
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The Developmental Origins of Health and Disease (DOHaD) hypothesis posits that in utero and early life conditions can disrupt normal fetal development and program susceptibility to later-life disease. Metastable epialleles are genomic loci in which CpG methylation patterning is responsive to maternal diet and conserved across time and tissues. Thus, these sites could serve as ‘signatures’ of gestational environment conditions. Here, we sought to determine if methylation of metastable epialleles was associated with changes in childhood body mass index (BMI) z-scores across ages one, two and ten in the Extremely Low Gestational Age Newborns (ELGAN) cohort. CpG methylation of 250 probes (corresponding to 111 genes) within metastable epiallele regions was measured in placental tissue. Linear mixed effects models were fit to evaluate the overall and sex-stratified associations between methylation and changes in BMI z-score over time. In total, 26 probes were associated (p < 0.05) with changes in BMI z-score overall, including probes within Mesoderm Specific Transcript (MEST) and Histone Deacetylase 4 (HDAC4), which have previously been associated with childhood obesity and adipogenesis. Sex-stratified analyses revealed a significant association, after adjusting for multiple comparisons (q < 0.05), within female placentas for one probe annotated to the imprinted gene PLAG1 Like Zinc Finger 1 (PLAGL1). These findings suggest epigenetic marks may be involved in programming susceptibility to obesity in utero and highlight the potential to use placental tissues in predicting growth rate trajectories among premature infants.
Introduction
The placenta plays a critical role in determining fetal health by transferring nutrients, waste, and growth promoting hormones [Citation1]. The placenta also serves as an incomplete barrier to exposures from the maternal environment which can disrupt fetal development and influence susceptibility to later-life health disorders, as posited by the Developmental Origins of Health and Disease (DOHaD) hypothesis [Citation2,Citation3]. Increasing evidence suggests that molecular mechanisms mediating this process include epigenetic mechanisms [Citation1,Citation3–Citation6], such as methylation of cytosines in cytosine-guanine (CpG) dinucleotides. During embryogenesis, CpG methylation patterning is highly sensitive to environmental insult because of the high rate of DNA synthesis and chromatin regulation required for organismal development [Citation7–Citation10]. Interestingly, genomic loci have been identified in which epigenetic patterning is variable between isogenic organisms and conserved across time andtissues [Citation11].
These sites, termed ‘metastable epialleles,’ were first characterized as variably expressed due to persistent, systemic (cross-tissue) epigenetic modifications established prior to gastrulation [Citation11]. In contrast to traditional alleles, in which methylation is erased and re-established in a predictable manner, the establishment of methylation at these sites is stochastic and responsive to environmental factors in a locus-specific manner. Kessler et al. extended the definition to incorporate genomic regions under partial, nondeterministic genetic influence in order to reflect the genetic heterogeneity observed in human populations [Citation12]. Consistency in methylation profiles has been observed across tissues, including the placenta, at imprinted genes [Citation13]. This is of note because many imprinted genes regulate fetal growth and development [Citation10,Citation14,Citation15], placental-specific imprinting preferentially maintains maternal germline-derived DNA methylation [Citation16], and several metastable epialleles overlap imprinted loci. Placental CpG methylation of metastable epialleles could serve both as a molecular biomarker of maternal environmental influences on development as well as a modification that is predictive of growth later in life.
A growing body of literature demonstrates metastable epiallele methylation is a molecular biomarker of environmental influences affecting development. In mouse models, maternal dietary methyl donor supplementation significantly increases methylation of metastable epialleles and is associated with the penetrance of the pseudoagouti coat color, tail kinking, and adult-onset obesity [Citation17–Citation19]. Notably, maternal dietary genistein supplementation reverses BPA-induced hypomethylation of the agouti viable yellow (Avy) metastable epiallele, causing a shift in the coat-color distribution and a decreased incidence of adult-onset obesity among offspring [Citation20]. Limited evidence suggests methylation of candidate human metastable epialleles is also influenced by maternal diet, as methyl-donor nutrient intake and gestational famine have been associated with methylation of these sites [Citation21–Citation23]. Moreover, the elevated rates of obesity observed in the Dutch Hunger Winter study were confined to individuals born to mothers exposed to famine during the early-gestational period [Citation5]. Taken together, these studies suggest that epigenetic programming may establish inter-individual susceptibility to obesity during critical periods of early gestation. Considering these findings, the relationship between methylation of metastable epialleles and later-life changes in body size warrants further investigation.
