top 4 methylation sites sorted by cord blood p-values based on the criteria described above; bottom 4 plots: methylation sites previously shown to be associated with asthma.
Sensitivity analysis
We additionally adjusted for cord blood DNA methylation when examining the association between Project Viva peripheral blood DNA methylation and childhood allergy at the same sites (N = 211). Our mid-childhood results were of the same signs of directionality and with relatively comparable magnitude of effect estimates after the adjustment of cord blood DNA methylation (Table S7).
In the mid-childhood meta-analysis, we identified multiple methylation sites annotated to genes related to eosinophilic activities (such as EPX, PRG2, SIGLEC8). To further ensure the associations we observed were due to variability in methylation levels, rather than shifts in eosinophil cell proportions, we controlled for cell-type heterogeneity using ReFACTor. Our findings were robust to the adjustment of cell proportions using ReFACTor (Table S8).
Among the top 30 methylation sites identified from the mid-childhood meta-analysis, most of them were not associated with known genetic variants in the cis-position (i.e. ± 500Kb) (Table S9). We identified 5 methylation loci with cis-mQTL in childhood peripheral blood for atopic sensitization, 3 for environmental allergen sensitization and 4 for food allergen sensitization, including cg26396815 (BANK1), cg07948085 (not annotated), cg24491618 (KCNH2), cg00114012 (SLC2A8), cg20885063 (ATPAF2), cg21919729 (CTSB), cg12198633 (KLHL24) and cg25738116 (PA2G4P4).
We replicated many of the FDR-significant methylation sites identified from a separate multi-ethnicity meta-analysis of IgE (Table S10) [Citation25].
Discussion
We performed comprehensive epigenome-wide DNA methylation analyses across major allergic phenotypes (739 children at mid-childhood and 1071 newborns) from two birth cohorts from the United States and the Netherlands. Our meta-analysis demonstrated differential methylation in mid-childhood peripheral blood associated with mid-childhood atopic sensitization, environmental and food allergen sensitizations, and some of these had nominal associations in cord blood (14% for atopic sensitization, 12% for environmental allergen sensitization and 7% for food allergen sensitization) and demonstrated a significant longitudinal time trend. Multiple associated methylation sites were annotated to genes that have been implicated in asthma pathways, which were consistent across all three atopy phenotypes, including EPX (eosinophil peroxidase), IL4 (interleukin 4), IL5RA (interleukin 5 receptor A) (PRG2 (proteoglycan 2), and SIGLEC8 (sialic acid-binding, Ig-like lectin-8). The EPX (eosinophil peroxidase) gene encodes for a pre-protein in eosinophils, which functions as a potent oxidant [Citation51,Citation52]. The IL4 and IL5RA are cytokine pathway genes associated with Th2 cell differentiation [Citation53]. The PRG2 (proteoglycan 2) gene product is the predominant constituent of the eosinophil granule, and displays anti-microbial activities against pathogens [Citation54]. SIGLEC8 (sialic acid-binding, Ig-like lectin-8) belongs to a family of single-pass transmembrane cell surface proteins on leukocytes; the SIGLEC8 gene product has a role in apoptosis of eosinophils [Citation56,Citation57,Citation63] and has been suggested recently as a therapeutic target [Citation64]. We did not find epigenome-wide statistically significant differential methylation of cord blood CpGs associated with mid-childhood allergic phenotypes, although we did observe a longitudinal time trend for a subset, suggesting a predominant role for post-natal environment in guiding epigenetic trajectories associated with allergic diseases of childhood. We did identify a number of methylation sites with nominal significance in the cord blood associated with mid-childhood allergic phenotypes, and some of those associations remained epigenome-wide significant in mid-childhood, suggesting that methylation patterns established at birth may mark allergic sensitization susceptibility in childhood.
In addition to the asthma pathway, we further identified multiple methylation sites associated with mTOR signaling and inositol phosphate metabolism for the mid-childhood cross-sectional analysis for atopic sensitization and environmental allergen sensitization. The mTOR pathway plays a central role in directing immune responses toward allergy, immune-disturbance of Th17/Treg and Th1/Th2 balance, and has been implicated in asthma onset, remission, and allergy [Citation65]; additionally mTOR inhibition through drugs such as rapamycin have been proposed as therapeutic options for allergic diseases [Citation66], suggesting further investigation of epigenetic plasticity of the mTOR pathway may have therapeutic potential. Inositol phosphate production is also involved in eosinophil degranulation and increased tracheal smooth muscle contractions; delineating mechanistic underpinnings of airway smooth muscle constriction may provide new therapeutic targets associated with the inhibition of bronchoconstriction in asthma [Citation67].
