
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
DNA methylation in CpGs dinucleotides is associated with high mutability and disappearance of CpG sites during evolution. Although the high mutability of CpGs is thought to be relevant for vertebrate evolution, very little is known on the role of CpG-related mutations in the genomic diversification of vertebrates. Our study analysed genetic differences in chickens, between Red Junglefowl (RJF; the living closest relative to the ancestor of domesticated chickens) and domesticated breeds, to identify genomic dynamics that have occurred during the process of their domestication, focusing particularly on CpG-related mutations. Single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) between RJF and these domesticated breeds were assessed in a reduced fraction of their genome. Additionally, DNA methylation in the same fraction of the genome was measured in the sperm of RJF individuals to identify possible correlations with the mutations found between RJF and the domesticated breeds. Our study shows that although the vast majority of CpG-related mutations found relate to CNVs, CpGs disproportionally associate to SNPs in comparison to CNVs, where they are indeed substantially under-represented. Moreover, CpGs seem to be hotspots of mutations related to speciation. We suggest that, on the one hand, CpG-related mutations in CNV regions would promote genomic ‘flexibility’ in evolution, i.e., the ability of the genome to expand its functional possibilities; on the other hand, CpG-related mutations in SNPs would relate to genomic ‘specificity’ in evolution, thus, representing mutations that would associate with phenotypic traits relevant for speciation.
Introduction
The emergence of novel genomic conformations is a fundamental process in evolution that has been the focus of recent high-profile studies [Citation1–Citation5]. One of the main challenges in evolution is to understand how genomes diversify. Domesticated organisms are ideal to study this process because domestication represents recent and trackable events of diversification that have radically changed phenotypes and genotypes over short time spans [Citation6]. Although domestication has been used as a proof-of-principle for evolution since Darwin, it has only recently started to be explored as a way to disentangle the genetic mechanisms underlying evolutionary processes. Particularly, chicken domestication has led to marked phenotypic differences between breeds and to the accumulation of mutations in each of them [Citation5]. Domesticated breeds of chickens are acknowledged to have originated from wild populations of Red Jungle Fowl (RJF) [Citation7,Citation8]. This domestication process is thought to have started recently, between 5,400 [Citation9] to 3,000 [Citation8] years ago in Southeast Asia. Currently, a wild form of RJF still inhabits Southeast Asia and regions of China and India [Citation8]. RJF is considered as the living variety of chickens that is the closest relative to the common ancestor of all known chicken breeds [Citation7,Citation8]. Considering the proximity of living RJF to the ancestor of all chickens and the ample variety of existing domesticated types [Citation8], the process of chicken diversification is a valuable model to study mechanistic aspects of genomic diversification in vertebrates.
Until recently it was common in evolutionary biology to assume genomic changes related to evolutionary novelties were mainly generated by random (stochastic) mutation events [Citation10,Citation11], which occurred independently from environmental influences [Citation12]. One line of thought to have challenged this assumption is the ability of epigenetic modifications to influence genomic variability [Citation13].
The influence of epigenetic state on mutation rate has, for example, been demonstrated in cytosines of 5ʹ to 3ʹoriented CG dinucleotides. These so-called CpG sites are prone to DNA methylation, an enzymatic reaction in which methyl groups (-CH3) are added to the carbon 5 of their cytosine ring [Citation14]. Interestingly, a methylated cytosine is one reaction (hydrolytic deamination) away from a complete mutation to a thymidine, a conversion that can even occur spontaneously [Citation15]. CpG to TpG transitions occur with much higher frequency than any other point mutation and depend on the methylation status. Estimations show that DNA methylation in CpGs increase CpG to TpG mutation rates by ~12 fold [Citation16–Citation19]. The increase is even higher in the male germ line [Citation20]. In Escherichia coli, experiments have shown CpGs are hotspots for mutations only when methylated [Citation15]. The hypermutability of CpGs is suggested to have influenced their deficiency in vertebrate genomes [Citation21,Citation22] and reported to be important in the evolution of transcription factor binding sites [Citation23]. In mammals, CpG-related mutations have influenced the well-known higher rate of transitions relative to transversions [Citation24] and contributed to the evolution of the BRCA1 gene [Citation16]. Additionally, CpGs are known to influence the mutation rates of neighbouring non-CpG DNA [Citation25].
CpG mutation rates in natural populations could be influenced by environmental exposures affecting their methylation status [Citation26]. Interestingly, since epigenetic alterations can be maintained for several generations (at least eight in plants) [Citation27,Citation28], there are many opportunities for CpGs with altered methylation patterns to mutate to TpGs. In addition to biasing point mutations, DNA methylation in CpGs is also involved in the generation of larger genomic rearrangements. DNA methylation regulates the activity of transposable elements [Citation29], which in turn impact the formation of new genomic arrangements such as insertions, deletions or duplications [Citation30]. Recent evidence shows that transposition of repetitive elements had a crucial role in the genome diversification resulting from the radiation of African cichlid fish [Citation1]. In spite of the known mechanistic connection between CpG methylation and genetic mutations, the precise role that CpG-related mutations play in evolution and genomic diversification is not known.
Studies in Darwin finches [Citation31] and darter fish [Citation32] show that differentiation between methylomes is greater than genetic divergence among closely related populations of animals. These studies, however, have the limitation of not addressing epigenetic changes solely in the germ line. Our previous study in Finches evaluated the methylome of red blood cells [Citation31], while Smith et al. [Citation32] investigated the methylome in ovaries, which contain both somatic and germ cells. DNA methylation differences in lungs of chickens from Fayoumi and Leghorn lines suggest the implication of DNA methylation in differential immune responses among these lines [Citation33]. Although it is interesting to find a correlation between somatic epigenetic marks and evolution, causation between epigenetic changes and genomic changes can only be established when the methylome is assessed in the germ line. Previous research comparing the sperm methylome obtained from one chicken to a publicly available human sperm methylome map suggests that sperm CpG hypermutability has strongly impacted the evolution of GC content of vertebrates [Citation34].
Using the model of chicken diversification, the present study addresses the role of CpG sites in generating genetic diversity in the form of Single Nucleotide Polymorphisms (SNPs) and Copy Number Variations (CNVs). We address in a number of chickens of different breeds the extent the chicken genome has been affected by CpG-related mutations. For this, we have analysed the dynamics of CpG-related mutations between RJF (in its condition of closest related to the ancestor) and four domesticated breeds, including broiler (BRL) and three heritage breeds from different regions of Sweden: Kindahöna (KIN), Hedemorahöna (HED) and Gotlandshöna (GOT).
Additionally, we investigated correlations between the germ line methylome of RJF and CpG-related mutations. Assuming the germ line methylome of current and past populations of RJF are highly similar, we evaluated whether the germ line methylome of current RJF specimens exhibits associations with the genomic variation observed between RJF and selected domesticated breeds. We focused on the germ line because germ line mutations can have transgenerational consequences, making them suitable to pinpoint the role of DNA methylation in generating genomic novelty. Both environmental exposures and DNA methylation changes have been associated with germ line mutations. For example, germ line mutations in rodents are shown to be influenced by environmental factors and subsequently carried over through generations, producing genomic variability representing evolutionary novelty [Citation35,Citation36]. Also in rodents, environmentally induced transgenerational changes in DNA methylation in the germ line associated with increased copy number variations (specifically, genomic gains) three generations after the exposure [Citation37]. In human sperm, Alu insertions exhibit CpG hypomethylation in their flanking regions [Citation29], suggesting CpGs are involved in regulating Alu transposition, which could induce CNVs.
From a biological and evolutionary perspective, it is fundamental to understand mutation dynamics in genomic regions that on one hand are susceptible to environmental influences, and on the other hand influence mutation rates, such as CpG sites. Taking advantage of the recent and well-studied diversification process that has originated domesticated chicken breeds, we evaluated how CpG-related mutations could have influenced genomic variability between RJF and four domesticated breeds. Additionally, we addressed correlations between germ line DNA methylation in RJF and the emergence of SNPs and CNVs in the domesticate breeds studied. The present study is the first to evaluate CpG mutation dynamics to this depth in the context of vertebrate genomic diversification.
Results
General sequencing parameters of genotyping
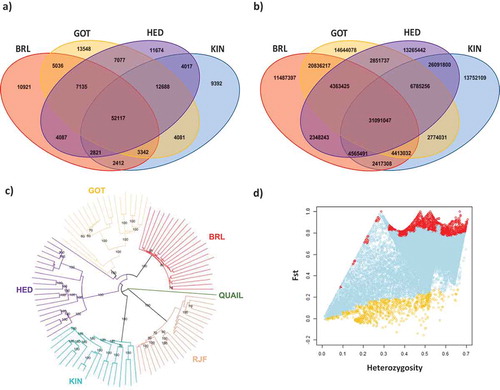
To identify SNPs and CNVs emerging between the domesticated chicken breeds and RJF we used Genotype-by-Sequencing, which we have recently described in chickens [Citation38]. This is an approach that enzymatically reduces the genome (with PstI) and is unbiased for CpG density. PstI digestion reduced the genome to a sequenced fraction of ~2.2%. Approximately 112 million reads were retained per library (3 libraries) after quality trimming by SeqyClean [Citation39]. Approximately 106 million reads were retained after application of the Tassel filter (reads >64 bp and properly identified with barcodes). The number of unique sequence tags that aligned against the chicken reference genome (Gallus gallus 4.0, NCBI) was ~1.1 million and 91.2% of them could be mapped (detailed coverage per breed is shown in ). We identified SNPs and CNVs common to all breeds (thus, unrelated to divergence), as well as breed-specific SNPs and CNVs that could be relevant for the diversification of the chicken breeds studied here. A total of 150,348 SNPs was identified when comparing the PstI-reduced genomes of individuals from the domesticated breeds to the same fraction in RJF (used as reference). Among these SNPs, 30.29% were breed specific. ) shows a Venn-diagram with the number of SNPs detected in each domesticated breed in relation to RJF. Additionally, CNVs were identified in the PstI-reduced genomes of individuals from the domesticated breeds compared to the same fraction in RJF (used as reference). These are presented as base pairs covered by CNVs emerging between RJF and the domesticated breeds, which allowed us to compare CNV regions to CpG locations. A total of 161,686,613 bp was identified in regions of CNVs, with 32.9% of these base pairs being breed specific. ) shows a Venn-diagram with the number of base pairs covered by CNVs emerging in each domesticated breed in relation to RJF.