To address whether CpG methylation of metastable epialleles influences growth rate, we sought to determine if placental methylation of putative metastable epialleles was associated with changes in childhood body mass index (BMI) z-scores across ages one, two and ten in the Extremely Low Gestational Age Newborns (ELGAN) cohort. Additionally, sex-stratified analyses were used to determine the associations among males and females separately, as sexual epigenetic dimorphism has been observed in the human placenta [Citation24]. The goal of this study was to investigate whether methylation patterns established during early development might be associated with later life health outcomes, in concordance with the DOHaD paradigm. It was hypothesized that placental methylation of metastable epialleles would be associated with growth rate trajectories and that probes would be annotated to genes involved in growth and metabolism. This is among the first studies to examine metastable epialleles in the context of the placenta as it relates to child health across multiple life stages.
Results
Study cohort
Demographic information for the ELGAN placental epigenetics sub cohort (n = 426) was collected at childbirth and is largely representative of the full cohort (). Data on children’s BMI were collected during follow-up at ages one, two and ten years. In the sub cohort for the current study, there were 203 (47.7%) females and 223 (52.4%) males, and the average gestational age was 26 weeks. At ages one, two and ten years, the average child’s BMI was 16.7, 16.0, and 17.7, respectively. The average maternal BMI and age were 25.4 and 29.4, respectively. Approximately 34% of the sub cohort was covered by public insurance, and mothers self-identified as White (62.1%), Black (28.4%) or other (9.5%). Further details on subject characteristics are presented in .
Table 1. ELGAN (n = 889) and placental epigenetics sub cohort (n = 426) characteristics.
Overall association between placental cpg methylation and changes in BMI z-score
In assessing the relationship between CpG methylation of metastable epialleles and changes in BMI z-score, linear mixed effects models were employed to account for within-subject correlation of BMI measurements. These models assessed whether an increase in placental CpG methylation was associated with changes in BMI z-score across ages one, two and ten. In the overall analysis, 26 probes, annotated to 14 genes, displayed significant associations (p < 0.05) with changes in BMI z-score (). The probe (cg13211302) displaying the strongest association (β = 0.0056, p = 0.0003) was not annotated to any genes. After adjusting for multiple comparisons at q < 0.05, none of the statistically significant associations between methylation and changes in BMI z-score were conserved. However, several probes were annotated to genes involved in obesity, adipogenesis and development, including PLAG1 Like Zinc Finger 1/Hydatidiform Mole Associated and Imprinted (PLAGL1/HYMAI), Mesoderm Specific Transcript (MEST), Histone Deacetylase 4 (HDAC4), and Engrailed Homeobox 1 (EN1).
Table 2. Probes associated with changes in BMI z-score overall (n = 26 probes, representing 14 genes).
Sex-stratified analyses reveal imprinted gene PLAGL1/HYMAI is associated with changes in BMI z-score among females
To evaluate the association between placental methylation of metastable epialleles and changes in BMI z-score among males and females separately, sex-stratified analyses were performed. In total, 26 probes, annotated to 15 genes, displayed significant associations (p < 0.05) with changes in BMI z-score among placentas derived from female pregnancies (). After adjusting for multiple comparisons (q < 0.05), one association between methylation and changes in BMI z-score was conserved at a probe (cg00702231) annotated to the imprinted gene PLAGL1/HYMAI (β = −0.0119, p < 0.0001, q < 0.01) (, ). Additionally, some probes were annotated to genes related to adipocyte differentiation and obesity, including MEST, Fatty Acid Desaturase 3 (FADS3), and Regulator of G Protein Signaling 14 (RGS14). Among placentas derived from male pregnancies, 18 probes, annotated to 10 genes, displayed significant associations (p < 0.05) with changes in BMI z-score (). However, no associations were conserved after adjusting for multiple comparisons.
Table 3. Probes associated with changes in BMI z-score within placentas derived from female pregnancies (n = 26 probes, representing 15 genes).
Table 4. Probes associated with changes in BMI z-score within placentas derived from male pregnancies (n = 18 probes, representing 10 genes).
