
ABSTRACT
Increasing numbers of studies implicate abnormal DNA methylation in cancer and many non-malignant diseases. This is consistent with numerous findings about differentiation-associated changes in DNA methylation at promoters, enhancers, gene bodies, and sites that control higher-order chromatin structure. Abnormal increases or decreases in DNA methylation contribute to or are markers for cancer formation and tumour progression. Aberrant DNA methylation is also associated with neurological diseases, immunological diseases, atherosclerosis, and osteoporosis. In this review, I discuss DNA hypermethylation in disease and its interrelationships with normal development as well as proposed mechanisms for the origin of and pathogenic consequences of disease-associated hypermethylation. Disease-linked DNA hypermethylation can help drive oncogenesis partly by its effects on cancer stem cells and by the CpG island methylator phenotype (CIMP); atherosclerosis by disease-related cell transdifferentiation; autoimmune and neurological diseases through abnormal perturbations of cell memory; and diverse age-associated diseases by age-related accumulation of epigenetic alterations.
Introduction
Changes in the amount and distribution of 5-methylcytosine (5mC) in human DNA are of major importance to development and disease although the details of the many and varied relationships between DNA methylation and expression are still being untangled. Genomic 5mC is the predominant genetically programmed modification of human DNA, and the vast majority of 5mC is located in CG dinucleotides (CpGs) in human tissues other than brain and embryonic stem cells (ESC) [Citation1]. The other naturally modified bases in the human genome are derived from 5mC, except for 6-methyladenine, which is present in extremely low amounts and mostly in mitochondrial DNA [Citation2,Citation3]. The second most common modified base in human DNA is 5-hydroxymethylcytosine (5hmC). Biochemical and biological properties of 5mC and 5hmC differ [Citation4–Citation6]. Like 5mC, the genomic distribution and global levels of 5hmC are tissue-specific and disease-specific [Citation1,Citation2,Citation7]. Vertebrate DNA methylation generally refers just to 5mC in DNA but 5mC and 5hmC are indistinguishable by many assay methods still currently in use [Citation8]. In many studies (e.g., various bisulfite-based assays), minor amounts of the detected changes in ‘DNA methylation’ are attributable to the usually much lower levels of 5hmC [Citation9]. This review focuses on a tiny fraction of the >9000 articles in the last 20 years that describe increases in genomic 5mC (DNA hypermethylation) in disease relative to appropriate control tissues or cell populations. Only minor mention of 5hmC will be made, mostly in its (non-exclusive) role [Citation2,Citation9–Citation11] as an intermediate in DNA demethylation [Citation12,Citation13]. In addition, disease-related DNA hypomethylation is equally important as DNA hypermethylation (e.g., [Citation7,Citation14–Citation18],) but only DNA hypermethylation in cancer and other diseases is the focus of this review.
DNA hypermethylation has many roles in tissue-specific transcription control
Various studies implicate tissue- or cell type-specific DNA hypermethylation or hypomethylation at cis-acting transcription regulatory regions in the control of gene expression during development (e.g., [Citation1,Citation19–Citation23],). Tissue-specific promoter hypermethylation influences gene expression most notably at promoter regions that are rich in CpGs [Citation24]. However, tissue-specific DNA hypermethylation occurs more frequently in actively transcribed gene bodies and in intragenic or intergenic enhancers than at promoters [Citation1,Citation25–Citation27]. Understanding the differentiation-related roles for tissue-specific and cell type or cell composition-specific DNA hypermethylation [Citation28] is important for evaluating the biological functionality of disease-related DNA hypermethylation.
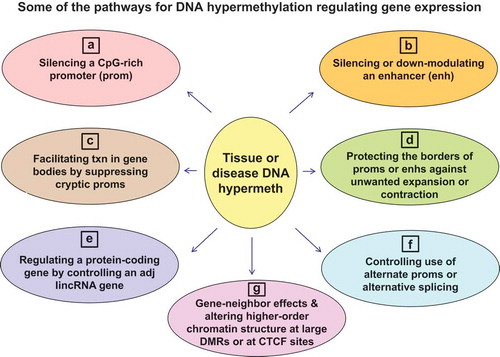
Known or likely roles of DNA hypermethylation in cis regulation of transcription include the following:
Silencing gene promoters, usually as a result of extensive DNA methylation near the transcription start site (TSS) in regions rich in CpGs [Citation19,Citation29] by regulating the binding of transcription factors (TFs) or other DNA-binding proteins and/or by altering chromatin modification or structure [Citation30–Citation32] (; note that the majority of human promoters are essentially unmethylated in all normal tissues);
Silencing enhancers or alternatively down-modulating them without complete loss of their activity [Citation33] ();
Facilitating transcription in gene bodies partly by silencing cryptic intragenic promoters [Citation9,Citation25] ();
Aiding transcription by maintaining the borders of active promoters or enhancers and thereby providing a barrier against repressive chromatin encroaching into the promoter or enhancer or activating chromatin spreading too far outward [Citation14,Citation34] ();
Indirectly regulating transcription of protein-encoding genes in cis by controlling expression of adjacent long intergenic non-coding RNA genes (lincRNA genes), which are often close to the 5ʹ end of the protein-encoding gene [Citation33] ();
Regulating in cis the nature of the transcript, rather than its levels, by controlling the use of alternative splice sites during co-transcriptional splicing or the choice of alternative promoters [Citation34,Citation35] ();
Controlling higher-order chromatin structure by inhibiting binding of CCCTC-binding factor (CTCF), a chromatin-looping protein, or by altering histone modifications in a large region of the DNA landscape [Citation14,Citation36,Citation37] ().