Table 1. Summary of sequencing coverage in the chicken breeds analysed, in relation to the reference genome.
Figure 1. Categorization of SNPs and CNVs found among domesticated breeds of chickens and RJF. (a) Venn diagram showing SNPs emerging between the domesticated breeds analysed and RJF. (b) Venn diagram showing base pairs covered by CNVs emerging between the domesticated breeds analysed and RJF. (c) SNP based phylogenetic tree of the chicken breeds studied here, constructed with neighbour joining and using the Japanese quail as the outgroup. (d) Schematic representation of the Fst analysis against heterozygosity performed among the breeds studied here; dots with different colours represent SNPs evolving under balancing selection (yellow), neutral evolution (light blue), or positive selection (red).
Relatedness analysis
To determine if RJF indeed locates in a position of ancestry in relation to the other domesticated breeds analysed, a relatedness cladogram was generated using the Japanese quail as the out group. Neighbour-joining clustering was performed on 34,219 common SNPs identified between the Japanese quail and the chicken breeds analysed.
As expected, the results indicate that the current RJF is the closest relative to quails, in a position of ancestry in relation to all other domesticated breeds included ()). Moreover, also expectedly, BRL appears as the most derived breed. The analysis correctly clustered each individual within the corresponding clade, which indicates the appropriateness of using GBS-generated sequencing data for inferring phylogenetic correlations. Our analysis also shows that among the Swedish breeds evaluated, KIN is the closest related to RJF, while GOT is the most distant (. Inbreeding coefficients, Fst, were estimated in the same subset of SNPs (common between the chickens and the Japanese quail) to identify genes that exhibit adaptive divergence in their allelic frequency, which is indicative of natural selection among sub-structured populations [Citation40]. The Fst coefficients obtained show that almost all SNPs detected between the derived breeds and RJF emerged under neutral evolution (97.5%), while only 2% evolved under balancing selection and 0.6% due to positive selection (.
Relation of CpGs to mutations
We then identified the SNPs and CNVs that occurred specifically in overlapping positions to CpGs observed in the sequenced PstI-reduced RJF genome and created the subgroups CpG-SNPs and CpG-CNVs, respectively. A total of 14,298 CpG-SNPs was found, representing 1.71% of all the CpGs analysed (835,182 CpGs). 9.51% of all SNPs are related to CpGs, which is 52.16% above the expected value (P < 0.001, Chi-Square; Suppl Table S1). We defined the expected value (1/16) as the probability that CpG dinucleotides would contribute the same as other dinucleotides to SNP formation (i.e., 1 out of all 16 possible dinucleotide combinations). Thus, our results show CG dinucleotides disproportionally influence SNP formation. Interestingly, the more genetically distant the breed is from RJF, the more CpG-SNPs are present above expectancy. In BRL, CpG-SNPs presence is as high as 100.7% above expectancy (P < 0.001, Chi-Square; . This suggests CpGs, in addition to being hotspots of mutations [Citation15–Citation19], are hotspots of SNPs that are relevant for genomic speciation. While 31.3% of CpG-SNPs are breed-specific, the fractions of breed-specific CpG-SNPs in relation to all CpG-SNPs are 8.05% in KIN, 9.2% in HED, 9.4% in GOT and 12.54% in BRL (Suppl Table S1). A Venn-diagram showing the number of CpG-SNPs detected in each domesticated breed in relation to RJF is shown in .
Figure 2. Categorization of CpG-SNPs and CpG-CNVs found among domesticated breeds of chickens and RJF. (a) Plot depicting the incidence of CpG-SNPs in the domesticated breeds in relation to RJF (* depicts P < 0.001; Chi-Square). (b) Venn diagram showing CpG-SNPs emerging between the domesticated breeds analysed and RJF. (c) Venn diagram showing base pairs covered by CpG-CNVs emerging between the domesticated breeds analysed and RJF.
CpG-CNVs were defined as counts of CpGs present in base pairs covered by CNVs emerging between RJF and the domesticated breeds. A Venn-diagram with the CpG-CNVs detected in each domesticated breed in relation to RJF is shown in . A total of 285,097 CpG-CNVs was identified, with 23.6% of these being breed-specific (Suppl Table S2). This represents 34.1% of all the CpGs analysed (835,182 CpGs). Thus, we identified nearly 20 times more CpG-CNVs than CpG-SNPs. In contrast to the pattern observed for CpG-SNPs, CpGs are highly under-represented in CNVs in relation to expected values (approximately 98% decrease; Suppl Table S2), and the percentage of decrease in CpG-CNVs expectancy per breed is independent of genetic relatedness to RJF (Suppl Table S2).
Types of mutations emerging in domesticated breeds
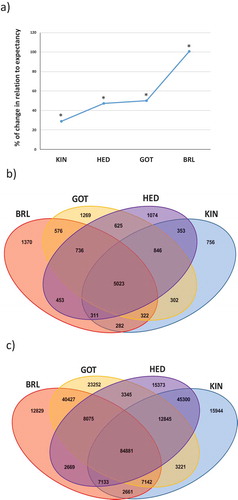
We investigated which point mutations emerged from SNPs occurring in C positions in general (C-SNPs) or in Cs neighbouring Gs (i.e., CpG sites). In each case we considered whether in RJF the C position involved a reference allele (i.e., when C is exclusive or majority in a specific position compared to other bases), represented by C/N, or an alternate allele (i.e., when C is minority in a specific position compared to other bases), represented by D/C. The nomenclature D/C was chosen instead of N to highlight that it represents a degenerate base with a minority presence Cs. Choosing N would have included the case when C is degenerate and majority. The nomenclature C/N, in turn, include Cs fixed as well as Cs present in majority. Remarkably, in all scenarios tested progressive changes in mutation patterns were observed that are concordant with the genetic relatedness of each breed to RJF. Mutation patterns are different depending on whether the mutation originated from C/N or D/C. When SNPs originate from C/N only C/T increases progressively with reduced genetic relatedness to RJF, while T/C, and fixed Cs and Gs decrease progressively ()). However, when SNPs originate from alternate Cs, both C/T and T/C increase progressively with reduced genetic relatedness to RJF, mainly at the expense of fixed Cs and Gs ()). Thus, our results demonstrate mutations in alternate Cs generate a different outcome than mutations in reference Cs.
Figure 3. Types of mutations emerging in domesticated breeds in relation to RJF. (a) SNPs emerging from Cs as reference base (i.e., C/N) in RJF. (b) SNPs emerging from Cs as an alternate base (i.e., D/C) in RJF. (c) SNPs emerging from CpGs with C as reference base (i.e., C/N) in RJF. (d) SNPs emerging from CpGs with C as an alternate base (i.e., D/C) in RJF.
The same analysis was performed considering SNPs related to true CpGs (CpG-SNPs) or to D/CpGs (D/CpG-SNPs). This was based on the expectation that methylation-prone Cs (i.e., in CpG sites) would have different mutation patterns than Cs that are not prone to be methylated. Marked differences were observed in the mutation patterns between CpG- and D/CpG-SNPs. In true CpG-SNPs C/T increases substantially as genetic relatedness to RJF decreases, which occurs at the expense of fixed Cs and Ts ()). A completely different pattern is observed in D/CpG-SNPs, which are progressively substituted by A/G (mainly) and C/T (to a lesser extent) as genetic relatedness to RJF decreases, which occurs at the detriment of all the fixed bases ()). Our results show it is not only important in genomic dynamics whether Cs are within CpG sites but also their status as alternate or reference base, since the mutation patterns differ considerably between these two statuses.