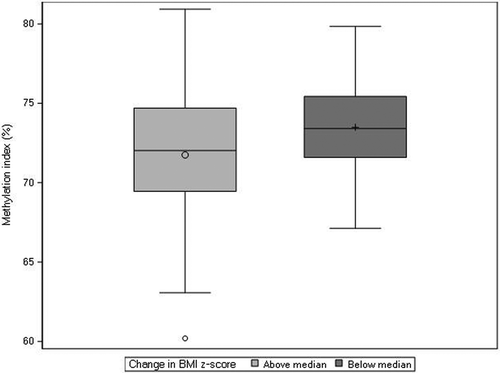
Figure 1. Distribution of CpG methylation at probe cg00702231, annotated to PLAGL1, within placentas derived from female pregnancies. Females were dichotomized as experiencing a change in BMI z-score that was above (n = 91) or below (n = 91) the median change in BMI z-score across ages one and ten (median = −0.08).
Sub-analyses across individual epochs were consistent with findings across ages one, two and ten
As growth is more rapid between ages one and two than two and ten, we evaluated the association between placental methylation of metastable epialleles and changes in BMI z-score across these epochs separately. Fewer associations were identified between ages two and ten (n = 9 CpG probes) than ages one and two (n = 25 CpG probes), but probes identified with p < 0.05 were largely consistent with the primary analysis across ages one, two and ten (Supplementary Table 2–7). After adjusting for multiple comparisons, we identified associations across ages one and two at q< 0.05 for cg11324748 (ANKRD20A11P) and cg13012494 (SPATC1L). Among male-derived placentas, cg11324748 (ANKRD20A11P) was significant at q < 0.05. Across ages two and ten, we did not identify associations after adjusting for multiple comparisons (q < 0.05). However, cg00702231 (PLAGL1) was identified in the overall analysis and within placentas derived from female pregnancies (p = 0.03 and 0.02, respectively).
Finally, we explored differentially methylated regions using the DMRScan package, but no significant results were identified (data not shown).
Discussion
In the present study we sought to determine if CpG methylation of metastable epialleles within the placenta was associated with changes in BMI z-score across ages one, two and ten among children born extremely preterm. Metastable epialleles are of interest because CpG methylation at these genomic loci is responsive to maternal diet and systemically maintained across tissues over time [Citation11]. Several of the identified probes were annotated to genes that have been implicated in development, adipogenesis and obesity. After stratifying the analysis by sex, one probe annotated to the imprinted gene PLAGL1 was significantly associated with changes in BMI z-score within placentas derived from female pregnancies. Methylation of this gene has previously demonstrated lability to maternal diet, consistent with the characterization of metastable epialleles. These findings support the DOHaD hypothesis, providing evidence for an association between in utero programming and later-life health outcomes in the ELGAN cohort.
Several probes associated with changes in BMI z-score were annotated to genes involved in development, obesity and adipogenesis [Citation25–Citation31]. MEST and HDAC4 were of special interest, as associations between their methylation profiles and obesity-related outcomes have been explored among children and adults. In the present study, DNA methylation of a probe annotated to MEST was positively associated with BMI z-score across childhood. In contrast, Nahm et al. [Citation32] found that methylation of MEST was negatively associated with anthropometric measures among females at ages one and three. Additionally, in assessing later-life associations between DNA methylation of MEST and body weight, Hajj et al. [Citation33] identified significantly decreased blood methylation of MEST among obese adults. Differences in findings between studies may be a result of differences in scientific analyses (e.g. those that focus on single time point associations versus those that examine changes in body size over time). This is further supported in that growth occurs most rapidly during the earliest stages of development. MEST is a known imprinted gene which has displayed similar methylation patterning between the placenta and other fetal somatic tissues from all three germ layers [Citation13]. Taken together, these data suggest that early-life epigenetic programming of MEST, as indicated by placental methylation, could predict susceptibility to later-life obesity regulated through somatic tissue responses.