Other gene-neighbour effects like tissue-specific DNA methylation of small protein-encoding genes embedded in an intron of a larger dissimilar gene [Citation38] or of a tissue-specific enhancer in one gene linked to expression of the neighbouring gene [Citation39] ().
Figure 1. Developmental or disease-associated DNA hypermethylation can modulate gene expression in various ways. (a) and (b), down-regulation of gene expression by DNA hypermethylation (either initiating or stabilizing gene repression). (c) and (d), positive associations of DNA hypermethylation with gene expression. (e) and (g), positive or negative associations with gene expression. (f), effects on the nature of the transcript (RNA isoform), which can occur without changes the steady-state levels of the transcript.
DNA methylation can act in concert with, prior to, or subsequent to chromatin epigenetic changes (especially histone modifications and stable binding of non-histone proteins) to regulate transcription [Citation1]. DNA methylation, as a more stable epigenetic modification than repressive chromatin marks, can silence or down-modulate promoters or enhancers even without concomitant histone repressive modifications. Differential DNA methylation may often help stabilize a transcriptional state (often in conjunction with repressive chromatin epigenetics) as in X-chromosome inactivation in eutherian mammals [Citation40], rather than establish it. However, that too is an essential function for normal development, homeostasis, and some long-lasting changes in cellular physiology.
Caveats in DNA methylation analyses
There are many possible confounding factors in assigning causal roles to DNA methylation changes in diseased tissues. Some of these are as follows:
Ascertaining that the examined tissue is meaningful to the analyzed phenotype (e.g., blood, buccal samples or saliva sometimes, but not always, provide DNA methylation markers relevant to other tissues [Citation41]);
Confirming the differential DNA methylation findings in biological replicates and by different assay methods (e.g., [Citation42,Citation43],) to rule out technical problems and individual-specific variation in methylation of the examined CpG sites, unless it is a population study looking at person-specific epigenetic variation [Citation44];
Compensating for cell composition changes (although these are being addressed with increasing precision [Citation45]) and considering possible allele-specific differences in DNA methylation [Citation14];
Ruling out biologically unimportant bystander effects or neutral changes in DNA methylation consequent to expression changes (e.g., [Citation46–Citation48],);
Determining if a specific DNA methylation pattern persists from an earlier stage in development, physiology or disease as a non-functional relic [Citation49] or as a permissive DNA environment for possible inducible transcriptional changes [Citation50] rather than as a biologically active mark in the studied tissue;
DNA hypermethylation as markers for a broad variety of diseases
Abnormal increases in methylation at specific DNA sequences can serve as biomarkers for a variety of diseases. However, most of the many hundreds of potential biomarker applications are still under development rather than already in clinical use, with some notable exceptions, such as, hypermethylation at eight genes in cancer biomarker assays included in clinical guidelines [Citation51] and 5ʹ FMR1 methylation in molecular assays for the fragile X syndrome [Citation52].
The major types of applications for DNA hypermethylation markers for clinical use are as follows:
Diagnostic markers [Citation51–Citation56];
Prognostic markers [Citation51,Citation57–Citation59];
Markers for tailoring the best treatment for a subclass of the disease [Citation51,Citation57,Citation59,Citation60];
Markers for monitoring treatment efficacy [Citation58];
Markers for identifying genes to be examined for development of genetically or epigenetically targeted therapies [Citation57].
DNA hypermethylation markers (as well as hypomethylation markers) have been studied most extensively in cancer but, recently, aberrant DNA hypermethylation is being implicated in more and more disparate diseases, as described further below with some examples.
