
ABSTRACT
Exercise training is continually challenging whole-body homeostasis, leading to improvements in performance and health. Adaptations to exercise training are complex and are influenced by both environmental and genetic factors. Epigenetic factors regulate gene expression in a tissue-specific manner and constitute a link between the genotype and the environment. Moreover, epigenetic factors are emerging as potential biomarkers that could predict the response to exercise training. This systematic review aimed to identify epigenetic changes that have been reported in skeletal muscle following exercise training in healthy populations.
A literature search of five databases (PUBMED, MEDLINE, CINHAL, SCOPUS and SportDiscuss) was conducted in November 2018. Articles were included if they examined epigenetic modifications (DNA methylation, histone modifications and non-coding RNAs) in skeletal muscle, following either an acute bout of exercise, an exercise intervention in a pre/post design, or a case/control type of study.
Twenty-two studies met the inclusion criteria. Several epigenetic markers including DNA methylation of genes known to be differentially expressed after exercise and myomiRs were reported to be modified after exercise.
Several epigenetic marks were identified to be altered in response to exercise, with potential influence on skeletal muscle metabolism. However, whether these epigenetic marks play a role in the physiological impact of exercise is unclear. Exercise epigenetics is still a very young research field, and it is expected that in the future the causality of such changes will be elucidated via the utilization of emerging experimental models able to target the epigenome.
Introduction
Exercise training results in many positive morphological and metabolic muscle adaptations leading to improvements in performance and health-related traits. Skeletal muscle is a plastic tissue that is able to adapt quickly in response to changes in metabolic homeostasis such as exercise [Citation1]. Repetitive muscle contractions result in increases in mitochondrial size and number, changes in substrate metabolism, enhanced angiogenesis and hypertrophy of cardiac and skeletal muscle fibres [Citation1]. Adaptations to exercise occur in a coordinated time frame and are mediated by a plethora of transcriptional, translational, and post-translational regulators [Citation2,Citation3]. In the last two decades, there have been significant advances in research regarding the cellular and molecular adaptations to exercise training in muscle [Citation4]. However, despite ongoing investigations, the molecular mechanisms responsible for these adaptations are yet to be fully understood [Citation4,Citation5]. Specifically, it is not well understood if and how epigenetic signals can mediate physiological adaptations to exercise training [Citation6,Citation7].
Epigenetic remodelling
Epigenetic mechanisms are thought to play a role in the complex adaptations to exercise. Epigenetics (epi: on top, genetics: genes) constitutes an important level of gene expression regulation, without changes in the DNA sequence. We define an epigenetic property as that of a cell, mediated by genomic regulators, conferring on the cell the ability to remember a past event [Citation8]. Epigenetic modifications often involve the addition of chemical groups to the DNA or to histone tails, and can be inherited through cell divisions [Citation9]. Here we consider two major epigenetic events, DNA methylation and histone modifications, along with an epigenetic regulator, non-coding RNAs (ncRNAs) (e.g. miRNA) (). We do not consider ncRNAs as epigenetic per se, as it is difficult to understand how small RNAs could by themselves be heritable through cell division in animals [Citation8]. However, ncRNAs can influence gene expression at both the transcriptional and post-transcriptional level without altering the DNA sequence [Citation10]. Furthermore, the maintenance of chromatin marks is a pivotal mechanism of epigenetic change, and ncRNA is thought to be a fundamental part of this process [Citation11,Citation12]. Therefore, we have included ncRNAs as epigenetic regulators for the purpose of this systematic review. Interestingly, studies have shown that epigenetic modifications can be modified in a tissue-specific manner [Citation9,Citation13,Citation14] by environmental stimuli such as diet, smoking and exercise [Citation5]. Importantly, some epigenetic changes are transient and may have a key function in skeletal muscle.
Figure 1. Epigenetic modifications after environmental stimuli (e.g. exercise).
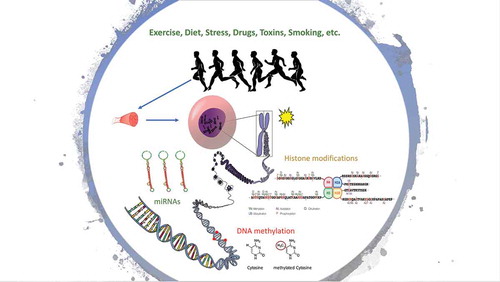
DNA methylation is the covalent modification of a cytosine base usually located in the dinucleotide sequence 5ʹCpG3ʹ (cytosine and guanine separated by a phosphate) [Citation6]. Global DNA methylation patterns are established during embryogenesis in mammals [Citation15], and is accurately replicated after cell divisions, and therefore it is often considered a form of cell memory [Citation16]. The enzymes DNA methyltransferases (DNMTs) specifically DNMT3A and DNMT3B are responsible for the addition of methyl group to the cytosine base during de novo methylation. The enzyme DNMT1 is then responsible for maintaining the methyl marks during subsequent cell divisions [Citation17]. Whether DNA methylation alters gene expression is highly dependent on the genomic location within a gene (i.e. promoter, gene body, or enhancer), and the density of CpGs. For example, increased DNA methylation at CpG-dense promoters tends to lead to a decrease in gene transcription [Citation18]. In addition, the silencing of a gene can lead to the accumulation of DNA methylation at the promoter of said gene, further locking it into a silent state [Citation19–Citation24]. DNA methylation can also be removed actively (demethylation) by Ten-eleven translocation (TET) enzymes [Citation23,Citation25]. Further, there is a cross-talk between DNA methylation and other epigenetic processes such as histone lysine methylation and acetylation [Citation17].