Functional annotation of mutations
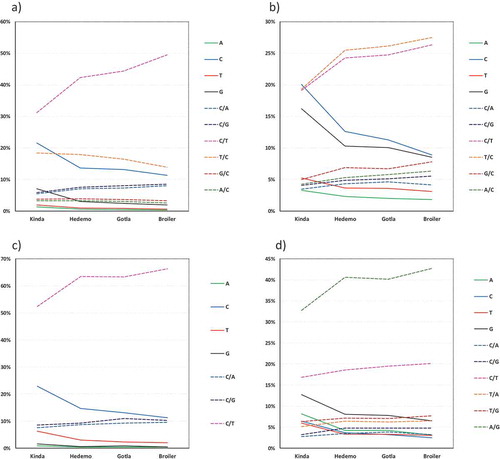
We performed functional genomic annotation of the SNPs, CpG-SNPs, CNVs and CpG-CNVs found, to determine which genomic regions are affected in each case. For this analysis, we created more subgroups in order to identify potential differences between mutations that are (i) common to all breeds (named breed-common SNPs, CpG-SNPs, CNVs or CpG-CNVs) or (ii) specific to each breed (named breed-specific SNPs, CpG-SNPs, CNVs or CpG-CNVs). A general observation that holds for both SNPs and CNVs is that the vast majority of them relate to intergenic regions, followed by intronic regions ( and . However, the regions that contribute the most to the Chi-square significance differ substantially across groups (). Association with coding regions increases for CpG-SNPs, particularly for breed-specific SNPs, compared to all SNPs (; ). As for CNVs, alterations in coding regions share importance with alterations in promoters. A large increase in the association with coding regions is observed for CpG-CNVs, particularly for breed common CpG-CNVs, compared to all CNVs (; ).
Table 2. Summary of functional annotation of CpG-related mutations observed between domesticated breeds and RJF.
Figure 4. Functional annotation of mutations found among domesticated breeds of chickens and RJF: (a) SNPs; (b) CNVs.
Relation of mutations to repetitive elements
We performed Repeat Masker analyses on the SNPs, CpG-SNPs, CNVs and CpG-CNVs found, in order to identify overlaps with categories of repetitive elements. In general, we found a slightly higher representation of repetitive elements in relation to CNVs than to SNPs ()). In the particular case of breed-common mutations, this over-representation is nearly twofold in CNVs compared to SNPs. In contrast, when focusing only on CpG-related mutations, a higher representation of repetitive elements in breed-specific CpG-SNPs is found compared to breed-specific CpG-CNVs ()).
Figure 5. Relation of mutations found among domesticated breeds of chickens and RJF to repetitive elements. (a) summarized comparison between SNPs and CNVs. (b) relation of SNPs to repetitive elements. (c) relation of CNVs to repetitive elements.
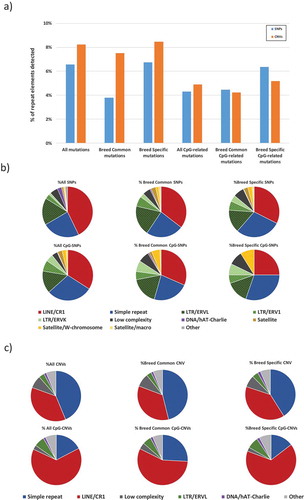
When analysing the detailed composition of repetitive elements overlapping with the SNPs obtained, we observed an overall decrease of LINE/CR1 in CpG-SNPs compared to SNPs. Also, an increased representation of some LTR elements (ERV1 and ERVK) is evidenced in CpG-SNPs compared to all SNPs. An increase in low complexity repeats is also observed in breed-specific SNPs compared to all SNPs and breed-common SNPs. When focusing only on CpG-SNPs, an increase in simple repeats in breed-specific CpGs-SNPs is observed compared to all CpGs and breed-common CpGs ()). It is also important to highlight that the LTR elements ERV1 and ERVK that are present in SNPs are absent in CNVs. One striking difference observed is the much higher presence of breed-specific CpG-SNPs in satellite elements in the W chromosome, compared to all SNPs and to CpG-SNPs.
By looking at the detailed composition of repetitive elements of the CNVs obtained, one noticeable observation is that LINE/CR1 are generally overrepresented in CpG-CNVs (particularly in breed-specific), which occurs at the expense of simple repeats. Also, an increase of LTR/ERVL in CpG-CNVs (particularly in breed-specific) is observed compared to all CNVs ()).
Relation of CpG-related mutations to DNA methylation in RJF sperm
Based on the observations that mutation dynamics differ between Cs in or outside CpGs, we inquired into potential correlations between DNA methylation levels in the sperm of our RJF population (from ejaculates) and CpG-related mutations. For the DNA methylation analysis, we used a novel combination of methods (Genotyping-by-Sequencing and Methylated DNA immunoprecipitation) that we developed previously [Citation41]. RJF sperm DNA methylation was also measured in the PstI-reduced genome, thus we compared them to mutations in the same genomic fraction. Although we cannot assert that DNA methylation in RJF sperm has directly influenced CpG mutation rates in the domesticated breeds evaluated here, we rationalized that the analysis of DNA methylation from sperm from our RJF population could give a good approximation of ancestral sperm DNA methylation patterns. We investigated whether two variables in RJF sperm DNA methylation, namely i) DNA methylation levels, and ii) associated inter-individual variability, correlated to the SNPs or CNVs observed between RJF and the domesticated breeds. The methylated fraction of the PstI-reduced sperm RJF genome was captured by Methylated DNA immunoprecipitation (PstI-MeDIP), as previously described [Citation41]. Sequencing of this PstI-MeDIP fraction resulted in an average of 1.4 million thousand reads per individual (N = 20) aligned against the chicken reference genome (Gallus_gallus 4.0, NCBI). This corresponds to 80.7% of the sequenced reads. In average, 227 million bps were sequenced, spanning 7 million unique genomic positions covered at 22.3 ± 11.3 X per individual. This corresponds to 0.6% of the total chicken genome per individual. To identify CpG sites to be analyzed, we merged the data from 20 RJF individuals. This merging resulted in 2.9 billion bps being analyzed, spanning 35 million unique genomic positions covered at 81.8X in average. This corresponds to 3.34% of the total chicken genome. The merging allowed to identify 835.182 CpGs, which corresponds to 7.8% of all CpGs within the chicken genome. Of these, 572,066 CpGs (68.5%) presented some degree of methylation in at least one individual and were therefore selected for further analyses.
The measured sperm DNA methylation was categorized according to i) the level of DNA methylation, and ii) the variation in DNA methylation across the individuals studied (inter-individual variation). ) shows the level of DNA methylation (represented by the normalized number of reads by which each CpG is covered) versus the inter-individual variation in DNA methylation in these CpGs.
Figure 6. Association of CpG-related mutations in domesticated breeds with DNA methylation in RJF sperm. (a) Inter-individual DNA methylation variation plotted against the level of DNA methylation, measured per window, and grid representation of the division of CpGs in nine DNA methylation categories. (b) Pie representation of the number of CpGs within each DNA methylation category in sperm RJF. (c) Representation of the subgroups of CpGs that relate to mutations (CpGs or CNVs) in each domesticated breed. (d) Pie representations of DNA methylation categories of the subgroups of CpGs measured for DNA methylation in sperm RJF that relate to SNPs (CpG-SNPs) or to CNVs (CpG-CNVs) in the domesticated breeds. (e) Bar plots summarizing how levels and inter-individual variation of DNA methylation in RJF sperm associate with mutations (SNPs or CNVs) in the domesticated breeds.
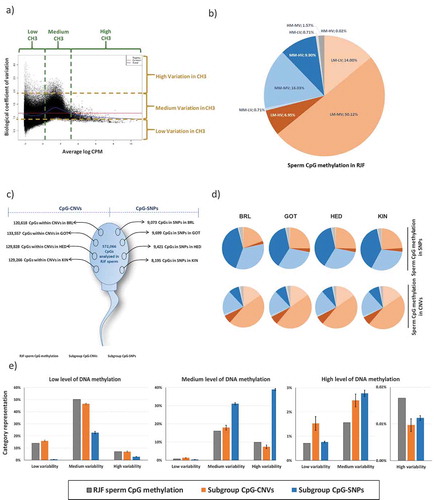
Nine categories were defined for these CpGs based on their DNA methylation levels and inter-individual variation: Low Methylation-Low Variation (LM-LV); Low Methylation-Medium Variation (LM-MV); Low Methylation-High Variation (LM-HV); Medium Methylation-Low Variation (MM-LV); Medium Methylation-Medium Variation (MM-MV); Medium Methylation-High Variation (MM-HV); High Methylation-Low Variation (HM-LV); High Methylation-Medium Variation (HM-MV); High Methylation-High Variation (HM-HV). The criteria for the definition of the limits of these categories are described in detail in the Materials and Methods section. CpG proportions across DNA methylation categories in RJF sperm are shown in ).
The majority of CpGs detected in the methylated fraction of the PstI-reduced fraction of the sperm genome are LM-MV (50.12%), followed by MM-MV (16.03%), and LM-LV (14%). We then investigated if DNA methylation status in the male germ line of RJF would associate to the mutations (SNPs and CNVs) observed between RJF and the domesticated breeds. To determine this, we selected CpG sites that were in the same genomic locations where SNPs (i.e., CpG-SNPs) or CNVs (i.e., CpG-CNVs) emerged between RJF and the domesticated breed. The number of CpGs in which DNA methylation was evaluated in the sperm of RJF, and the number of CpG-SNPs and CpG-CNVs obtained in each breed are shown in ).