Recently, transcription factors have been implicated in the pathogenesis of allergic diseases and asthma in part through impacting the differentiation of Th1/Th2 cells and promoting inflammation through regulation of inflammatory pathways [Citation68]. One functional feature of variable DNA methylation that associates with allergy may be through DNA methylation of transcription factors or DNA methylation within transcription factor binding sites, with downstream impacts on gene expression. Our evaluation of overlapping methylation marks for the allergic phenotypes () revealed that the majority of the CpGs were located within putative TF binding sequences, including multiple sequences that occur in the GATA and SMAD families of transcription factors. Both of these families of transcription factors have been implicated in allergy. For example, GATA1 and GATA2 have been implicated as transcription factors for eosinophil lineage commitment [Citation69,Citation70]; in addition, GATA pathways have been suggested as potential therapeutic targets [Citation71]. The SMAD family members are key regulators of TGF beta signaling [Citation72], with SMAD3 [Citation4,Citation73] and low SMAD2 expression were associated with both increased asthma susceptibility and exacerbations [Citation74]. Although this is an in silico, analysis it highlights the potential ways that EWAS identified targets may have coordinated impact on resulting disease phenotypes, and offers targets to consider for functional evaluation and therapeutic development.
We performed regional analysis based on meta-analysis p-values obtained from the mid-childhood cross-sectional analysis (comb-p) [Citation47]. We identified 8 methylation regions associated with mid-childhood atopic sensitization and 26 regions associated with environmental allergen sensitization. All 8 regions associated with atopic sensitization overlapped with environmental allergen sensitization and were annotated to VKORC1L1, TLDC2, ACOT7, LOC339524, SIGLEC8, ZFPM1, NHLRC4, and ZNF862. Interestingly, several methylation sites identified from the regional analysis also showed nominal significance in the cord blood, which included methylation sites annotated to ACOT7 and VKORC1L1. The VKORC1L1 gene encodes for an enzyme in the vitamin K cycle, which catalyzes the de-epoxidation of vitamin K. The VKORC1L1 gene product protects cell membrane proteins, and upregulation of this gene is involved in oxidative damage [Citation75]. ACOT7 is a hub gene involved in macrophage activation [Citation76]. SIGLEC8 and ZFPM1 influence eosinophil apoptosis and production [Citation56,Citation57,Citation63,Citation77].
Previous epigenome-wide association studies have linked blood DNA methylation with childhood food allergy. In a case-control study of 12 allergic children and 12 non-allergic controls, multiple genes (CACNA1B, DUSP3, RPS6KA2, ZAK) involved in MAPK signaling pathway were identified in purified CD4 + T cells [Citation23]. A separate study of 2,197 children of European ancestry showed differential DNA methylation of the HLA-DQB1 and HLA-DRB1 gene partially mediates the genetic risk associated with peanut allergy [Citation24]. Further, an epigenome-wide association study of cow’s milk allergy also demonstrated multiple hypo-methylated sites in allergic children in comparison to their matched controls [Citation78]. Top significant differentially methylated sites were mapped to NDFIP2, EVL, TRAPPC9, RPS6KA2, IL1RL1, IL5RA, STAT4, IL4, CLL18 in the discovery dataset. However, those associations were less robust in the replication dataset [Citation78]. To our knowledge, no epigenome-wide association studies have been performed for environmental allergy. Candidate gene studies identified ORMDL1 and STAT6 hypomethylation and RAD50 and IL13 hypermethylation in children whose mothers were living on or running a farm with livestock during pregnancy compared to children whose mothers were living in rural areas but not being exposed to farms or livestock during pregnancy [Citation79], indicating that maternal exposures to farm environment may be associated with differential methylation in distinct genes in children. Early life microbial exposure has also been shown to be related to interferon gamma gene demethylation in naïve T-cells, and associated with subsequent risk for allergic diseases [Citation80].
Neither the cohort-specific nor the meta-analysis results identified differential methylation in cord blood associated with childhood atopic sensitization and environmental allergen sensitization at a stringent statistical threshold, although there were suggestive nominal associations. Among those nominal associations, a number of methylation sites encode genes that are involved in cell migration and adhesion. For example, ITGAL (integrin subunit alpha L) is a lymphocyte co-stimulatory protein and plays a key role in cellular adhesion [Citation81]. ACTN3 (alpha actinin-3) is an actin-binding protein responsible for myofibrillar actin filament anchoring [Citation82]. APC2 (Adenomatous polyposis coli 2) downregulates beta-catenin and interacts with E-cardherin [Citation83]. Adenomatous polyposis coli proteins are also known to regulate cell polarization, attachment and migration [Citation83]. Allergen-associated airway remodeling has been repeatedly observed in animal experiments, in vitro studies and in silico models [Citation84–Citation87]. Although airway remodeling is thought to have a genetic component [Citation88], to our knowledge, no study to date has reported differential methylation sites in cord blood associated with genes involved in airway remodeling. At nominal significance level, our study identified a number of methylation sites that regulate cytoskeletal remodeling, cell adhesion and polarization across the three allergy phenotypes, which may suggest that epigenetic marks in cord blood DNA – reflecting the in utero environment – may serve as early biomarkers of allergen-associated airway remodeling susceptibility in childhood.