Cancer-linked DNA hypermethylation
Cancer epigenetics background
Very many studies indicate that changes in DNA methylation contribute to cancer (e.g., [Citation7,Citation19,Citation48,Citation53,Citation59–Citation66]). Cancers characteristically display extensive hypomethylation of DNA repeats and large regions of DNA as well as frequent focal DNA hypermethylation [Citation7,Citation14,Citation29,Citation37,Citation67–Citation69]. A DNA sequence that is normally partly methylated can even be hypermethylated in some specimens of a given cancer type and hypomethylated in others [Citation70]. Analogously, it was found that a gene promoter region could be hypermethylated in one neurological disease and hypomethylated at the same DNA sequence in a different neurological disease [Citation56].
Cancer-linked promoter and non-promoter hypermethylation
Cancer DNA hypermethylation has been studied most extensively at promoters (), which often are embedded in CpG-rich regions of ~1–2 kb called CpG islands or CGIs [Citation37,Citation48,Citation71–Citation73]. About 50% of human promoters (both DNA sequences immediately upstream and downstream of the TSS) overlap CGIs, and another ~22% of mammalian promoters are CpG rich without meeting the definition of a CGI [Citation58]. About 95% of CGI promoter regions are essentially unmethylated in normal human tissues irrespective of the expression level of the associated gene. Although CGIs are preferentially found near TSSs and partly overlapping promoters, many occur in other positions including in gene bodies, intergenic regions, at CGI shores (i.e., within 2 kb of another CGI), and even sometimes in enhancers [Citation58,Citation74]. Like CGI promoters, CGIs in enhancers are highly susceptible to cancer-associated DNA hypermethylation [Citation74].
Cancer-associated hypermethylation at non-promoter regions as well as at CGI promoters was seen in a quantitative methylation analysis (bisulfite pyrosequencing) of 19 cancer-associated hypermethylation markers in invasive breast cancers, adjacent histologically normal tissue and reduction mammoplasty controls [Citation48]. In this study, cancer-linked hypermethylation was observed at four CGI-promoter regions, two intronic CGIs, two CpG-enriched regions that did not meet the definition of a CGI but were within CGI shores, and one far-upstream CGI. The latter was 5.6 kb upstream of the 5ʹ end of EN1, which encodes a homeobox TF. This far-upstream CGI was also hypermethylated in normal EN1-expressing myoblasts and skeletal muscle relative to various non-expressing normal cell cultures or tissues. This result indicates a positive relationship between gene expression and DNA methylation at this intergenic CGI in normal samples, possibly through the mechanism shown in . In contrast, in breast cancer, this hypermethylation was associated with gene silencing probably because of spreading of the methylation through to the promoter itself [Citation48].
Cancer-linked CGI hypermethylation in already repressed promoters
Introduction of high levels of DNA methylation throughout an active CpG-rich promoter region of protein-coding or non-coding genes (ncRNA genes) is almost always associated with repression of that gene’s transcription unless the gene contains an alternative unmethylated promoter ( [Citation19,Citation33,Citation58,Citation59]. However, most promoters that display cancer-associated hypermethylation are linked to genes that were already silenced in control tissues by repressive chromatin marks (polycomb or PcG repression, enrichment in histone H3 lysine-27 trimethylation, H3K27me3) [Citation29,Citation75,Citation76]. In control cells, especially in ESC, these promoters usually display so-called bivalent (poised) chromatin, namely, both repressive chromatin marks (enrichment in H3K27me3) and active promoter-like marks (H3K4me3), and the associated genes are silent or only very weakly transcribed [Citation77]. In cancers, the disease-linked promoter DNA hypermethylation (which usually occurs with the loss of H3K27me3) might prevent induction of transcription under otherwise inducing conditions [Citation29,Citation78], as seen in some bivalent promoters upon differentiation of ESC [Citation79]. Nonetheless, occasionally CpG-rich promoters, including those encoding certain tumour suppressor proteins or miRNAs, are active in normal tissues but become repressed upon hypermethylation in cancer [Citation57]. Even the cancer-linked promoter methylation events that are without biological effect and are just bystanders in waves of oncogenesis-linked epigenetic dysregulation can serve as very useful markers for cancer.
CpG island methylator phenotype as a cancer biomarker
The CpG island methylator phenotype (CIMP) is exhibited by cancers that have mostly coordinate hypermethylation of small sets of CGI promoters which tend to be hypermethylated together in a given tumour subtype [Citation57]. In addition, oncogenesis-specific but sporadic DNA methylation in other CGI promoters is observed in both CIMP-positive and CIMP-negative cancers [Citation57].
In non-familial colon cancers, three distinct CIMP subgroups have been described as follows [Citation57]:
CIMP-high, where a set of CGI promoters, including those of tumour suppressor genes CDKN2A (cell cycling gene) and MLH1 (mismatch repair gene) are often coordinately hypermethylated in the cancers;
CIMP-low, where only partial and infrequent hypermethylation at CIMP-diagnostic promoters is seen;
Non-CIMP, where there are only very low rates of hypermethylation at this set of promoters.