In eukaryotes, DNA tightly coils around proteins called histones to form the chromatin [Citation26–Citation28]. Histones have (N)- and (C)- terminals tails that protrude from the centre of the nucleosome and can interact with adjacent nucleosomes and linker DNA [Citation28]. These histone tails can undergo post-translational modifications (acetylation, phosphorylation, methylation and ubiquitylation) that alters chromatin structure and modifies the accessibility of transcription factors and machinery to the DNA [Citation29]. Histone tails can also serve as a binding site for other proteins (non-histones) to chromatin [Citation28]. Active genes typically display high levels of lysine acetylation on the tails of histones H3 and H4, trimethylation of H3 lysines 4, 36 and 79, and ubiquitylation of H2B [Citation28]. Conversely, gene that has been silenced typically displays trimethylation of H3 lysine 9 and 27, and ubiquitylation of H2A lysine 119 [Citation28].
Gene expression can also be regulated by ncRNAs [Citation12]. The best-characterized ncRNAs are microRNAs (miRNAs) that are ~22 nucleotides long and mediate post-transcriptional gene silencing [Citation10]. miRNAs are non-protein coding molecules that act by base-pairing to the 3ʹ-untranslated regions of the target mRNAs and repress protein synthesis [Citation30]. Approximately 50% of the protein-coding genes are regulated by miRNAs [Citation31].
While epigenetic patterns are partly heritable [Citation32–Citation34], they are also influenced by environmental factors. A seminal study in genetically identical monozygotic twins showed that those with comparable lifestyles had similar epigenetic patterns across multiple tissues (lymphocytes, epithelial mouth cells, intra-abdominal fat and skeletal muscle) when compared with monozygotic twins with different lifestyles [Citation35]. Thus, epigenetics can be considered the crossroads between genetics (nature) and the environment (nurture) [Citation36]. Epigenetics holds great promise to explain exercise-related phenomena such as the inter-individual variability to similar exercise training, and skeletal muscle memory. Inter-individual variability refers to the observation that following exercise training, some individuals improve their fitness significantly after the intervention (‘responders’) while other individuals show only limited improvements after the intervention (‘low-responders’).
Exercise is considered one of the most financially viable and under-utilised form of management and prevention of almost any chronic disease. Exercise training can induce many positive molecular changes and reduce the prevalence of cognitive, metabolic, musculoskeletal, and bone disorders, and increase general well-being and health [Citation4,Citation37]. Epigenetic programming can be modulated by exercise in multiple tissues such as skeletal muscle [Citation38] and adipose tissue [Citation39]. After a strenuous exercise bout, promoter regions of genes important in exercise metabolism (PGC-1α, PDK4, and PPAR-δ) were hypomethylated and this was followed by a concomitant increase in mRNA levels. Interestingly, 3-hour post-exercise, these promoters became re-methylated, demonstrating the dynamic epigenetic response to exercise in skeletal muscle [Citation38]. In addition, expression of some miRNAs change after exercise, which might influence skeletal muscle regeneration, gene transcription and mitochondrial biogenesis [Citation40].
This systematic review aimed to identify epigenetic changes reported in skeletal muscle following both acute (e.g. a single session of exercise) and few weeks or months of exercise in healthy populations.
Methods
This systematic review protocol is reported in accordance with the Preferred Reporting Items for Systematic Review and Meta-Analyses Protocols (PRISMA-P) 2015 statement [Citation41], and its protocol was submitted on PROSPERO for registration. Registration number: CRD42018101975.
Eligibility criteria
We used the Participant, Intervention, Comparison, Outcomes and Studies (PICOs) framework for this systematic review (). We included studies in English or Portuguese that reported epigenetic changes (DNA methylation, histone modifications or miRNA expression) in skeletal muscle following an exercise intervention in healthy human populations. We only included interventional studies, including randomised controlled trials, cohort studies and case–control studies. We included any acute, short- or long-term exercise intervention, either supervised or unsupervised comparing epigenetic patterns before and after exercise. Any exercise interventions combined with a pharmaceutical treatment were excluded. Qualitative studies, case studies, editorials, narrative reviews and systematic reviews were excluded.
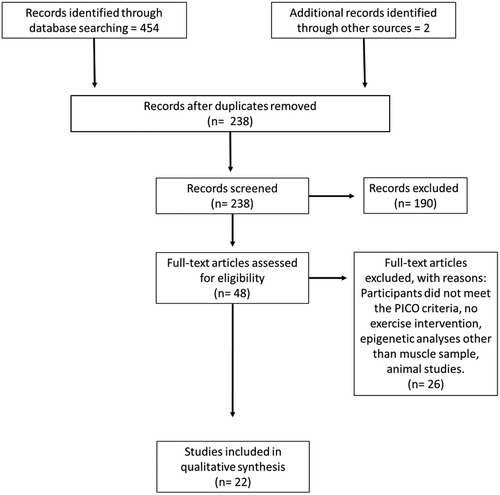
Figure 2. Identification and selection summary for systematic review and meta-analyses.