We observed multiple methylation sites that showed nominal significance in the cord blood analysis and that became epigenome-wide significant in the mid-childhood analysis. Among those sites, changes in methylation values from cord till mid-childhood were also nominally significantly associated with mid-childhood allergy. We observed significant methylation – time interaction among sites related to allergic and asthma pathways. Although under powered, these pieces of evidence suggest that methylation patterns established during fetal development and measured at birth may set the epigenetic trajectory and mark the susceptibility for allergic sensitization in childhood. Further longitudinal studies with larger sample sizes are warranted to further verify our hypothesis, as these may be targets for both diagnostics and therapeutics for allergic processes that start in childhood.
Our study has a number of strengths [Citation1]. For the mid-childhood concurrent analysis, our top associations were consistent in magnitude and direction across two relatively large birth cohorts from the United States and Europe, which suggests that our findings were unlikely to be cohort specific effects [Citation2]. We identified methylation sites implicated in several pathways (asthma, focal adhesion, mTOR signaling, and inositol phosphate metabolism), and these may highlight plastic pathways to leverage for prevention and therapeutics [Citation3]. Our results were robust to the adjustment by cell counts using the Houseman’s method [Citation39] or by a stringent reference-free method, ReFACTor [Citation50] – a modified PCA method to control for cellular heterogeneity [Citation4]. Many of the FDR-significant methylation sites identified from a separate multi-ethnicity meta-analysis of IgE also replicated in our cohort (Table S10) [Citation25] [Citation5]. We observed longitudinal time trends for a number of methylation sites that were nominally significant at birth and became epigenome-wide significant in mid-childhood, supporting the hypothesis that both in utero and postnatal environment are important for the development of allergy and the allergic march. Lastly, although exploratory, most of the overlapping methylation marks occur within predicted transcription factor motifs; this observation may provide a mechanistic link for the EWAS results for these allergic phenotypes.
Our study also has several limitations [Citation1]. Peripheral blood childhood DNA methylation and mid-childhood allergic outcomes are measured at the same time. Hence, it is difficult to disentangle the temporal relationships between DNA methylation and allergic sensitizations, and our results may be impacted by reverse causation; however this does not limit these findings as biomarkers of atopic disease. On the other hand, our longitudinal prospective cord blood analysis identified nominal significant methylation sites (in the range of 10−6 to 10−5) associated with mid-childhood allergy. We also identified a number of methylation sites that showed nominal significance (p < 0.05) at birth and epigenome-wide significance in mid-childhood, and a suggestive longitudinal time trend, suggesting that methylation patterns at birth may mark allergic vulnerability later in childhood. Larger longitudinal studies would be informative. (2 [Citation2] We analyzed atopic sensitization, and two composite intermediate phenotypes (environmental and food allergy). These phenotypes are correlated and hence not three independent sets of EWAS findings [Citation3]. Project Viva and the Generation R Study used different methods to assess allergy sensitizations. In Project Viva, atopic sensitization was defined using serum specific IgEs, while the Generation R Study used skin prick testing. Serum specific IgE testing is sensitive but not very specific for true atopy, whereas skin testing is more specific [Citation89,Citation90]. A more sensitive, but less specific test will yield more false positive but less false negative case diagnosis, while a less sensitive, but more specific test will yield less false positive but more false negative case diagnosis. The differences in allergic sensitization definitions may result in heterogeneous findings [Citation4]. However, we contend that the genes and pathways we have identified are robust despite this concern for contributions of phenotypic heterogeneity. DNA methylation variability does not always translate to phenotypic differences; functional analyses are needed to examine whether the methylation sites we identified influence gene expression or chromatin accessibility; this is a direction for future work.