We hypothesized that if no correlation between RJF sperm DNA methylation and SNPs or CNVs exists, then the distribution of CpGs across the nine DNA methylation categories in the RJF sperm (all sperm CpGs measured; reference pattern) is expected to be the same as in the CpG-SNPs or CpG-CNVs subgroups (in each domesticated breed). Interestingly, our results show major differences in the distribution of CpGs across the nine DNA methylation categories between the reference pattern (); all CpGs measured in RJF sperm) and the CpG-SNPs subgroups (), top panel). For instance, when considering all CpGs in RJF sperm, the largest category of CpG methylation is LM-LV, while in the subgroups of CpG-SNPs in the domesticated breeds the largest category is MM-HV. Furthermore, the distribution is altered in relation to the reference value in a consistent manner in all domesticated breeds, which represent independent replicates. In contrast, in the CpG-CNVs subgroups, the distribution of CpGs across methylation categories in the domesticated breeds is similar to the reference value (), lower panel). Thus, patterns of CpG methylation differ considerably between the CpG-SNPs and CpG-CNVs subgroups, with the latter being similar to the reference value.
We then tested which of the DNA methylation variables studied (i.e., DNA methylation levels or inter-individual variation) were altered the most between all RJF sperm CpGs (reference pattern) and the subgroups of CpGs related to mutations (SNPs or CNVS). For this, we compared the amount of CpGs in each DNA methylation category within the CpG-SNPs or CpG-CNVs subgroups against the reference value in RJF sperm (.
We found that CpGs in RJF sperm with medium levels of DNA methylation together with medium (P < 0.001) to high levels (P < 0.001) of inter-individual variation are substantially overrepresented in the CpG-SNPs subgroups, as well as CpGs with high levels of DNA methylation and medium variation (P < 0.001) (. Conversely, CpGs in RJF sperm with high levels of DNA methylation and low (P < 0.05) to medium (P < 0.05) variation are overrepresented in the CpG-CNVs subgroups, as well as CpGs with medium levels of DNA methylation and low variation (P < 0.005) ()). Interestingly, the combination of high methylation with medium variation is overrepresented in both subgroups. For both CpG-SNPs and CpG-CNVs the significant increases in some categories are compensated by decreases in other categories. In summary, the majority of CpG-SNPs associate with medium levels of methylation and medium/high variation in RJF sperm, while the majority of CpG-CNVs associate with high levels of methylation and low/medium variability in RJF sperm.
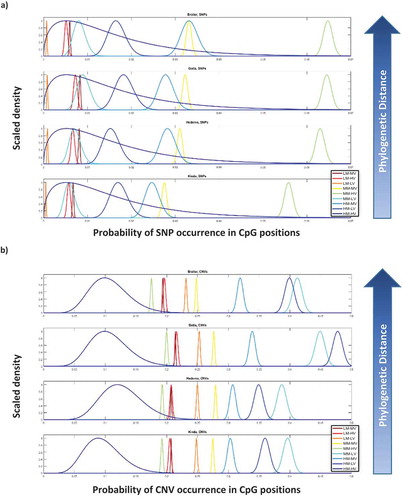
Based on these associations between DNA methylation features in RJF sperm and mutations emerging in specific breeds, we inquired about the mutation probabilities of CpGs within each DNA methylation category. We implemented Bayesian inference and calculated posterior distributions of mutation probabilities (separately for SNPs and CNVs) for the RJF sperm CpGs methylation categories, and observed substantial differences among them, both in relation to SNPs ()) and CNVs (. Additionally, the patterns of these mutation probabilities are different when related to SNPs or CNVs. A very interesting finding is that in two of the methylation categories, MM-HV and MM-MV, the CpG to SNP mutation rate increases as genetic relatedness to RJF decreases (, while none of the categories exhibit this pattern in CpGs related to CNVs (.
Figure 7. Posterior distributions (scaled to give equal rights to all density curves) of mutation probabilities of CpGs within the sperm RJF DNA methylation categories, in relation to (a) SNPs; (b) CNVs.
Discussion
Genomic divergence related to speciation has historically been associated with changes in allele frequencies in response to adaptation [Citation42,Citation43]. New knowledge in genomics, however, portraits a much more complex scenario of genomic dynamics that occur during speciation in which many other factors, including genetic drift and biased mutations, have an important role in shaping genetic diversity [Citation44]. Many fundamental questions remain unanswered in speciation genetics, such as which genetic elements are relevant? how many loci are involved? where are the genomic determinants located? or what is the role of rearrangements, gene conversions and other molecular changes? [Citation43] In the present paper, we studied genomic dynamics related to CpG sites by focusing on the recent process of chicken diversification and using a reduced genome approach unbiased for CpG density (GBS). Importantly, CpG sites are on the one hand susceptible to undergo methylation [Citation45], and on the other hand relate to biased mutations [Citation16–Citation19]
Our study identified SNPs and CNVs common to all breeds (thus, unrelated to their divergence), as well as breed-specific SNPs and CNVs that could be relevant for the diversification of the chicken breeds studied here. RJF was confirmed as the most ancestral of the breeds analysed, while BLR was found to be the most derived. Among the Swedish breeds, KIN was classified as the closest to RJF, while GOT was the most distant. The fact that the neighbour joining analysis correctly clustered each individual within the corresponding clade indicates the appropriateness of using GBS-generated sequencing data for inferring phylogenetic correlations. GBS has been used previously to infer domestication scenarios in Lima Bean (Phaseolus lunatus L.) in Mesoamerica [Citation46]. The vast majority of the SNPs identified between our chicken breeds and the Japanese quail are shown to have evolved under neutrality (97.5%), while only 0.6% evolved due to positive section. This is concordant with previous estimates in humans showing that virtually all of the genome has evolved under neutrality, while the occurrence of positive selection is rare [Citation47].
We then inquired about the contribution of CpGs in relation to the SNPs and CNVs identified. We found that 9.51% of all SNPs are related to CpGs, which is above the expected value of 1/16 (i.e., 6.25%). This is the probability of occurrence of CG dinucleotides among all dinucleotides, which is what is expected in case all dinucleotides would contribute equally to SNP formation. Thus, CG dinucleotides disproportionally influence SNP formation. Moreover, the more distant the breed is from RJF, the more SNPs emerge from CpGs, reaching the value of 100.7% above expectancy for broilers. This suggests CpGs, in addition to being hotspots of mutations [Citation15–Citation19], are hotspots of SNPs that are relevant for genomic speciation. CpGs in CNVs, on the other hand, are highly under-represented, being present at less than 2% of their expected value, and their presence is independent of genetic relatedness to RJF.
Next, we investigated if SNPs mutation dynamics are different depending on whether mutations emerge from fixed or alternate bases. Overall, as genetic relatedness to RJF decreases, substitutions emerging from either C-SNPs or CpG-SNPs tend to produce a progressive disappearance of fixed Cs correlated and the appearance of degenerated C/T bases. This is consistent with the tendency of CpGs to mutate to TpGs [Citation16–Citation19] and indicates C to T substitutions are very prone to occur in a context of diversification, being influenced by other factors in addition to the hypermutability of CpG sites. The observed increased C to T mutability related to phylogenetic distance could have an important causative role in species diversification, a possibility that would need further investigation.
However, our results show it is not only important in genomic dynamics whether Cs are within CpG sites or not but also their status as alternate or reference base. Mutations in alternate Cs generate a different outcome than mutations in reference Cs. Although both reference and alternate Cs tend to mutate to T, in positions where Cs are alternate Gs substantially disappear by mutating to C, which is reflected in the concomitant raise of degenerate G/C bases. Although it is expected that C and G mutations trend together due to being complementary bases, it is somehow surprising that Gs tend to generate Cs. This substitution represents a transversion, which are less common than transitions [Citation48]. Since transitions are known to be influenced by CpGs [Citation24], it is possible that other epigenetic mechanisms might induce transversions.
We also observed mutation differences between true CpGs and D/CpGs. Mutations from true CpGs expectedly produce degenerated C/T positions, while mutations from D/CpGs are mainly related to A/G degenerate bases that progressively increase at the expense of fixed As and Gs. This confirms previous observations showing that CpGs mutate significantly different than other positions, being important for the over-representation of transitions relative to transversions in mammalian genomes [Citation24]. Overall, our mutation analyses reveal a general trend of disappearance of fixed Cs and Gs as genetic diversification from RJF increases. It remains to be seen if this pattern occurs in the genetic diversification of other vertebrates. This trend is i) concomitant with the appearance of degenerate bases (i.e., C/T, G/C and A/G), ii) dependent on the alternate/reference status of the base, and iii) dependent on whether mutations relate to CpG sites. In the particular case of CpGs, mutations in true CpGs expectedly produce degenerated C/T positions, while mutations in D/CpGs are mainly related to A/G degenerate bases that progressively increase at the expense of fixed As and Gs. The observed overall increase in degenerate bases due to the higher mutability of Cs in relation to phylogenetic distance could have an important, and maybe causative, role in genome diversification associated with speciation. The possibility that this trend would also occur in the genomic evolution of other organisms needs to be investigated.
Functional annotation of the mutations observed revealed that SNPs and CNVs are mostly present in intergenic regions, followed by intronic regions. Intergenic regions include regulatory elements such as exons, splice junctions, and proper genes [Citation49]. Mutations in intergenic regions may affect so-called ‘Dark Matter’ transcripts, which are mostly located near genes and associate with regulatory elements such as alternative cleavage or polyadenylation sites, promoter- and terminator-associated transcripts, as well as alternative exons [Citation50]. Meanwhile, mutations in intronic regions may affect splicing with consequences for RNA processing [Citation51]. Mutations in splicing sites are associated with human diseases such as cancers [Citation52], and neurological [Citation53,Citation54] and metabolic [Citation55] disorders. The evolutionary relevance of mutations in intergenic and intronic regions of the genome deserves further investigation, since mutations in these regions might relate with the emergence of new genes or altered genomic regulation and function [Citation56].