Information sources & search strategy
A comprehensive database search was conducted in November 2018. The following databases were used to select the relevant studies: PUBMED, MEDLINE, CINHAL, SCOPUS and SportDiscuss databases. We developed our search strategy using medical subheadings (MeSH) and keywords. The keywords and MeSH terms were: (((epigenetic* OR DNA methylation OR histone modification OR chromatin remodelling OR microRNA OR miRNA)) AND (exercise* OR physical activity* OR sport* OR fitness* OR aerobic* OR resistance* OR strength*)) AND skeletal muscle. To eliminate any discrepancies or inconsistencies in inclusion and exclusion of studies, two independent reviewers (MJ and DH) conducted the search to identify titles and abstracts and selected the full texts to evaluate.
Study selection
Studies that met the inclusion criteria for DNA methylation and miRNAs are reported in and . Only one study that met the criteria for histone modifications and is described in the results. Mendeley reference manager and Endnote were used to detect and remove duplicates.
Table 1. Summary of DNA methylation study’s findings.
Table 2. Summary of miRNAs study’s findings.
Table 3. Summary of miRNAs that were up- or down-regulated after exercise.
Results
Systematic search
The search returned 454 publications (). After duplicate removal, 238 publications remained, after screening based on the a priori PICO selection criteria 48 publications remained for the full-text analysis. Of the 48 publications screened, 26 did not meet the inclusion criteria (Supplementary Table 1) leaving 22 papers to be included in the qualitative analysis. We divided our report between candidate genes and genome-wide. Candidate gene studies are based on selected genes or miRNAs that are known to be associated with skeletal muscle metabolism. Genome-wide studies are hypothesis free, scanning the entire genome for possible markers related to skeletal muscle metabolism which generate much larger and comprehensive results. However, genome-wide studies are an expensive approach and therefore not as widely used in the research community.
Qualitative analysis – systematic review
DNA methylation and exercise – candidate gene approach
Early studies on DNA methylation, and exercise investigated candidate genes involved in exercise adaptations (). Most candidate studies focused on peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1a), the master regulator of mitochondrial biogenesis and fat metabolism [Citation3]. Alibegovic et al. investigated the effect of forced bed rest (10 days) on DNA methylation levels at three CpG sites located in the promoter of PGC-1a, in the vastus lateralis muscle of 20 participants [Citation42]. DNA methylation at two of those sites negatively correlated with PGC-1a mRNA expression at baseline (site 816: r = −0.65, P = 0.03; site 783: r = −0.59, P = 0.04), and methylation at site 816 increased by ~7% following bed rest (P = 0.04). After four weeks of aerobic retraining, DNA methylation levels decreased, but did not return to their baseline levels [Citation42]. Barres et al. investigated DNA methylation in vastus lateralis biopsies after an acute bout of exercise and also identified the PGC-1a promoter as differentially methylated after an exercise intervention. In addition, the promoter regions of key genes involved in exercise response (i.e. PGC-1a, TFAM, MEF2A and PDK4) were hypomethylated by ~10% immediately after a strenuous bout of cycling and became remethylated 3 h after exercise (n = 14, p < 0.05). It should be noted that some genes showed a delayed hypomethylation (i.e. PPAR-d, 3 h after exercise), and these changes were exercise intensity dependent. Hypomethylation of the promoters was accompanied by increases in mRNA levels either immediately after or 3 h after exercise [Citation38]. Bajpeyi et al. divided 11 healthy young men into high- and low-responders based on their DNA methylation response at an important regulatory region in PGC-1a following an single bout of exercise [Citation43]. Only high-responders, who had decreased DNA methylation after exercise, showed nucleosome repositioning in the promoter of PGC-1a along with an associated increase in PGC-1a mRNA expression (1.05 ± 0.08 to 1.29 ± 0.11 fold-change). The dichotomising of only 11 subjects included in this study was arbitrary which reduced statistical power and leads to the question whether the conclusions would have been similar using a continuous spectrum of responses. Nonetheless it can be concluded that these candidate-gene studies demonstrated that exercise alters DNA methylation levels at genes involved in muscle metabolism and are associated with a concomitant change in mRNA expression.
DNA methylation and exercise – genome-wide approach
Four studies conducted Epigenome-Wide Association Studies (EWAS) investigating genome-wide DNA methylation changes following an exercise training intervention in healthy populations [Citation44–Citation47] (). The studies consisted of a 6-month endurance training intervention in 28 middle-aged men with and without family history of Type 2 Diabetes (T2D) [Citation45], a 3-month unilateral endurance training in 17 young men and women [Citation44]; a 7-week resistance training intervention in eight young men [Citation46]; 3 months of endurance, resistance or combined training in 34 young and 26 old men and women [Citation47]. Results from these studies were highly heterogeneous, due to differences in exercise mode (resistance [Citation46,Citation47], endurance [Citation44,Citation45,Citation47] or both [Citation47]), length of intervention (from 7 weeks to 6 months), genome coverage (gene promoters only [Citation45,Citation47] or all genomic regions [Citation46,Citation48]), density coverage (2–4% of all CpGs with Illumina 450 k or 850 k arrays [Citation44,Citation45,Citation47] or 8% of all CpGs with MeDIP-chip [Citation45]), sample size, age and sex.