In summary, our study identified differential methylation in mid-childhood peripheral blood DNA associated with mid-childhood atopic sensitization, environmental allergen and food allergen sensitization. Multiple DNA methylation sites here associated with atopy have been implicated in allergy and asthma. Further, we observed that epigenetic perturbations in focal adhesion, mTOR signaling, and inositol phosphate metabolism pathways are likely relevant to allergy and the allergic march, and that mechanistic implications may include gene regulatory pathway perturbations due to variable methylation in regions of transcription factor binding. Most compelling is the suggestion that methylation patterns established during fetal development may set the epigenetic trajectory and mark the susceptibility for allergic sensitization in childhood. Our study provides evidence for the epigenetic regulation as a marker of childhood allergy, and may inform future efforts for prevention and treatment of childhood allergy that leverage the plasticity of the epigenome.
Data availability statement
The datasets for the current project are not publicly available because this study began in 1999, and we did not obtain consent for such public release of genetic data. Data are available from the responding author or Project Viva study team ([email protected]) upon request.
Supplemental Material
Download Zip (705.3 KB)Acknowledgments
The Project Viva study is supported by grants from the National Institutes of Health (NIH R01 HL 111108, R01 NR013945, R01 HD 034568, UG3OD023286, K23 ES022242, R01 AI102960). DLD is additionally supported by P01 HL132825. The Generation R Study is made possible by financial support from the Erasmus Medical Center (MC), Rotterdam, the Erasmus University Rotterdam and the Netherlands Organization for Health Research and Development. V.W.V.J. received an additional grant from the Netherlands Organization for Health Research and Development (VIDI 016.136.361) and a Consolidator Grant from the European Research Council (ERC-2014-CoG-64916). The EWAS data was funded by a grant to V.W.V.J. from the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO)/Netherlands Consortium for Healthy Aging (NCHA; project nr. 050-060-810), by funds from the Genetic Laboratory of the Department of Internal Medicine, Erasmus Medical Center, and by a grant from the National Institute of Child and Human Development (R01HD068437). L.D. received funding from the European Union’s Horizon 2020 co-funded programme ERA-Net on Biomarkers for Nutrition and Health (ERA HDHL) (ALPHABET; project nr. 696295/ZonMW The Netherlands project nr. 529051014). The project received fundings from the European Union’s Horizon 2020 research and innovation program (DynaHEALTH; project nr. 633595, and Life Cycle; project nr. 733206). The researchers are independent from the funders. The study sponsors had no role in the study design, data collection, data analysis, interpretation of data, and preparation, review or approval of the manuscript. Dr Kimberly Glass also has addition funding from K25HL133599.
The authors would like to thank Project Viva staff and participants. The Generation R Study is conducted by the Erasmus Medical Center (MC) in close collaboration with the School of Law and Faculty of Social Sciences of the Erasmus University Rotterdam, the Municipal Health Service Rotterdam area, Rotterdam, the Rotterdam Homecare Foundation, Rotterdam and the Stichting Trombosedienst & Artsenlaboratorium Rijnmond (STAR-MDC), Rotterdam. We gratefully acknowledge the contribution of children and parents, general practitioners, hospitals, midwives and pharmacies in Rotterdam. The generation and management of the Illumina 450K methylation array data (EWAS data) for the Generation R Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Erasmus University Medical Center, Rotterdam, the Netherlands. We thank Mr. Michael Verbiest, Ms. Mila Jhamai, Ms. Sarah Higgins, Mr. Marijn Verkerk and Dr. Lisette Stolk for their help in creating the EWAS database.
Disclosure statement
None of the authors has any actual or potential competing financial interests. The views expressed in this article do not necessarily represent the views of the US Government, the Department of Health and Human Services or the National Institutes of Health.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol. 2017;18(10):1076–1083. PubMed PMID: 28926539.
- Pawankar R, Canonica GW, Holgate ST, et al. World allergy organization (WAO) white book on allergy: update 2013. In Organization WA:editor. 2013.
- Paternoster L, Standl M, Waage J, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–1456. PubMed PMID: 26482879; PubMed Central PMCID: PMCPMC4753676.
- Torgerson DG, Ampleford EJ, Chiu GY, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43(9):887–892. PubMed PMID: 21804549; PubMed Central PMCID: PMCPMC3445408.
- Hirota T, Takahashi A, Kubo M, et al. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat Genet. 2011;43(9):893–896. PubMed PMID: 21804548; PubMed Central PMCID: PMCPMC4310726.
- Van Eerdewegh P, Little RD, Dupuis J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418(6896):426–430. PubMed PMID: 12110844.
- Zhang Y, Leaves NI, Anderson GG, et al. Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat Genet. 2003;34(2):181–186. PubMed PMID: 12754510.
- Allen M, Heinzmann A, Noguchi E, et al. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet. 2003;35(3):258–263. PubMed PMID: 14566338.