A constellation of characteristics distinguishes CIMP-high, CIMP-low and non-CIMP cancers. For example, only CIMP-high sporadic colon cancers display high microsatellite instability, low frequencies of chromosome instability, and high frequencies of a pro-oncogenic BRAF mutation (V600E), and susceptibility to 5-fluorouracil chemotherapy [Citation57]. In CIMP-high colon cancers, only ~7% of the cancer-hypermethylated promoters had accompanying transcription down-regulation (at least two-fold) in cancer vs. adjacent histologically normal tissue [Citation76]. Importantly, these genes are often driver genes for the formation of colorectal cancer and include the TF-encoding FOXD2 gene, inhibitors of Wnt signalling SFRP1 and SFRP2, and MLH1, whose inactivation by promoter hypermethylation or by mutation causes microsatellite instability and increased rates of point mutations [Citation57,Citation76].
Cancer hypermethylation and gene silencing in the Wnt signalling pathway
The Wnt signalling pathway plays central roles in determining cell fate decisions in early embryogenesis and in the development and maintenance of stem cells [Citation80]. Pathogenic activation of Wnt signalling is often involved in early stages of carcinogenesis [Citation80]. DKK3 and DKK4 encode Wnt signalling inhibitors and frequently undergo epigenetic silencing in colon cancers [Citation81], like the above-mentioned Wnt-repressive SFRP1 and SFRP2 genes. Hypermethylation of the DKK3 and DKK4 promoters in cancer is associated with decreased expression of TET1, which itself is a common feature of colon cancer [Citation81,Citation82]. DKK3 and DKK4 hypermethylation was reversed by experimental up-regulation of TET1 [Citation81]. As described below in the section on TET enzymes, these enzymes protect CGIs from accumulating abnormal DNA methylation. DKK3 and DKK4 promoter hypermethylation due to cancer-linked decreases in TET1 levels is likely to be an early event in colon carcinogenesis [Citation81]. Using publicly available whole-genome bisulfite sequencing data (WGBS) [Citation1,Citation83,Citation84], I illustrate DKK3 promoter hypermethylation in two colon cancers and even stronger promoter hypermethylation in two breast cancer cell lines vs. seven diverse normal tissues or cell cultures, including colon and an untransformed DKK3-expressing breast epithelial cell line (). The consistent lack of DNA methylation in the CGI at the 5ʹ end of DKK3 in normal samples (even in non-expressing tissues) contrasts with the considerable-to-high levels of methylation at this promoter region in the cancers and cancer cell lines (, dotted box). For MCF-7, one of the breast cancer cell lines, transcription data were available and showed that, as expected, promoter hypermethylation was associated with repression of DKK3 ( and ).
Figure 2. Illustration of cancer-associated promoter hypermethylation and tissue-specific enhancer hypomethylation that correlate with expression. (a) Isoforms of the DKK3 tumour suppressor gene, which encodes a repressor of Wnt signalling (chr11:11,981,590–12,036,438, hg19, as seen in the UCSC genome browser [Citation84]). Broken arrow, TSS; tall blue boxes, coding exons; short boxes, 5ʹ or 3ʹ non-coding exonic regions. (b) CpG density and the one CpG island (CGI) in this region. (c) DNA methylation levels from WGBS (gold bars) and regions of significantly low DNA methylation relative to the rest of the same genome (horizontal blue bars [Citation83]). (d) Publicly available genome-wide mapping of 5hmC in prefrontal cortex (PFC), as determined by TET-assisted bisulfite sequencing [Citation11]. (e) Predicted features of chromatin segments determined from histone modifications [Citation1]. Str, Wk, Biv Promoter and Str, Wk, Biv Enhancer, chromatin with the histone marks characteristic of strong (active), weak, or bivalent (poised) promoters or enhancers. Enh/Prom, chromatin with histone modifications found in both active promoters and enhancers; Txn-chrom, chromatin with the histone modification of genes actively involved in transcription; Low signal, chromatin with little or no H3 lysine-4, 9, or 27 modifications; Repr, chromatin with repressive H3 lysine-27 trimethylation. (f) RNA-seq for two cell cultures (malignant, MCF-7, vs. non-malignant breast cancer epithelial cells, HMEC) and for tissues (bar graph for median values from hundreds of normal samples [Citation110]). Orange lettering, cancer tissues or cell lines; PBMC, peripheral blood mononuclear cells; HMEC, human mammary epithelial cell cultures (untransformed); TPM, transcripts per million.