As for features of CpG-related mutations, specifically, CpG-SNPs are overrepresented in coding regions, while CpG-CNVs are overrepresented in coding regions and promoters. This suggests different functional consequences depending on whether CpGs are involved in the appearance of SNPs or CNVs. Although in both cases most probably intergenic regions will be affected, CpG-CNVs will have more chances of affecting promoters and coding regions than CNVs, CpG-SNPs or SNPs. CNVs in promoters can alter protein length and introduce frameshifts and are strongly associated with protein binding, followed by morphogenesis [Citation57]. A famous case of the effect of CNV in a coding region is the role of CAG repeats in the etiology of Huntington’s Disease [Citation58]. Interestingly, recent research has shown that CNVs can be directed by environmental factors, regulated by histone modifications [Citation59].
Next, we inquired into associations between the mutations found and repetitive elements. In general, repetitive elements were found to be marginally more associated with CNVs than with SNPs. When looking at specific categories, however, noticeable particularities emerged. CpG-CNVs are much more associated with LINE/CR1 elements than CNVs in general, which occurs at the expense of simple repeats. LINE elements are well known to be suppressed by DNA methylation [Citation60] and involved in genomic rearrangements produced due to their ‘cut-and-paste’ and ‘copy-and-paste’ activities [Citation61]. Moreover, LINE elements are highly active during germ line development, which is crucial for the maintenance of the transgenerationally transmitted genomic integrity [Citation62]. CR1 stands for ‘Chicken repeat 1ʹ LINE elements, which were initially discovered in chickens but have been identified in many bird species [Citation63]. Considering this background, the association found between LINE/CR1 elements and CNVs suggests that changes in DNA methylation (particularly in the germ line) in LINE elements would have a role in allowing their retro-transposition for the consequent generation of CNVs that will be inherited. Interestingly, we have previously reported in rats that developmental exposure to an environmental toxicant (vinclozolin) generates DNA methylation changes and CNVs (particularly duplications [Citation37]) in the male germ line three generations after the exposure [Citation37,Citation64]. This combined information calls for further research on the role of germ line methylation and activity of LINE elements in generating CNVs.
In parallel, ‘simple’ and ‘low complexity’ repeats seem to be increased in breed-specific SNPs in comparison to all the SNPs, particularly in CpG-SNPs, at the expense of LINE elements. Interestingly, simple repeats in flanking regions of introns exhibit high mutation rates, making introns a rapidly evolving type of genomic element [Citation65]. Coincidentally, as mentioned above, many of the mutations found in the present study were indeed in intronic regions. Our results raise questions about the role of simple repeats in speciation and on their relation to SNPs. Important differences were also observed in the composition of LTR elements between the SNPs and CNVs identified; ERV1 and ERVK elements are overrepresented in CpG-SNPs, while ERVL is overrepresented in CpG-CNVs, in relation to reference values. Although not many studies exist on the role of LTR elements in evolution, in plants LTR elements are generally associated with genomic expansion, and have recently been found to be a major diversification force in the speciation of Capsicum spp, where new proteins were created by their retroduplication [Citation66]. One of the most striking findings, however, was the much higher presence of breed-specific CpG-SNPs in satellite elements of the W chromosome, suggesting that this sex-chromosome could have more relevance in speciation than previously thought. Although the W chromosome has received much less attention than the Z in birds’ speciation, recent evidence suggests that the W chromosome is highly involved in speciation [Citation67].
We then investigated the relation of CpG-related mutations to DNA methylation in RJF sperm, the closest living relative to the ancestor. Although we cannot assert that DNA methylation in RJF sperm have directly influenced CpG mutation rates in the domesticated breeds evaluated here, the associations found can provide clues about this connection. We studied two traits related to DNA methylation in RJF sperm (levels and variability across individuals) to build nine categories representing combinations of these variables. We hypothesized that a non-association of sperm CpG methylation and mutations would mean to observe the same distribution of these categories in all the CpGs in which DNA methylation was measured in the sperm, and in subgroups of those CpGs related to mutations (SNPs or CNVs). Although a similar pattern of methylation is observed between all sperm CpGs and CpGs-CNVs, striking differences are observed with CpGs-SNPs, suggesting that medium levels of CpG methylation in sperm combined with medium/high interindividual variability are important variables influencing SNP formation. Moreover, while the majority of CpG-SNPs associate with medium levels of methylation and medium/high variation in RJF sperm, the majority of CpG-CNVs associate with high levels of methylation and low/medium variability in RJF sperm. This combined information suggests different DNA methylation features in the germ line (e.g., levels and inter-individual variability) can lead to the emergence of different types of mutations. Current knowledge points towards an important role for CpG methylation in biasing mutations, however, we show that other aspects need to be taken into consideration such as DNA methylation variability in a population. Moreover, the high mutation rates of CpGs are usually considered in relation to SNPs but not to CNVs. A recent study observed CNV breakpoints in nearly half of the genes of great tits (Parus major) [Citation68]. These CNV breakpoints were prominent at repetitive (segmental duplications) and regulatory regions, overlapped with transcription start sites, and were CpG rich [Citation68]. These results combined with ours show the importance of addressing the specific mechanisms in which CpG methylation regulate retro-transposition to promote genomic instability related to evolution.
Through Bayesian analyses, we showed that the probability of mutating to SNPs or CNVs varied substantially among RJF sperm CpG methylation categories, with each category exhibiting well-defined and specific ranges of mutation probabilities. Interestingly, for some of these categories (MM-HV and MM-MV; ) the mutation rate increases as genetic relatedness to RJF decreases. This finding suggests CpGs with medium levels of methylation and high/medium inter-individual variability in the sperm are related to SNPs of relevance for speciation. Interestingly, this pattern was not observed in CNVs. Thus, our sperm DNA methylation analysis suggests CpG-CNVs are not related to speciation, while CpGs-SNPs would be relevant for the emergence of genomic features involved in speciation. However, the bulk of CpG-related mutations found in our study are related to CNVs (~20 times more CpG-CNVs than CpG-SNPs). Based on this combined information we suggest the majority of CpGs in the genome, those related to CNVs, provide a source of genomic ‘flexibility’ in evolution, i.e., the ability of the genome to expand its functional possibilities. Meanwhile, a small fraction of CpGs, those related to SNPs, will provide genomic ‘specificity’ in evolution, thus, representing mutations related to phenotypic traits relevant for speciation. The germ line has previously been proposed as a catalyst for genomic evolution [Citation56,Citation69], by propitiating the emergence of novel genes that subsequently adopt functions in somatic tissues. It will be important in the future to know the role of CpG methylation in regulating the activity of repeat elements in the germ line and its relation to genome integrity and variability in descendants.
Materials and methods
Animals
The domesticated breeds analysed for SNP and CNV variability were Broilers (BRL; n = 24), and three heritage breeds from different regions of Sweden, namely Kindahöna (KIN; n = 19), Hedemorahöna (HED; n = 20) and Gotlandshöna (GOT; n = 20). All three heritage breeds were created before specialized layer breeds, such as White leghorn, entered Sweden in the later part of the nineteenth century. They have traditionally been used both for egg and meat production and exhibit a large variation in plumage colours. KIN originates from the Östergötland County and exhibits variations in plumage colors, in the number of toes, and in a comb shape. HED originates from the Dalarna County and has a dense plumage suit, which makes individuals well adapted to a cold climate. GOT originates from the small Fårö island, which is close to the Gotland island in the Baltic sea, and is the largest of the three Swedish heritage breeds included in this study. Ross 308 broiler breeder were obtained from a commercial farm. RJF individuals (n = 24 for genotyping; n = 20 for sperm DNA methylation) were from a population maintained in-house for several generations.
Ethics statement
The experiments reported in this paper were carried out in accordance with ethical guidelines approved by the Linköping Animal Ethics Committee, license no 122‑10.
Tissues used for genotyping
For genotyping DNA was obtained from different sources. In RJF and BRL animals, DNA was collected from whole blood. In animals from the HED and GOT pure line breeds DNA was obtained from blood. In animals from KIN, however, DNA was obtained mostly from bulb tips of archived feathers, and in two samples DNA was obtained from blood.
Separation of sperm for DNA methylation analyses
The methylation status in the sperm of RJF was measured in a number of animals (n = 20) in order to determine DNA methylation levels and inter-individual variability in CpGs across the genome. This was further compared to SNPs and CNVs identified between domesticated breeds and RJF. In order to measure DNA methylation in the sperm of RJF, ejaculates were collected from live specimens after cloacal massaging. Sperm samples were then frozen at −20°C until further processing. Sperm cells were then purified. For this, sperm samples were re-suspended in 100 µL PBS and 100 µl of collagenase (850u/mL) and incubated at 37°C for 1 h under rotation. After incubation, the samples were added 1 mL PBS and then sonicated for 5 s at 60% amplitude (Fisher ultra-sonicator attached to a cooling chamber, cup horn, with capacity for eight microfuge tubes). Samples were the subjected to three series of vortexing (30 s), centrifugation (3 min; 4000 g; RT), discarding of the supernatant, and re-suspension in 1 mL PBS. The last re-suspension, however, was in 820 µL of digestion buffer (prepared by mixing 5 mL of 1 M Tris–HCl pH 8.0, 2 mL of 0.5 M EDTA, 5 mL of 10% SDS and 88 mL of DNAse free water). DTT was then added (80 µL; 0.1 M) and the mixture was incubated at 65°C for 15.