- Hollams EM, de Klerk NH, Holt PG, et al. Persistent effects of maternal smoking during pregnancy on lung function and asthma in adolescents. Am J Respir Crit Care Med. 2014;189(4):401–407. PubMed PMID: 24251622.
- Burke H, Leonardi-Bee J, Hashim A, et al. Prenatal and passive smoke exposure and incidence of asthma and wheeze: systematic review and meta-analysis. Pediatrics. 2012;129(4):735–744. PubMed PMID: 22430451.
- Thacher JD, Gruzieva O, Pershagen G, et al. Pre- and postnatal exposure to parental smoking and allergic disease through adolescence. Pediatrics. 2014;134(3):428–434. PubMed PMID: 25136039.
- Saulyte J, Regueira C, Montes-Martinez A, et al. Active or passive exposure to tobacco smoking and allergic rhinitis, allergic dermatitis, and food allergy in adults and children: a systematic review and meta-analysis. PLoS Med. 2014;11(3):e1001611. PubMed PMID: 24618794; PubMed Central PMCID: PMCPMC3949681.
- Bowatte G, Lodge C, Lowe AJ, et al. The influence of childhood traffic-related air pollution exposure on asthma, allergy and sensitization: a systematic review and a meta-analysis of birth cohort studies. Allergy. 2015;70(3):245–256. PubMed PMID: 25495759.
- Gruzieva O, Bergstrom A, Hulchiy O, et al. Exposure to air pollution from traffic and childhood asthma until 12 years of age. Epidemiology. 2013;24(1):54–61. PubMed PMID: 23222555.
- Molter A, Simpson A, Berdel D, et al. A multicentre study of air pollution exposure and childhood asthma prevalence: the ESCAPE project. Eur Respir J. 2015;45(3):610–624. PubMed PMID: 25323237.
- Nishimura KK, Galanter JM, Roth LA, et al. Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188(3):309–318. PubMed PMID: 23750510; PubMed Central PMCID: PMCPMC3778732.
- Bunyavanich S, Rifas-Shiman SL, Platts-Mills TA, et al. Prenatal, perinatal, and childhood vitamin D exposure and their association with childhood allergic rhinitis and allergic sensitization. J Allergy Clin Immunol. 2016;137(4):1063–70 e2. PubMed PMID: 26874366; PubMed Central PMCID: PMCPMC4826831.
- Bunyavanich S, Rifas-Shiman SL, Platts-Mills TA, et al. Peanut, milk, and wheat intake during pregnancy is associated with reduced allergy and asthma in children. J Allergy Clin Immunol. 2014;133(5):1373–1382. PubMed PMID: 24522094; PubMed Central PMCID: PMCPMC4004710.
- Litonjua AA, Carey VJ, Laranjo N, et al. Effect of prenatal supplementation with vitamin d on asthma or recurrent wheezing in offspring by age 3 years: the VDAART randomized clinical trial. JAMA. 2016;315(4):362–370. PubMed PMID: 26813209.
- Chawes BL, Bonnelykke K, Stokholm J, et al. Effect of vitamin D3 supplementation during pregnancy on risk of persistent wheeze in the offspring: a randomized clinical trial. JAMA. 2016;315(4):353–361. PubMed PMID: 26813208.
- Bisgaard H, Stokholm J, Chawes BL, et al. Fish oil-derived fatty acids in pregnancy and wheeze and asthma in offspring. N Engl J Med. 2016;375(26):2530–2539. PubMed PMID: 28029926.
- Everson TM, Lyons G, Zhang H, et al. DNA methylation loci associated with atopy and high serum IgE: a genome-wide application of recursive Random Forest feature selection. Genome Med. 2015;7:89. PubMed PMID: 26292806; PubMed Central PMCID: PMCPMC4545869.
- Martino D, Joo JE, Sexton-Oates A, et al. Epigenome-wide association study reveals longitudinally stable DNA methylation differences in CD4+ T cells from children with IgE-mediated food allergy. Epigenetics. 2014;9(7):998–1006. PubMed PMID: 24762976; PubMed Central PMCID: PMCPMC4143415.
- Hong X, Hao K, Ladd-Acosta C, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Commun. 2015;6:6304. PubMed PMID: 25710614; PubMed Central PMCID: PMCPMC4340086.
- Liang L, Willis-Owen SAG, Laprise C, et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature. 2015;520(7549):670–674. PubMed PMID: 25707804; PubMed Central PMCID: PMCPMC4416961.
- Chen W, Wang T, Pino-Yanes M, et al. An epigenome-wide association study of total serum IgE in hispanic children. J Allergy Clin Immunol. 2017;140(2):571–577. PubMed PMID: 28069425; PubMed Central PMCID: PMCPMC5500449.