![Figure 2. Illustration of cancer-associated promoter hypermethylation and tissue-specific enhancer hypomethylation that correlate with expression. (a) Isoforms of the DKK3 tumour suppressor gene, which encodes a repressor of Wnt signalling (chr11:11,981,590–12,036,438, hg19, as seen in the UCSC genome browser [Citation84]). Broken arrow, TSS; tall blue boxes, coding exons; short boxes, 5ʹ or 3ʹ non-coding exonic regions. (b) CpG density and the one CpG island (CGI) in this region. (c) DNA methylation levels from WGBS (gold bars) and regions of significantly low DNA methylation relative to the rest of the same genome (horizontal blue bars [Citation83]). (d) Publicly available genome-wide mapping of 5hmC in prefrontal cortex (PFC), as determined by TET-assisted bisulfite sequencing [Citation11]. (e) Predicted features of chromatin segments determined from histone modifications [Citation1]. Str, Wk, Biv Promoter and Str, Wk, Biv Enhancer, chromatin with the histone marks characteristic of strong (active), weak, or bivalent (poised) promoters or enhancers. Enh/Prom, chromatin with histone modifications found in both active promoters and enhancers; Txn-chrom, chromatin with the histone modification of genes actively involved in transcription; Low signal, chromatin with little or no H3 lysine-4, 9, or 27 modifications; Repr, chromatin with repressive H3 lysine-27 trimethylation. (f) RNA-seq for two cell cultures (malignant, MCF-7, vs. non-malignant breast cancer epithelial cells, HMEC) and for tissues (bar graph for median values from hundreds of normal samples [Citation110]). Orange lettering, cancer tissues or cell lines; PBMC, peripheral blood mononuclear cells; HMEC, human mammary epithelial cell cultures (untransformed); TPM, transcripts per million.](/cms/asset/a51588aa-88ea-4df9-abd1-afbca83d92b8/kepi_a_1638701_f0002_c.jpg)
Cancer-linked enhancer hypermethylation
Recent whole-genome methylation analyses of cancer vs. analogous control DNAs have shown that enhancers often display oncogenesis-linked decreases or increases in DNA methylation [Citation74,Citation85,Citation86]. Changes in DNA methylation at enhancers in cancers have been studied much less often than at promoters, partly because enhancers are more difficult to map experimentally in the appropriate control samples for the cancers. Nonetheless, enhancer hypermethylation is beginning to be implicated in tumorigenesis or tumour progression [Citation51]. For example, Glass et al. [Citation85] compared methylomes (enhanced reduced representation bisulfite sequencing) of 14 subtypes of acute myeloid leukaemia (AML) to those of normal hematopoietic stem and progenitor cells (HSPC). Two of the 14 subtypes, including the IDH2-mutant subtype, had frequent leukaemia-associated hypermethylated CpGs at regions that displayed enhancer chromatin in the HSPC controls. Enhancer chromatin was defined by peaks of histone H3 lysine-27 acetylation (H3K27ac) and H3K4me1 in chromatin immunoprecipitation/DNA-seq (ChIP-seq) profiles. Enhancer methylation profiles in AML were better able to predict AML subtype than were DNA methylation changes at promoters, gene bodies, CGIs or CGI-adjacent sequences.
Data downplaying the importance of cancer-related enhancer methylation was provided by Hon et al. [Citation14] in an analysis of a breast cancer cell line (HCC1954) and control breast epithelial cell line (HMEC) by WGBS. Their results supported the hypothesis that cancer-hypermethylated enhancers were usually the result of methylation at a partially methylated domain (PMD), at which one allele was marked by PcG repression without DNA methylation while the other allele was highly methylated but without PcG repression. They also concluded that DNA methylation and PcG methylation on a given allele can be mutually exclusive although other studies show that in some DNA contexts both epigenetic marks coexist in a local region [Citation87,Citation88]. In contrast to the study of cultured breast cancer cells by Hon et al., two studies of breast cancer tissues provided genome-wide evidence for enhancer hypermethylation being significantly correlated with low expression levels [Citation74,Citation86]. The DNA, chromatin, cell, and cancer subtype contexts probably determine the extent to which enhancer or promoter hypermethylation is contributing to cancer formation or tumour progression.
Cancer-linked gene-body or large-block hypermethylation
Cancer-linked changes in DNA methylation in gene-bodies (other than intragenic enhancers) might pathogenically alter transcription elongation or alternative splicing by affecting binding of the methylation-sensitive CTCF chromatin-looping protein ( and ) [Citation64,Citation89–Citation91]. In addition, large-scale changes in DNA methylation in blocks (5 kb to 10 Mb) are frequent in cancer [Citation14,Citation19,Citation69]. They contribute to global DNA hypomethylation seen often in cancer [Citation92] and could affect higher-order chromatin structure and regulation of gene expression. Only a small percentage of aberrantly methylated large blocks of DNA are cancer-hypermethylated rather than hypomethylated [Citation14,Citation69]; nonetheless, this hypermethylation too could influence higher-order structure of chromatin (). Epigenetic breakdown in cancer of normal DNA methylation compartments denoted by low DNA methylation surrounded by borders of high methylation () [Citation13,Citation69] could also change higher-order and local chromatin structure and thereby alter gene expression.