DNA isolation
Proteinase K was added to each sample (80 mL; 20 mg/mL) and incubation was performed under rotation for 1 h at 55°C. After incubation, 300 µL of protein precipitation solution (Promega) was added and samples were incubated for 15 min on ice. Samples were centrifuged at max speed for 30 min at 4°C and then 1 mL of the supernatant was transferred to a new tube. Isopropanol (1 mL) and glycogen (3 µL; 5mg/mL) were then added. The samples were incubated at 4°C under rotation for 30 min and then centrifuged at max speed for 30 min at 4°C. The supernatant was then discarded and 500 µL of 70% ethanol was added. Centrifugation was performed again for 10 min at 4°C. The supernatant was discarded, the samples were dried at the bench for at least 20 min, and then re-suspended with 150 µL of DNAse free water. The DNA concentration was measured with nanodrop (Thermofisher).
Genotyping
Genotype by Sequencing was used for the assessment of genetic variability observed between RJF and the domesticated breeds. We have previously optimized SNP detection through genotype by sequencing using the PstI enzyme [Citation38]. For this genotyping, DNA was collected from animals from different breeds, reduced with PstI digestion, barcoded (to individualize the fragmented DNA), pooled among individuals from the same breeds, and then sequenced in Illumina platform (SciLife Lab, Uppsala, Sweden), following our previously described protocol [Citation38]. Genetic variability in the form of single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) were assessed between the RJF and the domesticated breeds.
DNA methylation
DNA methylation was measured in RJF sperm in the same fraction of the genome in which SNPs and CNVs were evaluated, i.e., the fraction of the genome digested with the PstI restriction enzyme. This enzymatic digestion is not affected by the methylation status of CpG sites because PstI cuts the AG dinucleotide in the recognition site 5ʹ CTGCAG 3ʹ [Citation38], as described by manufacturer information (New England Biolabs). Another important feature of genome restriction by PstI is that the resulting digested genome is already enriched for CpG sites [Citation38], capturing CpG regions in CpG-rich micro-chromosomes [Citation70]. Genome restriction by PstI allowed to compare the same fraction of reduced genome across all individuals tested. Once the RJF sperm genome was PstI fragmented, the methylated DNA fraction of sperm cells was captured through methylated DNA immunoprecipitation (MeDIP) by an anti-methyl-cytosine antibody (2 µg/µl; catalogue number C15200006, Diagenode, Denville, NJ, USA), following a protocol previously optimized for chicken DNA in our lab [Citation71]. This DNA, which represents a reduced fraction genome that is enriched for methylation, was then barcoded in order to individualize the DNA fragments, and pooled for sequencing in Illumina platform (SciLife Lab, Uppsala, Sweden). We have recently used this approach for the determination of differential DNA methylation in a reduced fraction of the chicken genome [Citation41].
Bioinformatic methods
Sequence processing
Quality trimming was performed in short sequences with SeqyClean tool v. 1.9.10 [Citation39] using a Phred quality score ≥24 and a fragment size ≥50. The quality of the reads was checked before and after the cleaning by FastQC v.0.11.3 [Citation72].
For the SNP assessment, the Tassel v.3.0 program was used to process the data [Citation73], while for CNV and methylation assessments, Stacks v.1.39 was used for de-multiplex the barcodes identifying individuals. For each sample stored in a FASTQ file, one identification map key file exists. This key file has the matching information of the sample, flow-cell and lane. The reads that begin with one of the expected barcodes (found in the key map) are followed by the expected cut site remnant (CTGCA for PstI). For SNP calling, fragments were then trimmed to 64 bases and grouped into a single list called “master” by the TASSEL-GBS Discovery Pipeline. For methylation analysis and CNV calling this FASTQ information of each individual sample was stored to be further analyzed.
Alignment and coverage
The alignment of quality-trimmed reads was performed using the Bowtie2 tool v.2.2.5 [Citation74] against the chicken reference sequence (Gallus_gallus 4.0, NCBI). For coverage analyses, Samtools v.0.1.19 [Citation75] with the ‘depth’ option was used in all the individuals that were de-multiplexed by the Stacks v.1.39 [Citation76] pipeline.
Identification of genetic variants
The aligned reads were then used as input in the Tassel v.3.0 default pipeline [Citation73] for SNP identification. We aligned the sequences of each individual (from each breed) against our sequenced Red Jungle Fowl (RJF) reference genome. We filtered the polymorphisms initially identified based on the sequencing quality criteria and on the genomic bases identified. After individual SNP calling, we merged all the SNPs called for each breed to be filtered together following the parameters: i) minimum taxon call rate (mnTCov) of 20%; ii) minimum site coverage (mnScov) of 70%; iii) mismatch rate (misMat) of 5%; iv) minimum minor allele frequency (mnMAF) of 0.01. A more detailed description of these filters parameters was provided by Glaubitz et al. [Citation73].
For CNV calling, the aligned sequence files (.bam) of each individual (from each breed) were merged into unique files representing CNVs emerging within each breed (regarding RJF). The “view“ option from Samtools v.1.3.14 was used to generate a “hit” file from each unique file containing the coverage information for each base pair sequenced from each breed. This ”hit” file was then used for CNV calling by the CNV-Seq tool [Citation77] across the chicken genome (Gallus_gallus 4.0, NCBI) using default parameters.
Determination of relatedness between breeds
A cladogram was elaborated to determine relatedness between the domesticated breeds and RJF, using the Japanese quail (Coturnix japonica) as the outgroup. The sequencing reads from the Japanese quail were retrieved from a previous study [Citation78]. The Japanese quail sequencing reads were processed in a similar way as for the chicken breeds in regard to the cleaning and filtering criteria. The only difference was that the SNP call was performed using the Samtools v.0.1.19 [Citation75] program with default parameters. After this, the SNPs were merged with those from the chickens. A relatedness cladogram was then generated using the neighbour joining clustering method [Citation79] using bootstrap = 100 to determine relatedness between the domesticated chicken breed and RJF. The Japanese quail (Coturnix japonica) was used as the outgroup. The results were then plotted using the ‘ape’ package from the R repository. Additionally, we performed a Fixed Index (Fst) analysis using the LOSITAN [Citation80] package (with default parameters) to identify SNPs evolving either under neutrality or balancing or positive selection. The Fst analysis was based on 34,218 SNPs that were common among our four domesticated breeds, RJF and the Japanese quail.
Determination of states of methylation in CpG sites
A combination of R packages from both the CRAN and the Bioconductor projects was used to handle the DNA methylation data. A reference set of CpGs that contained all the CpG sites within the chicken (Gallus gallus) genome (UCSC version galGal4) was built to be compared with the CpGs obtained by our PstI-MeDIP method. Based on this, the number of CpGs covered by our approach was defined. For the normalization of CpG coverage, we used the ‘calcNormFactors’ function from the edgeR package [Citation81]. With the normalized data, we performed a quasi-likelihood pipeline for differential methylation using negative binomial generalized linear models [Citation82] with F-tests instead of likelihood ratio tests [Citation83]. Dispersion estimates were then calculated based on the normalized data to determine the biological coefficient of variation (BCV), which represents inter-individual variability in DNA methylation. The BCV was then plotted using the edgeR function against the level of DNA methylation measured per window, represented by the average of CpGs coverage (expressed as the log of counts per million, logCPM) and termed ‘tags’ [Citation84] ()).
We then proceeded to calculate tag-wise dispersion trends to determine how DNA methylation levels relate to BCV in general. As DNA methylation levels have non-identical and dependent distribution between windows, we employed an Empirical Bayes strategy that generated two kinds of trends [Citation85]. One trend line (red line in )) assumes a normal distribution of all the tags (common dispersion), while the other (green line in )) corresponds to the distribution of tags assuming non-identical methylation levels across the genome (adjusted dispersion) [Citation84]. In addition, we generated a distribution based on the ‘square root of the trended dispersion’ (blue line in )). Information from these different distributions was used to establish the limits of our categories of CpG methylation levels and inter-individual variability in the sperm of RJF. The two intersection points between the ‘common’ and the ‘trended’ dispersions (coordinates −0.25; 1.35 Y and 2.40; 1.35) were calculated to define the X-axis thresholds dividing three coverage states, which correspond to levels of DNA methylation categorized as low (containing 71.1% of all tags), medium (containing 26.6% of all tags) or high (containing 2.3% of all tags).
To define the Y-axis thresholds that divide states of inter-individual variation, we calculated the lowest and highest values of the ‘square root of the trended dispersion’, which corresponds, respectively, to the Y-axis values 0.86 and 1.83. These thresholds delimited three categories of inter-individual variation, namely low (containing 15.4% of all tags), medium (containing 67.7% of all tags) and high (containing 16.9% of all tags).
Comparison of SNPs and CNVs within CpG states
Using R, we made tables with the positions of all the SNPs by breeds according to our reference or alternative alleles. We merged the SNPs of each breed with the CpGs coverage and dispersion states table. For the CNVs, we took the individual positions of each CNV range within each breed and merged these positions with the CpG table. From these merged files we were able to make all comparisons described in this manuscript in order to determine differences between the observations and expected values.