- Xu CJ, Soderhall C, Bustamante M, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med. 2018;6(5):379–388. PubMed PMID: 29496485.
- Baccarelli A, Rusconi F, Bollati V, et al. Nasal cell DNA methylation, inflammation, lung function and wheezing in children with asthma. Epigenomics. 2012;4(1):91–100. PubMed PMID: 22332661; PubMed Central PMCID: PMCPMC3297414.
- Yang IV, Pedersen BS, Liu AH, et al. The nasal methylome and childhood atopic asthma. J Allergy Clin Immunol. 2017;139(5):1478–1488. PubMed PMID: 27745942; PubMed Central PMCID: PMCPMC5391298.
- Kim YJ, Park SW, Kim TH, et al. Genome-wide methylation profiling of the bronchial mucosa of asthmatics: relationship to atopy. BMC Med Genet. 2013;14:39. PubMed PMID: 23521807; PubMed Central PMCID: PMCPMC3616917.
- Oken E, Baccarelli AA, Gold DR, et al. Cohort profile: project viva. Int J Epidemiol. 2015;44(1):37–48. PubMed PMID: 24639442; PubMed Central PMCID: PMCPMC4339753.
- Jaddoe VW, Mackenbach JP, Moll HA, et al. The generation R Study: design and cohort profile. Eur J Epidemiol. 2006;21(6):475–484. PubMed PMID: 16826450.
- Kooijman MN, Kruithof CJ, van Duijn CM, et al. The generation R study: design and cohort update 2017. Eur J Epidemiol. 2016 12;31:1243–1264. PubMed PMID: 28070760; PubMed Central PMCID: PMCPMC5233749.
- Peng C, Cardenas A, Rifas-Shiman SL, et al. Epigenome-wide association study of total serum immunoglobulin E in children: a life course approach. Clin Epigenetics. 2018;10:55. PubMed PMID: 29692868; PubMed Central PMCID: PMCPMC5905182.
- Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. PubMed PMID: 23175756; PubMed Central PMCID: PMCPMC3546795.
- Lehne B, Drong AW, Loh M, et al. A coherent approach for analysis of the illumina humanmethylation450 beadchip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015;16:37. PubMed PMID: 25853392.
- Schmiemann V, Bocking A, Kazimirek M, et al. Methylation assay for the diagnosis of lung cancer on bronchial aspirates: a cohort study. Clin Cancer Res. 2005;11(21):7728–7734. Epub2005 11 10. PubMed PMID: 16278393.
- van der Valk JP, Gerth van Wijk R, Hoorn E, et al. Measurement and interpretation of skin prick test results. Clin Transl Allergy. 2015;6:8. Epub 2016 02 26. PubMed PMID: 26909142; PubMed Central PMCID: PMC4763448.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. Epub 2012 05 10. PubMed PMID: 22568884; PubMed Central PMCID: PMC3532182.
- Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–2191. PubMed PMID: 20616382; PubMed Central PMCID: PMCPMC2922887.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B. 1995;57:289–300.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. PubMed PMID: 19131956.
- Weirauch MT, Yang A, Albu M, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158(6):1431–1443. PubMed PMID: 25215497; PubMed Central PMCID: PMCPMC4163041.
- Sonawane AR, Platig J, Fagny M, et al. Understanding tissue-specific gene regulation. Cell Rep. 2017;21(4):1077–1088. PubMed PMID: 29069589; PubMed Central PMCID: PMCPMC5828531.
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–1018. PubMed PMID: 21330290; PubMed Central PMCID: PMCPMC3065696.
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. PubMed PMID: 20110278; PubMed Central PMCID: PMCPMC2832824.
- Pedersen BS, Schwartz DA, Yang IV, et al. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28(22):2986–2988. PubMed PMID: 22954632; PubMed Central PMCID: PMCPMC3496335.
- Bakulski KM, Feinberg JI, Andrews SV, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11(5):354–362. PubMed PMID: 27019159; PubMed Central PMCID: PMCPMC4889293.
- Gaunt TR, Shihab HA, Hemani G, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17:61. PubMed PMID: 27036880; PubMed Central PMCID: PMCPMC4818469.
- Rahmani E, Zaitlen N, Baran Y, et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443–445. PubMed PMID: 27018579; PubMed Central PMCID: PMCPMC5548182.
- Nair P, Ochkur SI, Protheroe C, et al. Eosinophil peroxidase in sputum represents a unique biomarker of airway eosinophilia. Allergy. 2013;68(9):1177–1184. PubMed PMID: 23931643; PubMed Central PMCID: PMCPMC3788081.