Two studies reported more hypomethylation than hypermethylation at the Differentially Methylated Positions (DMPs) [Citation45,Citation46] following an exercise training intervention, one study reported a similar number of hypo- and hyper-methylated DMPs [Citation44], and the last study did not find any DMP [Citation47] after 3 months of exercise. Interestingly, a moderate effect size was consistent across the studies (< 10% methylation change after the intervention [Citation44–Citation47]), suggesting that exercise training may alter the DNA methylation state of multiple genes, in an exercise dose-dependent manner. While the biological relevance of such small changes in methylation is questionable, a direct correlation between DNA methylation levels and the resulting expression level of mRNA of selected genes was demonstrated by gene reporter assay [Citation45]. Three of the studies identified a consistent inverse relationship between DNA methylation and gene expression changes [Citation44–Citation46]. Robinson et al., who used an absolute cut-off of 5% methylation change following exercise found no DMPs however only focused on promoter regions [Citation47]. Lindholm et al., investigated the distribution of these DMPs and reported an enrichment of DMPs in enhancers and regulatory regions which may give an explanation to why no DMPs were observed in the Robinson et al. study [Citation44]. It is worth noting that the magnitude of DNA methylation changes following training was smaller than after acute exercise [Citation38], indicating that DNA methylation changes in response to exercise are a dynamic process activated in the early phase of gene expression. Yet residual DNA methylation changes are retained after the training stimulus is gone, indicating that these changes are accumulated over multiple exercise sessions.
Differentially methylated genes were enriched for pathways such as retinol metabolism and calcium signalling [Citation45], structural remodelling of the muscle, inflammatory/immunological processes and transcriptional regulation [Citation44]. Three of the EWAS studies found DMPs enriched for pathways linked to glucose and/or insulin metabolism [Citation44–Citation47]. However, whether DNA methylation changes result in downstream changes in phenotype has not been investigated in depth. In fact, only one study found that a higher number of hypomethylated sites was associated with hypertrophy in the muscle following a repeated intervention of 7 weeks [Citation46]. This indicates DNA methylation changes might correlate with exercise trainability.
Histone modifications and exercise
Only one study focused on histone modifications following exercise. This study investigated changes in H3K36 and H3K9/14 acetylation in nine men following an acute bout of endurance exercise [Citation49]. While H3K9/14 acetylation was not altered, H3K36 acetylation increased by 64% from baseline (P < 0.05) immediately after exercise. As H3K36 acetylation regulates transcriptional elongation, these results suggest that exercise-induced chromatin remodelling is associated with enhanced transcription. While there was no change in global HDAC activity (P = 0.31), two kinases that can induce nuclear export of HDAC4 and 5 (AMPK and CaMKII) showed signs of activation. These data delineate a signalling pathway that might mediate gene transcription in human skeletal muscle in response to exercise [Citation49]. However, since this information is based on one study, further investigation is required to validate these findings and to uncover novel exercise-related histone modifications.
MiRNAs and exercise – candidate gene approach
MiRNAs act in a tissue-specific manner and when exclusively expressed in skeletal muscle are called myomiRs. A total of 10 miRNA studies were included and divided into a candidate and high-throughput studies ( & ). A vast majority of studies focused on candidate miRNA and expression following exercise training as it is a simple and cost-effective way to analyse miRNAs. Six of the papers focused on the effect of an acute bout of exercise (endurance [Citation40,Citation50,Citation51], resistance [Citation52,Citation53] and concurrent exercise [Citation54]), two studies conducted both an acute intervention and chronic exercise training intervention [Citation40,Citation50] (10 days and 12 weeks of training, respectively). In addition, three studies investigated the effect of chronic exercise training on miRNA expression [Citation55–Citation57], and one study compared powerlifting athletes to healthy controls [Citation58]. Keller et.al. [Citation55] conducted 6 weeks of endurance training, while Zhang et. al. [Citation56] and Mueller et. al[Citation57]. conducted 20 weeks and 12 weeks of resistance training, respectively. Seven studies were done in men, in addition, Zhang et. al. [Citation56] and Mueller et. al. [Citation57] included women in their cohorts. Each study had a small sample size (8–28 participants), with the largest combined cohort of 35 participants undergoing resistance training [Citation56,Citation57]. MiRNA expression changes following exercise training was dependent on the mode and length of the intervention. After acute exercise, only miR-1 and miR-133a, known modulators of muscle proliferation and differentiation [Citation59], were consistently upregulated in candidate miRNA studies (p < 0.05) [Citation40,Citation50,Citation53]. However after chronic exercise, miR-1 and miR-133b were downregulated in a majority of studies [Citation50,Citation55–Citation57]. Only two studies reported increased expression of miR-133b and miR-181 which is thought to be associated with increased glucose homeostasis [Citation40,Citation60], and another study found a decrease in miR-23 [Citation51] involved in myogenic processes [Citation61]. A case–control study comparing powerlifters to healthy controls [Citation58] reported a unique miR expression profiles that were able to distinguish powerlifters from healthy controls based on a five miR signature (miR-126, −23b, −16, −23a, −15a). While multiple miRNAs were identified to be associated with exercise, the results were heterogeneous. Discrepancies between studies could be due to variability in biopsy time, low statistical power, and differences in exercise intensity and duration. In addition, variable amount of total RNA can influence cDNA synthesis efficiency [Citation62], and the use of different housekeeping genes (small RNA to 18s) for normalization in studies could also generate variable findings [Citation63].