Bayesian analysis of CpG-related mutation rates
To assess the mutation probabilities per CpG category and breeds, we treated each SNP or CNV as a Bernoulli trial, where each location is either mutated or not. Using a Bayesian approach with the prior distribution for the mutation rate expressed by the beta distribution, the posterior density of the mutation probability of CpG category i and breed j, denoted , is given by
, where
if location k in category i and breed j is mutated and otherwise
,
the corresponding total number of locations, and
and
the prior parameters. We wanted to make inference primarily based on the data, and used Jeffreys prior to express a vague prior with
.
Data access
The dataset supporting the conclusions of this article is available in the European Nucleotide Archive (ENA) repository (EMBL-EBI), under accession PRJEB29249, which can be reached through the following link: http://www.ebi.ac.uk/ena/data/view/PRJEB29249.
Author contributions
Conceptualization: C.G.-B. P.J. and F. P.; Methodology and sample collection: F.P., C.G.-B.; Animal Resources: A.M.J., P.J.; Data analysis: F.P., V.S., T. L., C.G.-B.; Writing – original draft: C.G.-B.; Writing – review & editing: F.P., V.S., L.L.C., A.M.J., T. L., D. W., P.J., C.G.-B.; Funding acquisition: L.L.C. and P.J.
Supplemental Material
Download PDF (1 MB)Acknowledgments
F.P. is grateful for funding from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; Brazilian government) for PhD research internship (PDSE, Programa de Doutourado-sanduíche no exterior; number: 99999.008097/2014-03) at Linköping University (Sweden), and for post-doc funding from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, project no. 2016/20440-3 and 2018/13600-0). L.L.C. is a recipient of a research productivity scholarship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Brazilian government) and receives funding from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; São Paulo State, Brazil; grant 2014/08704-0). C.G.-B. and P.J. appreciate funding from the European Research Council advanced grant 322206 ‘Genewell’ to P.J. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Brawand D, Wagner CE, Li YI, et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature. 2014 Sep 18;513(7518):375–381. PubMed PMID: 25186727.
- Carneiro M, Albert FW, Afonso S, et al. The genomic architecture of population divergence between subspecies of the European rabbit. PLoS Genet. 2014 Aug;10(8):e1003519. PubMed PMID: 25166595; PubMed Central PMCID: PMC4148185.
- Carneiro M, Rubin CJ, Di Palma F, et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science. 2014 Aug 29;345(6200):1074–1079. PubMed PMID: 25170157.
- Guenther CA, Tasic B, Luo L, et al. A molecular basis for classic blond hair color in Europeans. Nat Genet. 2014 Jul;46(7):748–752. PubMed PMID: 24880339.
- Rubin CJ, Zody MC, Eriksson J, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010 Mar 25;464(7288):587–591. PubMed PMID: 20220755.
- Price EO. Animal domestication and behavior. Wallinford: CABI; 2003.
- Jensen P. Behavior genetics and the domestication of animals. Annu Rev Anim Biosci. 2014 Feb;2:85–104. PubMed PMID: 25384136.
- Storey AA, Athens JS, Bryant D, et al. Investigating the global dispersal of chickens in prehistory using ancient mitochondrial DNA signatures. PLoS One. 2012;7(7):e39171. PubMed PMID: 22848352; PubMed Central PMCID: PMCPMC3405094.
- Underhill AP. Current Issues in Chinese Neolithic Archaeology. J World Prehistory. 1997;11:103–160.
- Brisson D. The directed mutation controversy in an evolutionary context. Crit Rev Microbiol. 2003;29(1): 25–35. PubMed PMID: 12638717.
- Lenski RE, Mittler JE. The directed mutation controversy and neo-Darwinism. Science. 1993;259(5092): 188–194. PubMed PMID: 7678468.
- Dobzhansky T, Ayala F, Stebbins GL, et al. Evolution. San Francisco, CA, USA: W.H. Freeman and Company; 1988.
- Guerrero-Bosagna C. Finalism in Darwinian and Lamarckian evolution: lessons from epigenetics and developmental biology. Evol Biol. 2012;9(3):283–300.
- Singal R, Ginder GD. DNA methylation. Blood. 1999 Jun 15;93(12):4059–4070. PubMed PMID: 10361102.
- Coulondre C, Miller JH, Farabaugh PJ, et al. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978 Aug 24;274(5673):775–780. PubMed PMID: 355893.
- Huttley GA. Modeling the impact of DNA methylation on the evolution of BRCA1 in mammals. Mol Biol Evol. 2004 Sep;21(9):1760–1768. . PubMed PMID: 15190129.
- Tomatsu S, Orii KO, Islam MR, et al. Methylation patterns of the human beta-glucuronidase gene locus: boundaries of methylation and general implications for frequent point mutations at CpG dinucleotides. Genomics. 2002 Mar;79(3):363–375. PubMed PMID: 11863366.
- Tsunoyama K, Bellgard MI, Gojobori T. Intragenic variation of synonymous substitution rates is caused by nonrandom mutations at methylated CpG. PubMed PMID: 11675605 J Mol Evol. 2001;534–5:456–464.
- Ying H, Huttley G. Exploiting CpG hypermutability to identify phenotypically significant variation within human protein-coding genes. Genome Biol Evol. 2011;3:938–949. PubMed PMID: 21398426; PubMed Central PMCID: PMC3184784.
- Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father‘s age to disease risk. Nature. 2012 Aug 23;488(7412):471–475. PubMed PMID: 22914163; PubMed Central PMCID: PMC3548427.
- Sved J, Bird A. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc Natl Acad Sci U S A. 1990 Jun;87(12):4692–4696. PubMed PMID: 2352943.
- Simmen MW. Genome-scale relationships between cytosine methylation and dinucleotide abundances in animals. Genomics. 2008 Jul;92(1):33–40. PubMed PMID: 18485662.
- Zemojtel T, Kielbasa SM, Arndt PF, et al. CpG deamination creates transcription factor-binding sites with high efficiency. Genome Biol Evol. 2011;3:1304–1311. PubMed PMID: 22016335; PubMed Central PMCID: PMC3228489.
- Rosenberg MS, Subramanian S, Kumar S. Patterns of transitional mutation biases within and among mammalian genomes. Mol Biol Evol. 2003 Jun;20(6):988–993. . PubMed PMID: 12716982.
- Walser JC, Furano AV. The mutational spectrum of non-CpG DNA varies with CpG content. Genome Res. 2010 Jul;20(7):875–882. . PubMed PMID: 20498119; PubMed Central PMCID: PMCPMC2892088.
- Guerrero-Bosagna C, Sabat P, Valladares L. Environmental signaling and evolutionary change: can exposure of pregnant mammals to environmental estrogens lead to epigenetically induced evolutionary changes in embryos?. Evol Dev. 2005;7(4): 341–350. PubMed PMID: 15982371.
- Cortijo S, Wardenaar R, Colome-Tatche M, et al. Mapping the epigenetic basis of complex traits. Science. 2014 Mar 07;343(6175):1145–1148. PubMed PMID: 24505129.
- Johannes F, Porcher E, Teixeira FK, et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009 Jun;5(6):e1000530. PubMed PMID: 19557164; PubMed Central PMCID: PMCPMC2696037.
- Fukuda K, Inoguchi Y, Ichiyanagi K, et al. Evolution of the sperm methylome of primates is associated with retrotransposon insertions and genome instability. Hum Mol Genet. 2017 Sep 15;26(18):3508–3519. PubMed PMID: 28637190.
- Macia A, Munoz-Lopez M, Cortes JL, et al. Epigenetic control of retrotransposon expression in human embryonic stem cells. Mol Cell Biol. 2011 Jan;31(2):300–316. PubMed PMID: 21041477; PubMed Central PMCID: PMC3019972.
- Skinner MK, Guerrero-Bosagna C, Haque MM, et al. Epigenetics and the evolution of Darwin‘s Finches. Genome Biol Evol. 2014 Aug;6(8):1972–1989. PubMed PMID: 25062919; PubMed Central PMCID: PMC4159007.
- Smith TA, Martin MD, Nguyen M, et al. Epigenetic divergence as a potential first step in darter speciation. Mol Ecol. 2016 Apr;25(8):1883–1894. PubMed PMID: 26837057.
- Li J, Li R, Wang Y, et al. Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genomics. 2015 Oct 23;16:851. 10.1186/s12864-015-2098-8. PubMed PMID: 26497311; PubMed Central PMCID: PMCPMC4619007.
- Mugal CF, Wolf JB, von Grunberg HH, et al. Conservation of neutral substitution rate and substitutional asymmetries in mammalian genes. Genome Biol Evol. 2010 Jan 6;2:19–28. PubMed PMID: 20333222; PubMed Central PMCID: PMCPMC2839347.
- Molaro A, Hodges E, Fang F, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell. 2011 Sep 16;146(6):1029–1041. PubMed PMID: 21925323; PubMed Central PMCID: PMC3205962.
- Olsen AK, Andreassen A, Singh R, et al. Environmental exposure of the mouse germ line: DNA adducts in spermatozoa and formation of de novo mutations during spermatogenesis. PLoS One. 2010;5(6):e11349. PubMed PMID: 20596530; PubMed Central PMCID: PMC2893163.
- Skinner MK, Guerrero-Bosagna C, Haque MM. Environmentally induced epigenetic transgenerational inheritance of sperm epimutations promote genetic mutations. PubMed PMID: 26237076; PubMed Central PMCID: PMCPMC4622673 Epigenetics. 2015;108:762–771.