A second gene of interest identified in the present study, HDAC4, showed increased CpG probe methylation with respect to BMI z-score across childhood. Similarly, in comparing obese and lean pre-school aged children, Li et al. [Citation34] showed hypermethylation of HDAC4 was associated with the obese phenotype, and that HDAC4 hypermethylation was inversely associated with gene expression. HDAC4 belongs to a group of histone deacetylases which play a critical role in transcriptional regulation; it is also involved in adipocyte expansion, energy expenditure regulation and may be protective of obesity [Citation30,Citation35,Citation36]. Thus, the functions of HDAC4 may shed light on the biological underpinnings of associations between this gene and body size.
Sexual dimorphism has been observed in the human placenta [Citation37–Citation40], and we have recently highlighted sexual epigenetic dimorphism at the level of CpG methylation [Citation24]. Here, sex-stratified analyses revealed two probes annotated to PLAGL1 and one probe annotated to MEST displayed associations to changes in BMI z-score within placentas derived from female pregnancies. Among these, a single probe annotated to the imprinted gene PLAGL1/HYMAI displayed a statistically significant negative association. Gonzalez-Nahm et al. [Citation32] identified methylation of both PLAGL1 and MEST in cord blood were negatively associated with weight-for-length z-scores among one year old females. In contrast, additional studies examining cord blood have identified that methylation of PLAGL1 is positively correlated with anthropometric measures at 32 weeks of gestation, birth weight and age one [Citation41,Citation42]. Interestingly, DNA methylation of PLAGL1 is also influenced by maternal diet, namely alcohol, folate and vitamin B2 intake [Citation41,Citation42]. PLAGL1 and MEST are both maternally-imprinted genes. This is of note because in identifying placenta-specific imprinting, Hanna et al. [Citation16] noted that the vast majority of novel DMRs had maternally inherited DNA methylation. Taken together, prior literature and the data from the present study suggest that methylation of PLAGL1 may be labile to maternal diet and associated with changes in body size over time, specifically among females.
The results from the current study add to the growing body of literature demonstrating associations between CpG methylation of candidate metastable epialleles and later-life outcomes and is among the first to highlight CpG methylation of these genomic loci within the placenta. Still, some factors should be considered in the interpretation of our findings. First, the ELGAN birth cohort is comprised of infants born extremely preterm (prior to 28 weeks gestation), which could limit the generalizability of our results. Second, BMI does not completely account for body composition or changes in body fat related to growth. Still, the use of BMI as a health metric is appropriate as it is largely used in epidemiological studies and clinical settings to define obesity. Third, weight is influenced by postnatal environmental exposures, genetics, and lifestyle factors. Still, despite the lack of a controlled environment, as with any human population-based study, we demonstrate evidence for a role of epigenetics in the early life programming of later-life body size. Finally, CpG methylation was assessed in the placenta at one time point. While it is hypothesized that methylation of metastable epialleles is stable across time and tissues [Citation11], collection of additional samples across time and tissues and the functional assessment of mRNA expression would be helpful in determining their influence on growth rate trajectories.
This is among the first studies to examine the association between placental methylation of candidate metastable epialleles and longitudinal measures of body weight. Our findings provide support for the DOHaD hypothesis, as several associations were identified among probes annotated to genes involved in growth and metabolism. A major finding from the study was the association between PLAGL1 and BMI in childhood, as this gene has previously been associated with early life BMI and demonstrated lability to maternal diet [Citation32,Citation41,Citation42]. Future studies should incorporate maternal dietary information and assess the cross-tissue and longitudinal stability of methylation of these sites, as they may be vestiges of intrauterine environmental conditions functionally linked to later-life obesity risk.
Materials and methods
Subject recruitment and sample collection
The ELGAN birth cohort was established to identify factors contributing to the risk of neurological disorders among infants born extremely preterm, which was defined as a live birth before 28 completed weeks of gestation. Study procedures were approved by the Institutional Review Board at each of the 14 participating ELGAN sites [Citation43], and methods of subject recruitment and sample collection have been described elsewhere [Citation43]. Briefly, from 2002 to 2004, women giving birth at one of the 14 participating sites before 28 weeks of gestation were asked to enroll in the ELGAN study. Their consent was provided either upon hospital admission prior to or shortly after delivery. In total, 1506 infants and 1249 mothers enrolled in the ELGAN study. The cohort consists of both singleton (65%) and non-singleton births (35%), however there are no genetically identical twins in this placental repository. At the time of delivery, placenta specimens were collected from over 90% of study participants. Eighty-two percent of the samples were obtained within 1 hour of delivery. For epigenetic analyses, we focused on placental samples from the subset of study participants who were evaluated at 10 years of age (n = 889). Within this subset of study participants, sufficient placental tissue, and thus, DNA methylation data, was available for 426. These 426 placentas and the corresponding offspring comprise the sample for the present analysis.