Normal developmental changes in DNA epigenetics gone awry in carcinogenesis
Our discovery of frequent global DNA hypomethylation in diverse human cancers was predicated on the idea that cancer is a kind of derangement of normal development [Citation92,Citation93], consistent with recent findings of cancer-linked aberrations in the epigenetic machinery that figure prominently in differentiation [Citation77,Citation94]. One of the most important players in cancer has been receiving much attention lately, namely, cancer stem cells (CSCs) [Citation95]. These cells may originate from normal stem/progenitor cells, other normal cells, or from partially dedifferentiated bulk cancer cells [Citation95,Citation96].
Cancer stem cells
Much evidence indicates that CSCs play a central role in oncogenesis. This includes their being responsible for much of the intra-tumour heterogeneity in tumour aggressiveness, differentiation, chemoresistance, and various markers of oncogenicity [Citation96]. There are two major models to explain the large extent of intra-tumour heterogeneity. The first proposes that it results from a buildup of stochastic alterations in many parts of the tumour without the intermediacy of a special set of cancer progenitor cells. The alternative, hierarchical model is that intra-tumour heterogeneity is due to a small set of independent CSCs acting as the main drivers of the formation of a given cancer [Citation96]. Much evidence in many of the more than 10,000 articles on CSCs in the last 10 years supports the latter model for most types of cancer. Typically, a cancer is thought to contain several CSC clones, which, in solid tumours, sometimes amount to only ~1% or less of the total tumour cells [Citation96]. In solid tumours, CSCs tend to be located in a small number of positions at the tumour periphery, whose microenvironment influences CSC formation and maintenance. CSC clones display some critical early embryonic or stem-like markers and signal transduction pathways but each clone within the tumour can be distinguished by epigenetic and genetic differences. CSC clones can drive the expansion of the number of cancer cells in the tumour and replenish themselves by periodic replicative self-renewal. CSC or related progeny cells can differentiate into cancer cells, which can de-differentiate to generate more CSCs. Importantly, CSCs are characteristically chemo- and radiation-resistant and have the highest metastatic potential within the tumour. Hence, CSC persistence is considered a major source of failure of various cancer treatments [Citation96].
Formation of CSCs and tumour progression frequently involves transdifferentiation, especially in solid cancers, the epithelial-to-mesenchymal transition (EMT), the endothelial-to-mesenchymal transition (EndMT), and/or the mesenchymal-to-epithelial transition (MET) [Citation96]. DNA hypermethylation and hypomethylation are implicated in these transitions, which are also driven by cell signalling pathways (e.g., Wnt and Notch signalling) [Citation80,Citation81]. Maintenance of CSC populations can be controlled, in part, by disease-associated DNA hypermethylation. For example, a study indicated that HOXC8, which encodes an early development-related homeobox TF, is a suppressor of a stem-like phenotype that can limit the size of the pool of breast CSCs, and HOXC8 can be downregulated with concomitant promoter hypermethylation in breast cancer [Citation97]. Genes encoding epigenetic regulators are frequently mutated or epigenetically dysregulated in CSCs, as they are in bulk cancers [Citation80,Citation98–Citation100], and experimental manipulation of these genes can be oncogenic [Citation101]. To better understand differences and similarities in the epigenetics of CSCs and those of the other cancer cells in the bulk tumour, there is a need for more comparisons of their DNA epigenetics (including hydroxymethylomes), chromatin epigenetics, and transcriptomics, preferably by single-cell techniques on in vivo-derived samples.
Similarities between DNA methylation profiles in the placental lineage and cancer
Both formation of solid cancers and the normal development of the placental lineage require cell invasion, vascular remodelling, and suppression of the immune system [Citation102]. Moreover, both human placenta and many cancers exhibit unusually low overall levels of DNA methylation [Citation92,Citation103]. A recent study of genome-wide DNA methylation in extraembryonic ectoderm (ExE, early placental/trophoblast lineage) vs. the epiblast (precursor of the main embryo) from mouse prenatal stage E6.5 suggests that carcinogenesis may involve co-opting epigenetic features of a normal specialized developmental program [Citation104]. Relative to the epiblast, ExE is often hypermethylated at CpGs within 2 kb of the TSS. In these TSS-adjacent regions, the epiblast usually has little or no methylation, as is the case in postnatal tissues, while the ExE generally displays considerable methylation, although often only in the range of about 25%. At interspersed DNA repeats, the epiblast generally has high methylation, as in postnatal tissues, while the much lower levels in ExE are reminiscent of cancer. Importantly, analysis of methylomes from 16 types of human cancer revealed frequent cancer-associated CGI hypermethylation near the TSS of many of the same genes that are hypermethylated in mouse ExE, although average DNA methylation levels in these regions in cancers were often much higher than in the mouse ExE. The ExE-hypermethylated promoter regions were enriched in PcG repressive chromatin in the epiblast, where they are silenced; therefore, why are these promoter regions subject to repressive de novo DNA methylation specifically in the ExE? Smith et al. [Citation104] propose that DNA hypermethylation, rather than just PcG chromatin, at these silenced promoters is needed for more permanent repression in the placental lineage than in the epiblast, similar to a proposal explaining some of the CGI-promoter hypermethylation in cancer [Citation46].