Very few studies attempted to link changes in miRNA expression to exercise trainability. Russel et al. [Citation40] reported correlations between VO2peak and Peak Power Output (PPO) with changes in miRNA expression. Baseline VO2peak positively correlated with miR-181 (r = 0.70, P = 0.03), while baseline PPO negatively correlated with miR-23a (r = −0.79, P = 0.012) and post-training PPO negatively correlated with miR-31 (r = −0.74, P = 0.042). Zhang et. al. [Citation56] reported a strong positive correlation between the change in knee strength following resistance training and the change in miR-133a, miR-133b and miR-206 expression (p < 0.01). In summary, expression changes of candidate miRNAs were more consistent in chronic than acute studies, and appear to be dependent on the exercise modality, intensity and duration. Changes in expression of miRNA following exercise training may underlie training adaptations, but more work is needed to confirm this.
MiRNAs and exercise – high-throughput analyses
With advances in technology, high-throughput miRNA expression analysis has become more readily available () with technologies ranging from microarrays, digital multiplex to miRNA-seq, allowing hundreds of miRNAs to be analysed simultaneously. However, with such different platforms, results may yield differences in expression that might simply be due to variability between techniques. All high-throughput studies have been conducted after an acute bout of exercise. Of the four studies, three used targeted miRNA arrays [Citation64–Citation66] and one study conducted miRNA sequencing [Citation67]. Three of the studies were based on resistance exercise (men, n = 26) [Citation65,Citation66,Citation68], and one following endurance exercise (men and women, n = 6) [Citation67]. The range of miRNAs that were differentially expressed across three studies varied from 26 to 102 after resistance exercise [Citation65,Citation66,Citation68]. Interestingly, Zacharewicz et.al. [Citation66] identified that 7 of the 26 miRNAs may regulate cellular growth and proliferation pathways and another nine miRNAs may regulate the Akt-mTOR signalling pathway, a central regulator of muscle protein synthesis and muscle growth [Citation69]. McLean et.al. [Citation67] found 13 miRNAs that increased after endurance exercise (p < 0.001), and several of those miRNAs belonged to the miR-378 family. This family of miRNAs is embedded in the first intron of PGC-1β [Citation70]. Ogasawara et al. was the only study that investigated genome-wide miRNA expression changes following chronic exercise and found that the expression levels of 102 miRNAs were altered after chronic resistance exercise training (p < 0.05) [Citation65]. Interestingly 26 miRNAs were differentially regulated in high vs low responders for hypertrophy [Citation65]. This further consolidates the candidate miRNA studies that specific miRNAs change following acute (and perhaps chronic) exercise, although the specific function of the miRNAs in exercise trainability remains to be elucidated.
Discussion
This systematic review aimed to summarize the epigenetic changes in skeletal muscle following either an acute bout of exercise, or few-weeks to few months of exercise training in healthy populations. This systematic review included 22 studies investigating epigenetic changes (DNA methylation, miRNAs and histone modifications) in skeletal muscle following exercise. The limited number of studies highlights how young the field of exercise epigenetics is, and that further investigation is warranted in how epigenetic signals mediate exercise responses.
Although only one study investigated how histones modifications are associated with exercise responses, multiple studies have looked at DNA methylation and miRNAs as potential epigenetic modulators of exercise responses. Most of the candidate DNA methylation studies focused on PGC-1a, with a general consensus that a decrease in PGC-1a DNA methylation was followed by an increase in PGC-1a mRNA expression following exercise. Genome-wide DNA methylation approaches (i.e. 450 k array, 850 k EPIC array and MeDIP-Chip) yielded hundreds of differentially methylated positions (DMPs), and these were generally associated with a concomitant change in gene expression.