Participating women gave permission for the collection of a sample of their placenta for the ELGAN study. Upon delivery, placentas were placed into a sterile exam basin and taken to the sampling room, at which point, biopsies of the placentas were collected as previously described [Citation44]. The area sampled was at the midpoint of the longest distance between the cord insertion and the edge of the placental disk. To expose the chorion, the amnion was pulled away from the underlying chorion using a sterile technique. A tissue sample was collected by applying traction to the chorion and the underlying trophoblast tissue and cutting a sample out at the base of this tissue structure. The chorion is comprised of fetal membrane surrounding the developing embryo. Hence, the tissue sample was restricted to the fetal side of the placenta. Tissue samples were subsequently placed into a sterile 2 mL cryo-vial that was immediately submerged in liquid nitrogen. Samples were shipped to the University of North Carolina at Chapel Hill and stored at −80 °C until processing [Citation44].
DNA extraction and assessment of DNA methylation
DNA sequences >18 nucleotides long were isolated using ALLPrep DNA/RNA/miRNA Universal Kit (Qiagen, Valencia, CA). Extracted DNA samples were then shipped on dry ice to Wayne State University where bisulfite conversion was performed using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA). The Illumina Infinium MethylationEPIC BeadChip (Illumina, San Diego, CA) was then used to profile methylation status at more than 850,000 CpG sites across the genome. Average methylation values (β values) were computed to represent the ratio of methylated to unmethylated signal intensities.
As previously described [Citation45], β values were background corrected using the normal-exponential out-of-band (noob) correction method and normalized with functional normalization. To evaluate batch effects, a principal component analysis (PCA) was performed. Plate was identified as a significant source of variation; thus, we performed a surrogate variable analysis via the ComBat package [Citation46]. PCA was then repeated to ensure this bias was sufficiently corrected.
Identification of illumina probes corresponding to metastable epialleles
Prior to analyses, a list of 252 Illumina probes, annotated to 112 genes, was derived from the publicly available Infinium Methylation EPIC v1.0 B4 Manifest file (https://support.illumina.com/downloads/infinium-methylationepic-v1-0-product-files.html) (Supplementary Table 1, Supplementary Figure 1). These probes corresponded to genomic loci identified as metastable epiallele regions and were identified using the chromosome number and map information provided in the most recent genome-wide screening for human metastable epialleles [Citation12]. Probes annotated to the Y chromosome (n = 2) were excluded from the analysis to minimize technical artifacts among placentas derived from female pregnancies.
BMI assessment at ages 1, 2, and 10
At ages one, two, and ten, body weight and length were obtained by study personnel. Body mass index (BMI) was then calculated using the following formula: BMI = Weight (in kilograms)/Height (in meters). BMI z-score and percentiles for age and sex were then determined using the Statistical Analysis Software program based on current CDC growth charts [Citation47,Citation48].
Covariates
To identify covariates, a directed acyclic graph (DAG) approach was used. Maternal pre-pregnancy BMI (z-score), maternal race, infant sex, insurance status, multiple gestations, and smoking while pregnant were selected as covariates based on their potential to confound associations between methylation of metastable epialleles and changes in BMI z-score. These indicators were collected at enrollment in the ELGAN study via a structured questionnaire.