- Pertille F, Guerrero-Bosagna C, Silva VH, et al. High-throughput and Cost-effective Chicken Genotyping Using Next-Generation Sequencing. Sci Rep. 2016 May 25;6:26929. PubMed PMID: 27220827; PubMed Central PMCID: PMCPMC4879531.
- Zhbannikov IY, Hunter SS, Settles ML SEQYCLEAN User Manual. cited 2016 Nov 2. Available from: https://githubcom/ibest/seqyclean. 2013
- Beaumont MA. Adaptation and speciation: what can F(st) tell us? Trends Ecol Evol. 2005 Aug;20(8):435–440. . PubMed PMID: 16701414.
- Pertille F, Brantsaeter M, Nordgreen J, et al. DNA methylation profiles in red blood cells of adult hens correlate to their rearing conditions. J Exp Biol. 2017 Aug 07 PubMed PMID: 28784681. DOI:10.1242/jeb.157891.
- Schluter D, Conte GL. Genetics and ecological speciation. Proc Natl Acad Sci U S A. 2009 Jun 16;106 Suppl 1:9955–9962.
- Wolf JB, Lindell J, Backstrom N. Speciation genetics: current status and evolving approaches. Philos Trans R Soc London, Ser B. 2010 Jun 12;365(1547):1717–1733. . PubMed PMID: 20439277; PubMed Central PMCID: PMCPMC2871893.
- Wolf JB, Ellegren H. Making sense of genomic islands of differentiation in light of speciation. Nat Rev Genet. 2017 Feb;18(2):87–100. . PubMed PMID: 27840429.
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007 Apr;8(4):253–262. . PubMed PMID: 17363974.
- Chacon-Sanchez MI, Martinez-Castillo J. Testing domestication scenarios of Lima Bean (Phaseolus lunatus L.) in Mesoamerica: insights from genome-wide genetic markers. Front Plant Sci. 2017;8:1551. PubMed PMID: 28955351; PubMed Central PMCID: PMCPMC5601060.
- Ponting CP, Lunter G. Signatures of adaptive evolution within human non-coding sequence. Hum Mol Genet. 2006 Oct 15;15(2):R170–5. . PubMed PMID: 16987880.
- Collins DW, Jukes TH. Rates of transition and transversion in coding sequences since the human-rodent divergence. Genomics. 1994 Apr;20(3):386–396. . PubMed PMID: 8034311.
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012 Sep 6;489(7414):101–108. PubMed PMID: 22955620; PubMed Central PMCID: PMCPMC3684276.
- van Bakel H, Nislow C, Blencowe BJ, et al. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 2010 May 18;8(5):e1000371. PubMed PMID: 20502517; PubMed Central PMCID: PMCPMC2872640.
- Caminsky N, Mucaki EJ, Rogan PK. Interpretation of mRNA splicing mutations in genetic disease: review of the literature and guidelines for information-theoretical analysis. F1000Res. 2014;3:282. PubMed PMID: 25717368; PubMed Central PMCID: PMCPMC4329672.
- Friedman LS, Ostermeyer EA, Szabo CI, et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994 Dec;8(4):399–404. PubMed PMID: 7894493.
- Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5‘-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998 Jun 18;393(6686):702–705. PubMed PMID: 9641683.
- Pennacchio LA, Lehesjoki AE, Stone NE, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science. 1996 Mar 22;271(5256):1731–1734. PubMed PMID: 8596935.
- Carvalho GA, Weiss RE, Refetoff S. Complete thyroxine-binding globulin (TBG) deficiency produced by a mutation in acceptor splice site causing frameshift and early termination of translation (TBG-Kankakee). J Clin Endocrinol Metab. 1998 Oct;83(10):3604–3608. . PubMed PMID: 9768672.
- Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010 Oct;20(10):1313–1326. . PubMed PMID: 20651121; PubMed Central PMCID: PMCPMC2945180.
- O’Dushlaine CT, Edwards RJ, Park SD, et al. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biol. 2005;6(8):R69. PubMed PMID: 16086851; PubMed Central PMCID: PMCPMC1273636.
- Myers RH. Huntington‘s disease genetics. NeuroRx. 2004 Apr;1(2):255–262. . PubMed PMID: 15717026; PubMed Central PMCID: PMCPMC534940.
- Hull RM, Cruz C, Jack CV, et al. Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol. 2017 Jun;15(6):e2001333. PubMed PMID: 28654659; PubMed Central PMCID: PMCPMC5486974.
- Adelson D, Buckley R, Ivancevic A, et al. Retrotransposons: genomic and trans-genomic agents of change. In: Pontarotti P, editor. Evolutionary biology: biodiversification from genotype to phenotype. Switzerland: Springer; 2015. p. 55–76
- Kazazian HH Jr., Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998 May;19(1):19–24. . PubMed PMID: 9590283.
- Newkirk SJ, Lee S, Grandi FC, et al. Intact piRNA pathway prevents L1 mobilization in male meiosis. Proc Natl Acad Sci U S A. 2017 Jul 11;114(28):E5635–E5644. PubMed PMID: 28630288; PubMed Central PMCID: PMCPMC5514719.
- St John J, Quinn TW. Identification of novel CR1 subfamilies in an avian order with recently active elements. Mol Phylogenet Evol. 2008 Dec;49(3):1008–1014. . PubMed PMID: 18929670.
- Guerrero-Bosagna C, Settles M, Lucker B, et al. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One. 2010;5(9). PubMed PMID: 20927350; PubMed Central PMCID: PMCPMC2948035. DOI:10.1371/journal.pone.0013100
- Lin CL, Taggart AJ, Fairbrother WG. RNA structure in splicing: an evolutionary perspective. RNA Biol. 2016 Sep;13(9):766–771. . PubMed PMID: 27454491; PubMed Central PMCID: PMCPMC5014005.
- Kim S, Choi D. New role of LTR-retrotransposons for emergence and expansion of disease-resistance genes and high-copy gene families in plants. BMB Rep. 2018 Feb;51(2):55–56. PubMed PMID: 29353598; PubMed Central PMCID: PMCPMC5836556.
- Irwin DE. Sex chromosomes and speciation in birds and other ZW systems. Mol Ecol. 2018 Feb 14. PubMed PMID: 29443419. DOI:10.1111/mec.14537
- Da Silva VH, Laine VN, Bosse M, et al. CNVs are associated with genomic architecture in a songbird. BMC Genomics. 2018 Mar 13;19(1):195. 10.1186/s12864-018-4577-1. PubMed PMID: 29703149.
- Guerrero-Bosagna C. Evolution with no reason: a neutral view on epigenetic changes, genomic variability, and evolutionary novelty. Bioscience. 2017;67(5):469–476.
- McQueen HA, Fantes J, Cross SH, et al. CpG islands of chicken are concentrated on microchromosomes. Nat Genet. 1996 Mar;12(3):321–324. PubMed PMID: 8589727.
- Guerrero-Bosagna C, Jensen P. Optimized method for methylated DNA immuno-precipitation. MethodsX. 2015;2:432–439. PubMed PMID: 26740923; PubMed Central PMCID: PMCPMC4678308.
- Andrew S. FASTQC. A quality control tool for high throughput sequence data. cited 2016 Nov 2. Available from: http://wwwbioinformaticsbabrahamacuk/projects/fastqc, 2010
- Glaubitz JC, Casstevens TM, Lu F, et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One. 2014;9(2):e90346. PubMed PMID: 24587335; PubMed Central PMCID: PMCPMC3938676.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012 Mar 04;9(4):357–359. . PubMed PMID: 22388286; PubMed Central PMCID: PMCPMC3322381.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078–2079. PubMed PMID: 19505943; PubMed Central PMCID: PMCPMC2723002.
- Catchen JM, Amores A, Hohenlohe P, et al. Stacks: building and genotyping Loci de novo from short-read sequences. G3 (Bethesda). 2011 Aug;1(3):171–182. PubMed PMID: 22384329; PubMed Central PMCID: PMCPMC3276136.
- Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009 Mar 06;10:80.
- Kawahara-Miki R, Sano S, Nunome M, et al. Next-generation sequencing reveals genomic features in the Japanese quail. Genomics. 2013 Jun;101(6):345–353. PubMed PMID: 23557672.
- Zheng X, Levine D, Shen J, et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012 Dec 15;28(24):3326–3328. PubMed PMID: 23060615; PubMed Central PMCID: PMCPMC3519454.
- Antao T, Lopes A, Lopes RJ, et al. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics. 2008 Jul 28;9:323. PubMed PMID: 18662398; PubMed Central PMCID: PMCPMC2515854.
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 Jan 01;26(1):139–140. . PubMed PMID: 19910308; PubMed Central PMCID: PMCPMC2796818.
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012 May;40(10):4288–4297. . PubMed PMID: 22287627; PubMed Central PMCID: PMCPMC3378882.
- Lund SP, Nettleton D, McCarthy DJ, et al. Detecting differential expression in RNA-sequence data using quasi-likelihood with shrunken dispersion estimates. Stat Appl Genet Mol Biol. 2012 Oct 22;11(5). Chapter 8. PubMed PMID: 23104842.
- Chen Y, Lun AT, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016;5:1438. PubMed PMID: 27508061; PubMed Central PMCID: PMCPMC4934518.
- Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007 Nov 01;23(21):2881–2887. . PubMed PMID: 17881408.