- Rank MA, Ochkur SI, Lewis JC, et al. Nasal and pharyngeal eosinophil peroxidase levels in adults with poorly controlled asthma correlate with sputum eosinophilia. Allergy. 2016;71(4):567–570. PubMed PMID: 26645423; PubMed Central PMCID: PMCPMC4803514.
- Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787–793. PubMed PMID: 8893001.
- Sordillo JE, Kelly R, Bunyavanich S, et al. Genome-wide expression profiles identify potential targets for gene-environment interactions in asthma severity. J Allergy Clin Immunol. 2015;136(4):885–92 e2. PubMed PMID: 25913104; PubMed Central PMCID: PMCPMC4763940.
- Nan H. Genome-wide association study identifies novel alleles associated with risk of cutaneous basal cell carcinoma and squamous cell carcinoma. Hum Mol Genet. 2011;20:3718–3724.
- Farid S, Mirshafiey A, Razavi A. Siglec-8 and Siglec-F, the new therapeutic targets in asthma. Immunopharmacol Immunotoxicol. 2012;34(5):721–726. PubMed PMID: 22324980.
- Hudson SA, Herrmann H, Du J, et al. Developmental, malignancy-related, and cross-species analysis of eosinophil, mast cell, and basophil siglec-8 expression. J Clin Immunol. 2011;31(6):1045–1053. PubMed PMID: 21938510; PubMed Central PMCID: PMCPMC3329870.
- De Cauwer A, Mariotte A, Sibilia J, et al. DICER1: a key player in rheumatoid arthritis, at the crossroads of cellular stress, innate immunity, and chronic inflammation in aging. Front Immunol. 2018;9:1647. PubMed PMID: 30087677; PubMed Central PMCID: PMCPMC6066587.
- Marenholz I, Esparza-Gordillo J, Ruschendorf F, et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. 2015;6:8804. PubMed PMID: 26542096; PubMed Central PMCID: PMCPMC4667629.
- Gao PS, Shimizu K, Grant AV, et al. Polymorphisms in the sialic acid-binding immunoglobulin-like lectin-8 (siglec-8) gene are associated with susceptibility to asthma. Eur J Hum Genet. 2010;18(6):713–719. PubMed PMID: 20087405; PubMed Central PMCID: PMCPMC2987348.
- Jia Y, Yu H, Fernandes SM, et al. Expression of ligands for siglec-8 and siglec-9 in human airways and airway cells. J Allergy Clin Immunol. 2015;135(3):799–810 e7. PubMed PMID: 25747723; PubMed Central PMCID: PMCPMC4355580.
- Choi GS, Trinh HKT, Yang EM, et al. Role of clusterin/progranulin in toluene diisocyanate-induced occupational asthma. Exp Mol Med. 2018;50(5):58. PubMed PMID: 29717106; PubMed Central PMCID: PMCPMC5938014.
- Na HJ, Hamilton RG, Klion AD, et al. Biomarkers of eosinophil involvement in allergic and eosinophilic diseases: review of phenotypic and serum markers including a novel assay to quantify levels of soluble siglec-8. J Immunol Methods. 2012;383(1–2):39–46. PubMed PMID: 22683541; PubMed Central PMCID: PMCPMC3411856.
- Kiwamoto T, Kawasaki N, Paulson JC, et al. Siglec-8 as a drugable target to treat eosinophil and mast cell-associated conditions. Pharmacol Ther. 2012;135(3):327–336. PubMed PMID: 22749793; PubMed Central PMCID: PMCPMC3587973.
- Zhang Y, Jing Y, Qiao J, et al. Activation of the mTOR signaling pathway is required for asthma onset. Sci Rep. 2017;7(1):4532. PubMed PMID: 28674387; PubMed Central PMCID: PMCPMC5495772.
- Yamaki K, Yoshino S. Preventive and therapeutic effects of rapamycin, a mammalian target of rapamycin inhibitor, on food allergy in mice. Allergy. 2012;67(10):1259–1270. PubMed PMID: 22913509.
- Salari H, Yeung M, Howard S, et al. Increased contraction and inositol phosphate formation of tracheal smooth muscle from hyperresponsive guinea pigs. J Allergy Clin Immunol. 1992;90(6 Pt 1):918–926. PubMed PMID: 1460198.
- Caramori G, Casolari P, Adcock I. Role of transcription factors in the pathogenesis of asthma and COPD. Cell Commun Adhes. 2013;20(1–2):21–40. PubMed PMID: 23472830.