Statistical analysis
To test the hypothesis that placental methylation of candidate metastable epialleles is associated with changes in BMI z-score, we implemented a linear mixed effects model at each of the probes. The general model form was as follows:
where BMI z-scoreij is the measure of the child’s BMI relative to children of the same sex and age for the ith child at time j. Xi0 is the average methylation level (β value) for a probe located within a metastable epiallele region, as measured in placental tissue. Methylation levels were first multiplied by 100 to estimate the change in BMI z-score associated with a 1% increase in methylation. Time is the duration between baseline and each BMI z-score measurement. The β coefficient for Xi0(Timeij) is the expected annual change in BMI z-score corresponding to a 1% increase in methylation at the respective CpG probe. Zi0 is a matrix of potential confounders, representing adjustments made for fetal sex (male, female), gestational age and maternal pre-pregnancy BMI z-score (continuous variables), maternal race (White, Black, and Other), and insurance (public insurance yes or no). μi0 is a random intercept for each child used to account for the within-subject correlation of the repeated BMI z-score measurements, and eij is the error term. Due to evidence of sexual epigenetic dimorphism in CpG methylation patterning of the human placenta [Citation24], we conducted sex-stratified analyses to determine the associations among males and females separately.
While our primary hypothesis was that metastable epialleles established a single growth rate trajectory that would be maintained over the life course, we also evaluated the epochs across ages one and two and ages two and ten separately. Additionally, metastable epiallele probes were analyzed in the context of differentially methylated regions (DMRs) using the DMRScan package (R version 3.6.0) [Citation49]. For all models, we used a post-hoc adjustment for multiple comparisons via the Benjamini-Hochberg (FDR) procedure and controlled for the false discovery rate of 0.05. Apart from the DMR analysis, all analyses were conducted in Stata/SE version 15 (College Station, TX).
Supplemental Material
Download MS Excel (42 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data can be accessed here.
Additional information
Funding
Related Research Data
References
- Maccani MA, Marsit CJ. Epigenetics in the placenta. Am J Reprod Immunol. 2009;62:78–89.
- Heindel JJ, Vandenberg LN. Developmental origins of health and disease: a paradigm for understanding disease cause and prevention. Curr Opin Pediatr. 2015;27:248–253.
- Barker DJ, Clark PM. Fetal undernutrition and disease in later life. Rev Reprod. 1997;2:105–112.
- Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31:363–373.
- Schulz LC. The dutch hunger winter and the developmental origins of health and disease. Proc Natl Acad Sci USA. 2010;107:16757–16758.
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–251.
- Dolinoy DC, Das R, Weidman JR, et al. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr Res. 2007;61:30R–37R.
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093.
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365.
- Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141:1805–1813.
- Rakyan VK, Blewitt ME, Druker R, et al. Metastable epialleles in mammals. Trends Genet. 2002;18:348–351.
- Kessler NJ, Waterland RA, Prentice AM, et al. Establishment of environmentally sensitive DNA methylation states in the very early human embryo. Sci Adv. 2018;4:eaat2624.
- Murphy SK, Huang Z, Hoyo C. Differentially methylated regions of imprinted genes in prenatal, perinatal and postnatal human tissues. PLoS One. 2012;7:e40924.
- Frost JM, Moore GE, Ferguson-Smith AC. The importance of imprinting in the human placenta. PLoS Genet. 2010;6:e1001015.
- Ishida M, Moore GE. The role of imprinted genes in humans. Mol Aspects Med. 2013;34:826–840.
- Hanna CW, Peñaherrera MS, Saadeh H, et al. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 2016;26:756–767.
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300.
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 2007;104:13056–13061.
- Waterland RA, Dolinoy DC, Lin J-R, et al. Maternal methyl supplements increase offspring DNA methylation at axin fused. Genesis. 2006;44:401–406.
- Dolinoy DC, Weidman JR, Waterland RA, et al. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–572.
- Dominguez-Salas P, Moore SE, Baker MS, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746.
- Waterland RA, Kellermayer R, Laritsky E, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252.
- Finer S, Iqbal MS, Lowe R, et al. Is famine exposure during developmental life in rural Bangladesh associated with a metabolic and epigenetic signature in young adulthood? A historical cohort study. BMJ Open. 2016;6:e011768.
- Martin E, Smeester L, Bommarito PA, et al. Sexual epigenetic dimorphism in the human placenta: implications for susceptibility during the prenatal period. Epigenomics. 2017;9:267–278.
- Gesta S, Blüher M, Yamamoto Y, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci USA. 2006;103:6676–6681.
- Zhang C, Weng Y, Shi F, et al. The engrailed-1 gene stimulates brown adipogenesis. Stem Cells Int. 2016;(2016:7369491.
- Kamei Y, Suganami T, Kohda T, et al. Peg1/Mest in obese adipose tissue is expressed from the paternal allele in an isoform-specific manner. FEBS Lett. 2007;581:91–96.