Cancer hypermethylation as diagnostic, prognostic, or treatment-related markers
DNA hypermethylation (or hypomethylation) at specific loci is among the best prognostic indicators for cancer patients [Citation49,Citation60,Citation63,Citation105]. Cancer-associated DNA hypermethylation and hypomethylation occur early in tumorigenesis and both typically increase with tumour progression [Citation37,Citation48,Citation53,Citation62,Citation73]. DNA hypomethylation at high-copy-number DNA repeats provides an especially sensitive cancer marker in diverse tumours [Citation7]. However, DNA hypermethylation at CGIs in cancer might be even better than repeat-DNA hypomethylation as a cancer marker because CGI hypermethylation offers the advantage of providing small targets with high concentrations of CpG for assays [Citation66]. Clinically applicable assays for promoter CGI hypermethylation of cancer biopsies or surgical samples are being optimized for cancer subtype designations that can increase prognostic accuracy and aid in making decisions about therapeutic strategies [Citation59]. Promoter hypermethylation markers for field effects (changes in histologically normal tissue adjacent to the cancer) are being developed to aid in very early detection of neoplastic changes predictive of malignant transformation [Citation48,Citation53]. DNA methylation tests might supplement cytological tests for intraoperative diagnosis [Citation63]. Personally tailored cancer therapy [Citation106] should include assays for cancer DNA methylation markers as well as genetic markers because aberrant DNA methylation can target different genes (or alleles) from those affected by mutation in an individual cancer. Such clinical testing can indicate which genes have mutations or DNA methylation changes that are driving formation or progression of a particular cancer and which cancer-altered genes might decrease the effectiveness of a given treatment protocol [Citation60].
Assays for cancer hypermethylation at single or multiple CGI-promoter targets using cell-free DNA released by dying cancer cells are being developed for early diagnosis, including of cancers that are difficult to detect at early stages, like ovarian carcinoma [Citation54]. These tests supplemented with DNA sequencing for somatic mutations could be used to screen either general or high-risk populations. Composite methylomes from heterogeneous cell-free plasma DNA can be subjected to deconvolution techniques (resolution of complex signals into constituent parts) to distinguish and quantitate the cancer and normal cells giving rise to the DNA mixture [Citation55]. Because cancer cells typically retain much of the cell type-specific methylome of the normal cells from which the cancer was derived [Citation19], even cancers of otherwise indeterminate origin can often be assigned to a tissue category by DNA methylation analysis. Body fluid, buccal or faecal tests for one or more informative CGI promoters can be done using either methylation markers associated with a given type of cancer or markers broadly associated with diverse cancers [Citation63,Citation65,Citation66]. For example, a test for colon cancer by assaying hypermethylation at a SEPT9 CpG island in plasma samples is in clinical use [Citation66]. This SEPT9 hypermethylation is an excellent diagnostic and prognostic colon cancer marker although its biological function in cancer (if any) is unkown [Citation57]. Caveats about diagnostic testing for disease-associated DNA methylation changes include initially ascertaining high specificity for pathogenicity as distinct from normal cell type- or age-related methylation changes, avoiding trace chemical contamination of PCR products, and ensuring that the optimal CpGs within a given region displaying cancer-linked DNA methylation changes are being analyzed [Citation51].
Neurological disease-linked alterations in DNA methylation
Brain epigenetics background
The unique characteristics of DNA methylation in the nervous system suggest that abnormal DNA methylation is likely to play critical roles in neurological disease. Brain has the highest overall level of DNA methylation of any human tissue other than thymus, as we reported in 1982 [Citation103]. However, it is now clear that a minor but appreciable fraction of that ‘DNA methylation’ is actually 5hmC and that brain (specifically nerve cells and especially Purkinje cells in the cerebellum) has by far the highest genomic content of 5hmC of any human tissue [Citation107]. In addition, human brain, unlike other postnatal tissues, has large amounts of 5mC outside of the CpG context (in mCpH; H, any base other than G). By adulthood, neurons have almost equal amounts of genomic 5mC in mCpG and mCpH. The mCpH levels increase rapidly from birth to mid-adolescence, a time of intense learning [Citation107]. There are differences among brain regions (especially cerebellum vs. other regions) in genomic 5mC, mCpH, and 5hmC content and levels of DNA methyltransferase (DNMT) and TET enzymes that catalyze genomic C modification [Citation107–Citation110]. However, these brain regional differences are probably influenced partly by differences in cell composition [Citation111]. Below are a few examples of the many studies that link neurological disease to DNA methylation as disease markers or as pathogenic changes.