- Hirasawa R, Shimizu R, Takahashi S, et al. Essential and instructive roles of GATA factors in eosinophil development. J Exp Med. 2002;195(11):1379–1386. PubMed PMID: 12045236; PubMed Central PMCID: PMCPMC2193540.
- McNagny K, Graf T. Making eosinophils through subtle shifts in transcription factor expression. J Exp Med. 2002;195(11):F43–7. PubMed PMID: 12045250; PubMed Central PMCID: PMCPMC2193544.
- Fulkerson PC. Transcription factors in eosinophil development and as therapeutic targets. Front Med (Lausanne). 2017;4:115. PubMed PMID: 28791289; PubMed Central PMCID: PMCPMC5522844.
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. PubMed PMID: 14534577.
- Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–1221. PubMed PMID: 20860503; PubMed Central PMCID: PMCPMC4260321.
- van Rosmalen MGM, Li C, Zlotnick A, et al. Effect of dsDNA on the assembly pathway and mechanical strength of SV40 VP1 virus-like particles. Biophys J. 2018;115(9):1656–1665. PubMed PMID: 30301514; PubMed Central PMCID: PMCPMC6225054.
- Westhofen P, Watzka M, Marinova M, et al. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. 2011;286(17):15085–15094. PubMed PMID: 21367861; PubMed Central PMCID: PMCPMC3083210.
- Xue J, Schmidt SV, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. PubMed PMID: 24530056; PubMed Central PMCID: PMCPMC3991396.
- Jahreis S, Trump S, Bauer M, et al. Maternal phthalate exposure promotes allergic airway inflammation over 2 generations through epigenetic modifications. J Allergy Clin Immunol. 2017. DOI:10.1016/j.jaci.2017.03.017. PubMed PMID: 28392331.
- Hong X, Ladd-Acosta C, Hao K, et al. Epigenome-wide association study links site-specific DNA methylation changes with cow’s milk allergy. J Allergy Clin Immunol. 2016;138(3):908–11 e9. PubMed PMID: 27236499; PubMed Central PMCID: PMCPMC5392112.
- Michel S, Busato F, Genuneit J, et al. Farm exposure and time trends in early childhood may influence DNA methylation in genes related to asthma and allergy. Allergy. 2013;68(3):355–364. PubMed PMID: 23346934.
- Vuillermin PJ, Ponsonby AL, Saffery R, et al. Microbial exposure, interferon gamma gene demethylation in naive T-cells, and the risk of allergic disease. Allergy. 2009;64(3):348–353. PubMed PMID: 19210359.
- Arana E, Harwood NE, Batista FD. Regulation of integrin activation through the B-cell receptor. J Cell Sci. 2008;121(Pt 14):2279–2286. PubMed PMID: 18596256.
- Beggs AH, Byers TJ, Knoll JH, et al. Cloning and characterization of two human skeletal muscle alpha-actinin genes located on chromosomes 1 and 11. J Biol Chem. 1992;267(13):9281–9288. PubMed PMID: 1339456.
- Bell SM, Lam WK, Carr IM, et al. Assignment of the murine adenomatous polyposis coli 2 (Apc2) gene to mouse chromosome band 10B5-C2 by in situ hybridisation. Cytogenet Cell Genet. 1999;86(1):81–82. PubMed PMID: 10516441.
- Fehrenbach H, Wagner C, Wegmann M. Airway remodeling in asthma: what really matters. Cell Tissue Res. 2017;367(3):551–569. PubMed PMID: 28190087; PubMed Central PMCID: PMCPMC5320023.
- Hirota N, Martin JG. Mechanisms of airway remodeling. Chest. 2013;144(3):1026–1032. PubMed PMID: 24008953.
- Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol. 2011;128(3):451–462. quiz 63–4. PubMed PMID: 21636119.
- Davies DE, Wicks J, Powell RM, et al. Airway remodeling in asthma: new insights. J Allergy Clin Immunol. 2003;111(2):215–225. quiz 26. PubMed PMID: 12589337.
- Holgate ST, Yang Y, Haitchi HM, et al. The genetics of asthma: ADAM33 as an example of a susceptibility gene. Proc Am Thorac Soc. 2006;3(5):440–443. PubMed PMID: 16799089.
- Wittig HJ, Belloit J, De Fillippi I, et al. Age-related serum immunoglobulin E levels in healthy subjects and in patients with allergic disease. J Allergy Clin Immunol. 1980;66(4):305–313. PubMed PMID: 7419833.
- Klink M, Cline MG, Halonen M, et al. Problems in defining normal limits for serum IgE. J Allergy Clin Immunol. 1990;85(2):440–444. PubMed PMID: 2303647.