- Takahashi M, Kamei Y, Ezaki O. Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am J Physiol Endocrinol Metab. 2005;288:E117–24.
- Voigt A, Ribot J, Sabater AG, et al. Identification of Mest/Peg1 gene expression as a predictive biomarker of adipose tissue expansion sensitive to dietary anti-obesity interventions. Genes Nutr. 2015;10:27.
- Paulo E, Wu D, Hecker PA, et al. Adipocyte HDAC4 activation leads to beige adipocyte expansion and reduced adiposity. J Endocrinol. 2018.
- Lear PV, González-Touceda D, Porteiro Couto B, et al. Absence of intracellular ion channels TPC1 and TPC2 leads to mature-onset obesity in male mice, due to impaired lipid availability for thermogenesis in brown adipose tissue. Endocrinology. 2015;156:975–986.
- Gonzalez-Nahm S, Mendez MA, Benjamin-Neelon SE, et al. DNA methylation of imprinted genes at birth is associated with child weight status at birth, 1 year, and 3 years. Clin Epigenetics. 2018;10:90.
- El Hajj N, Pliushch G, Schneider E, et al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes. 2013;62:1320–1328.
- Li Y, Zhou Y, Zhu L, et al. Genome-wide analysis reveals that altered methylation in specific CpG loci is associated with childhood obesity. J Cell Biochem. 2018;119:7490–7497.
- Fang M, Fan Z, Tian W, et al. HDAC4 mediates IFN-γ induced disruption of energy expenditure-related gene expression by repressing SIRT1 transcription in skeletal muscle cells. Biochim Biophys Acta. 2016;1859:294–305.
- Ozcan L, Ghorpade DS, Zheng Z, et al. Hepatocyte DACH1 is increased in obesity via nuclear exclusion of HDAC4 and promotes hepatic insulin resistance. Cell Rep. 2016;15:2214–2225.
- Rosenfeld CS. Sex-specific placental responses in fetal development. Endocrinology. 2015;156:3422–3434.
- Gabory A, Roseboom TJ, Moore T, et al. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol Sex Differ. 2013;4:5.
- McCarthy NS, Melton PE, Cadby G, et al. Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns. BMC Genomics. 2014;15:981.
- De Coster S, van Leeuwen DM, Jennen DGJ, et al. Gender-specific transcriptomic response to environmental exposure in Flemish adults. Environ Mol Mutagen. 2013;54:574–588.
- Azzi S, Sas TCJ, Koudou Y, et al. Degree of methylation of ZAC1 (PLAGL1) is associated with prenatal and post-natal growth in healthy infants of the EDEN mother child cohort. Epigenetics. 2014;9:338–345.
- Hoyo C, Daltveit AK, Iversen E, et al. Erythrocyte folate concentrations, CpG methylation at genomically imprinted domains, and birth weight in a multiethnic newborn cohort. Epigenetics. 2014;9:1120–1130.
- O’Shea TM, Allred EN, Dammann O, et al., The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Hum Dev. 2009;85:719–725.
- Onderdonk AB, Delaney ML, DuBois AM, et al., Detection of bacteria in placental tissues obtained from extremely low gestational age neonates. Am J Obstet Gynecol. 2008;198:110.e1–7.
- Santos HP, Bhattacharya A, Martin EM, et al. Epigenome-wide DNA methylation in placentas from preterm infants: association with maternal socioeconomic status. Epigenetics. 2019.
- Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883.
- Centers for disease control and prevention SAS program (ages 0 to < 20 years) | resources | growth chart training | nutrition | DNPAO | CDC. Centers for disease control and prevention. [cited 2019 February 23]. Available from: https://www.cdc.gov/nccdphp/dnpao/growthcharts/resources/sas.htm
- Centers for disease control and prevention growth charts - data table of BMI-for-age charts. Centers for disease control and prevention. [ cited 2019 February 23]. Available from: https://www.cdc.gov/growthcharts/html_charts/bmiagerev.htm
- Page CM, Vos L, Rounge TB, et al. Assessing genome-wide significance for the detection of differentially methylated regions. Stat Appl Genet Mol Biol. 2018 Sep 19;17(5).