To date, only eight miRNAs have been classified as myomiRs including: miR-1, miR-133a, miR-133b, miR-206, miR-208a, miR-208b, miR-486 and miR-499 [Citation71,Citation72], and all were hypothesised to be altered after exercise. After acute exercise, only miR-1 and miR-133a were consistently upregulated in candidate studies [Citation40,Citation50,Citation53]. However were downregulated after chronic exercise in most studies [Citation50,Citation55–Citation57], suggesting that miRNA expression is dependent on the length and type of exercise. This is in agreement with previous studies where miRNA expression also changed in a dose/intensity-dependent manner [Citation40,Citation73]. miR-1, miR-133a and miR-133b promote myoblast differentiation and regeneration, regulate angiogenesis, are pro-apoptotic, control oxidative stress and reduce cell migration [Citation71]. miR-1/miR133a and miR-133b/miR-206 are organised in a bicistronic cluster and are transcribed together [Citation71,Citation74], which may in part explain why miR-1 and miR-133a were both upregulated in the same samples [Citation40,Citation50,Citation53]. The expression of miR-1 and miR-206 is repressed by the same transcriptional factor (YYI) [Citation75], perhaps explaining why miR-1 and miR-133b were both down-regulated after chronic exercise [Citation50,Citation55–Citation57]. Interestingly, there was no commonly altered miRNA across high-throughput studies after acute or chronic exercise. One possible explanation is that candidate miRNA studies used the same technique (PCR), while high-throughput analyses can vary from different arrays to sequencing. Such techniques can produce ‘noise’ and if not correctly processed and normalised data make it difficult to compare between studies [Citation76].
Importantly, few studies have linked epigenetic changes to physiological or health-related outcomes following exercise training. Only two studies addressed this question and found that miRNAs that were changed after exercise were not correlated with changes in physiological measures. These findings indicate that exercise does change epigenetic profiles but how such changes affect the phenotype is yet to be fully understood. Overall, epigenetic studies in exercise generally have small sample sizes, limiting the statistical power of these studies. Larger sample sizes, longer or more intense interventions are necessary to unravel the full spectrum of pleiotropic effects of exercise training on epigenetic programming. In addition, current research in exercise epigenetics is limited to whole muscle and to date no study has investigated fibre-type specific epigenetic changes. This is a major limitation since skeletal muscle is infiltrated by immune cells after exercise [Citation77,Citation78], which means that changes in epigenetic patterns observed after exercise may reflect changes in cell type proportions rather than true epigenetic changes in muscle cells. Further research utilising tools such as single-cell epigenetics and CRISPR (dCas9) analyses would provide novel information on the inheritance of chromatin states or epigenetic signatures at specific loci during cell division elucidating stability through generations [Citation79] and to uncover the muscle-specific response to exercise.
Conclusion and future directions
Our systematic review supports that exercise is a powerful tool to alter the gene expression profile of human skeletal through epigenetic mechanisms. Epigenetic programming may alter levels of gene expression and improve metabolism, which is one of the many health benefits of exercise. However, the research on epigenetic modifications in skeletal muscle following exercise is still in its infancy, and the downstream physiological consequences of such epigenetic changes are yet to be fully investigated. We have systematically reviewed all published articles on the topic and intended to conduct a comprehensive random effect meta-analysis [Citation80]. However, due to the small number of studies and the strong heterogeneity in study designs (differences in cohorts (sex, age), methodology (candidate-gene, genome-wide), exercise stimulus (acute/chronic, endurance/resistance, high/low intensity, long/short training programs), we were not able to conduct a quantitative analysis.
With the development of whole-genome epigenetic sequencing and analytical methods, the future in this field holds exciting promises. Future studies should include key tissues influenced by exercise (i.e. skeletal muscle, adipose tissue), at multiple time points during exercise training interventions to build a time-course of epigenetic modifications during training adaptations and should ideally be of larger size (n > 50) and duration (>6 months). In addition, it is necessary to conduct functional work to show the causal involvement of exercise-induced epigenetic modifications in physiological adaptations. In summary, we have systematically reviewed the literature regarding epigenetics modifications in skeletal muscle following exercise and found promising epigenetic biomarkers for validation and replication in future studies. Multi-centre and collaborative initiatives are required to advance the field, and to uncover the biological meaning of epigenetic changes in the exercising skeletal muscle.
Key points
Epigenetics is emerging as a potential mechanism underpinning the complexity of exercise adaptations.
Epigenetic changes are transient and may have a key function in skeletal muscle.
Supplemental Material
Download MS Word (15.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material:
Supplemental data for this article can be accessed here.
Related Research Data
References
- Coffey VG, Hawley JA. The molecular basis of training adaptation. Sport Med. 2007;37:737–763.
- Hargreaves M. Exercise and gene expression. Prog Mol Biol Transl Sci. 2015. p. 457–469.
- Bishop DJ, Granata C, Eynon N. Can we optimise the exercise training prescription to maximise improvements in mitochondria function and content? Biochim Biophys Acta Gen Subj. 2014;1840:1266–1275.
- Hawley JA, Hargreaves M, Joyner MJ, et al. Review integrative biology of exercise. Cell. 2014;159:738–749.
- Soci UPR, Melo SFS, Gomes JLP, et al. Exercise training and epigenetic regulation: multilevel modification and regulation of gene expression. Adv Exp Med Biol.. 2017. p. 281–322.
- Voisin S, Eynon N, Yan X, et al. Exercise training and DNA methylation in humans. Acta Physiol. 2015;213:39–59.
- Landen S, Voisin S, Craig JM, et al. Genetic and epigenetic sex-specific adaptations to endurance exercise. Epigenetics. 2019; DOI:10.1080/15592294.2019.1603961.
- Lappalainen T, Greally JM. Associating cellular epigenetic models with human phenotypes. Nat Rev Genet. 2017;18:441–451.
- Weinhold B. Epigenetics: the science of change. Environ Heal Perspect. 2006;114:160–167.
- Huang B, Zhang R. Regulatory non-coding RNAs: revolutionizing the RNA world. Mol Biol Rep. 2014;41:3915–3923.
- Tao H, Yang -J-J, Shi K-H. Non-coding RNAs as direct and indirect modulators of epigenetic mechanism regulation of cardiac fibrosis. Expert Opin Ther Targets. 2015;19:707–716.
- Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9:3–12.
- Rooney J Further thoughts on mercury, epigenetics, genetics and amyotrophic lateral sclerosis. 2011;523–524.
- van Dijk SJ, Molloy PL, Varinli H, et al. Epigenetics and human obesity. Int J Obes (Lond). 2015;39:85–97.
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254.
- Sharples AP, Stewart CE, Seaborne RA. Does skeletal muscle have an ’epi’-memory? The role of epigenetics in nutritional programming, metabolic disease, aging and exercise. Aging Cell. 2016;15:603–616.
- Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;1;6:a019133.
- Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389.
- Ohm JE, McGarvey KM, Yu X, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242.
- Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236.
- Gal-Yam EN, Egger G, Iniguez L, et al. Frequent switching of polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105:12979–12984.
- Strunnikova M, Schagdarsurengin U, Kehlen A, et al. Chromatin inactivation precedes de novo DNA methylation during the progressive epigenetic silencing of the RASSF1A promoter. Mol Cell Biol. 2005;25:3923–3933.
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492.
- Pacis A, Mailhot-Leonard F, Tailleux L, et al. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc Natl Acad Sci U S A. 2019;116:6938–6943.
- Guibert S, Weber M. Functions of DNA methylation and hydroxymethylation in mammalian development. Curr Top Dev Biol. 2013;104:47–83.
- Bentley GA, Lewit-Bentley A, Finch JT, et al. Crystal structure of the nucleosome core particle at 16A resolution. J Mol Biol. 1984;176:55–75.
- McGee SL, Hargreaves M. Histone modifications and exercise adaptations. J Appl Physiol. 2011;110:258–263.
- Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16:1467–1481.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395.
- Hussain MU. Micro-RNAs (miRNAs): genomic organisation, biogenesis and mode of action. Cell Tissue Res. 2012;349:405–413.
- Widmann M, Nieß AM, Munz B. Physical exercise and epigenetic modifications in skeletal muscle. Sport Med. 2019.
- McClay JL, Shabalin AA, Dozmorov MG, et al. High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol. 2015;16:291.
- Hannon E, Spiers H, Viana J, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2015;19:48.
- Banovich NE, Lan X, McVicker G, et al. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 2014;10:e1004663.
- Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10–15.
- van Dongen J, Nivard MG, Willemsen G, et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun. 2016;7:11115.
- Zimmer P, Bloch W. Physical exercise and epigenetic adaptations of the cardiovascular system. Herz. 2015;40:353–360.
- Barrès R, Yan J, Egan B, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–411.
- Fabre O, Ingerslev LR, Garde C, et al. Exercise training alters the genomic response to acute exercise in human adipose tissue. Epigenomics. 2018;10:1033–1050.
- Russell AP, Lamon S, Boon H, et al. Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J Physiol. 2013;591:4637–4653.
- Moher D, Shamseer L, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4:1–19.
- Alibegovic AC, Sonne MP, Højbjerre L, et al. Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am J Physiol - Endocrinol Metab. 2010;299:752–763.
- Bajpeyi S, Covington JD, Taylor EM, et al. Skeletal muscle PGC1alpha −1 nucleosome position and −260nt DNA methylation determine exercise response and prevent ectopic lipid accumulation in men. Endocrinology. 2017;158:2190–2199.
- Lindholm ME, Marabita F, Gomez-Cabrero D, et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9:1557–1569.
- Nitert MD, Dayeh T, Volkov P, et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes. 2012;61:3322–3332.
- Seaborne RA, Strauss J, Cocks M, et al. Human skeletal muscle possesses an epigenetic memory of hypertrophy. Sci Rep. 2018;8:1898.
- Robinson MM, Dasari S, Konopka AR, et al. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab. 2017;25:581–592.
- Lindholm ME, Giacomello S, Werne Solnestam B, et al. The impact of endurance training on human skeletal muscle memory, global isoform expression and novel transcripts. PLOS Genet. 2016;12:e1006294.
- McGee SL, Fairlie E, Garnham AP, et al. Exercise-induced histone modifications in human skeletal muscle. J Physiol. 2009;587:5951–5958.
- Nielsen S, Scheele C, Yfanti C, et al. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J Physiol. 2010;588:4029–4037.
- Ringholm S, Biensø RS, Kiilerich K, et al. Bed rest reduces metabolic protein content and abolishes exercise-induced mRNA responses in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E649–58.
- Rivas DA, Lessard SJ, Rice NP, et al. Diminished skeletal muscle microRNA expression with aging is associated with attenuated muscle plasticity and inhibition of IGF-1 signaling. Faseb J. 2014;28:4133–4147.
- D’Souza RF, Markworth JF, Aasen KMM, et al. Acute resistance exercise modulates microRNA expression profiles: combined tissue and circulatory targeted analyses. PLoS One. 2017;12e0181594.
- Fyfe JJ, Bishop DJ, Zacharewicz E, et al. Concurrent exercise incorporating high-intensity interval or continuous training modulates mTORC1 signaling and microRNA expression in human skeletal muscle. Am J Physiol - Regul Integr Comp Physiol. 2016;310:R1297–311.
- Keller P, Vollaard NBJ, Gustafsson T, et al. A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J Appl Physiol. 2011;110:46–59.
- Zhang T, Birbrair A, Wang ZM, et al. Improved knee extensor strength with resistance training associates with muscle specific miRNAs in older adults. Exp Gerontol. 2015;62:7–13.
- Mueller M, Breil FA, Lurman G, et al. Different molecular and structural adaptations with eccentric and conventional strength training in elderly men and women. Gerontology. 2011;57:528–538.
- D’Souza RF, Bjørnsen T, Zeng N, et al. MicroRNAs in muscle: characterizing the powerlifter phenotype. Front Physiol. 2017;8:1–12.
- Chen J-F, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233.
- Sun X, Lin J, Zhang Y, et al. MicroRNA-181b improves glucose homeostasis and insulin sensitivity by regulating endothelial function in white adipose tissue. Circ Res. 2016;118:810–821.
- Mercatelli N, Fittipaldi S, De Paola E, et al. MiR-23-TrxR1 as a novel molecular axis in skeletal muscle differentiation. Sci Rep. 2017;7:7219.
- Brunet-Vega A, Pericay C, Quílez ME, et al. Variability in microRNA recovery from plasma: comparison of five commercial kits. Anal Biochem. 20154;88:28–35.
- Whiley PJ, De La Hoya M, Thomassen M, et al. Comparison of mRNA splicing assay protocols across multiple laboratories: recommendations for best practice in standardized clinical testing. Clin Chem. 2014.
- Russell AP, Lamon S. Exercise, skeletal muscle and circulating microRNAs. Prog Mol Biol Transl Sci. 2015;135:471–496.
- Ogasawara R, Akimoto T, Umeno T, et al. MicroRNA expression profiling in skeletal muscle reveals different regulatory patterns in high and low responders to resistance training. Physiol Genomics. 2016;48:320–324.
- Zacharewicz E, Della Gatta P, Reynolds J, et al. Identification of microRNAs linked to regulators of muscle protein synthesis and regeneration in young and old skeletal muscle. PLoS One. 2014;9(12):e114009.
- McLean CS, Mielke C, Cordova JM, et al. Gene and microRNA expression responses to exercise; relationship with insulin sensitivity. PLoS One. 2015;10:1–14.
- Russell AP, Wallace MA, Kalanon M, et al. Striated muscle activator of Rho signalling (STARS) is reduced in ageing human skeletal muscle and targeted by miR-628-5p. Acta Physiol. 2017;220:263–274.
- Hodson N, Philp A. The importance of mTOR trafficking for human skeletal muscle translational control. Exerc Sport Sci Rev. 2019;47(1):46–53.
- Krist B, Florczyk U, Pietraszek-Gremplewicz K, et al. The role of miR-378a in metabolism, angiogenesis, and muscle biology. Int J Endocrinol. 2015;2015:281756.
- Horak M, Novak J, Bienertova-Vasku J. Muscle-specific microRNAs in skeletal muscle development. Dev Biol. 2016;410:1–13.
- Siracusa J, Koulmann N, Banzet S. Circulating myomiRs: a new class of biomarkers to monitor skeletal muscle in physiology and medicine. J Cachexia Sarcopenia Muscle. 2018;9:20–27.
- Ramos AE, Lo C, Estephan LE, et al. Specific circulating microRNAs display dose-dependent responses to variable intensity and duration of endurance exercise. Am J Physiol Heart Circ Physiol. 2018;315:H273–83.
- Nohata N, Hanazawa T, Enokida H, et al. microRNA-1/133a and microRNA-206/133b clusters: dysregulation and functional roles in human cancers. Oncotarget. 2012;3:9–21.
- Lu L, Zhou L, Chen EZ, et al. A novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PLoS One. 2012;7: e27596.
- Tam S, Tsao MS, McPherson JD. Optimization of miRNA-seq data preprocessing. Brief Bioinform. 2015;16:950–963.
- Chazaud B. Inflammation during skeletal muscle regeneration and tissue remodeling: application to exercise-induced muscle damage management. Immunol Cell Biol. 2016;94:140–145.
- Malm C, Nyberg P, Engstr M, et al. Muscular adaptation to physical stress is of significant importance for normal muscular development and function. without stimulation from physical activity. 2000;243–262.
- Bheda P, Schneider R. Epigenetics reloaded: the single-cell revolution. Trends Cell Biol. 2014;24:712–723.
- Riley RD, Higgins JPT, Deeks JJ. Interpretation of random effects meta-analyses. Bmj. 2011;342:964–967.