Alzheimer’s disease and DNA hypermethylation
In a microbead-based methylome profiling of 73 post-mortem samples from late-onset Alzheimer’s disease patients, brain region-specific differences in disease-associated hypermethylation and hypomethylation were found by Semick et al. [Citation112]. In this study, which compared cerebellum, hippocampus, entorhinal cortex, and dorsolateral prefrontal cortex DNA in diseased and control individuals, the authors found that differences in cellular composition among the studied brain regions could not have accounted for the results. Alzheimer’s-hypermethylated DNA sites, but not the almost equally common hypomethylated sites, were enriched in disease-relevant genes such as those involved in cell adhesion, immunity and calcium binding. Some of the genes associated with Alzheimer’s disease in this study have also been implicated in other neurodegenerative diseases, such as Parkinson’s disease [Citation113]. Therefore, as noted by Semick et al., the disease-related DNA methylation differences might be related to secondary disease processes, like neurodegeneration, or might be epiphenomena. Nonetheless, given the experimental evidence for the involvement of DNA methylation and hydroxymethylation in neurological function [Citation109,Citation114], some of the 130 genes that had disease-linked expression changes, as well as associated DNA methylation changes, may well contribute to Alzheimer’s disease pathogenesis.
Schizophrenia, depression, epilepsy, borderline personality disorder, and DNA hypermethylation
In a study of schizophrenia, single nucleotide polymorphisms (SNPs) associated with variable methylation at a local CpG (cis-mQTLs) in 166 human fetal brain samples were identified [Citation115]. Many stringently defined mQTLs were significantly enriched in schizophrenia-associated SNPs, as determined from genome-wide association studies (GWAS). Therefore, some CpG sites with SNP-associated DNA methylation levels might contribute to the genetic risk of this disease. The functional importance of some of the schizophrenia-associated mQTLs was suggested by their highly significant enrichment in DNA sequence motifs recognized by the chromatin looping protein CTCF.
In an experimental rat model for epilepsy, the disease phenotype was associated with global DNA hypermethylation in the hippocampus [Citation116]. Transient treatment of the rats with adenosine augmentation therapy led to decreases in global DNA hypermethylation in the brain as well as suppression of seizures. Adenosine augmentation therapy has been proposed to function partially through the down-modulation of DNA methylation.
Among the complex neurological diseases with evidence for the involvement of DNA methylation changes is borderline personality disorder. Bisulfite pyrosequencing of saliva DNA sequences was tested for its ability to detect borderline personality disorder-linked hypermethylation at a promoter of the brain-related BDNF gene [Citation41]. This gene, which encodes a nerve growth factor implicated in mood disorders, showed small but significant increases in DNA methylation in patients, and these increases were dampened by psychotherapeutic treatment.
Clinically applicable DNA methylation assays for neurological diseases are being developed. Aref-Eshghi et al. [Citation56] are evaluating the use of microarrays for genome-wide DNA methylation profiles of blood samples to screen for and discriminate between nine neurodevelopment syndromes. These studies have the caveat of the question of the relevance of DNA methylation changes in blood (or other accessible tissues) to abnormal DNA methylation in brain disease, given the very tissue-specific DNA methylation profile of brain [Citation28]. For example, Story Jovanova et al. [Citation42] used DNA methylation-based epigenetic genome-wide association studies (EWAS) to look for associations of blood DNA methylation (microbead array assay) with depressive symptoms in thousands of individuals. Three CpGs displayed significant associations (P ≤ 6 x 10−8) and were replicated in a second population. However, in a database comparison of methylation of the three CpG sites in blood and in four regions of brain, only one of these CpGs showed a significant correlation with the same directionality in brain and in blood. Nonetheless, it has been postulated that in some neurological diseases there is multi-tissue disruption of the epigenetic machinery [Citation56], which could validate some of these surrogate-tissue assays.
The fragile X syndrome and pathogenic DNA methylation targeted to the 5ʹ end of FMR1
The pathological contribution of DNA hypermethylation to brain disease has been most fully elucidated for the fragile X syndrome (FXS), a single-locus genetic disease that is responsible for the most frequent type of inherited cognitive impairment [Citation52]. This syndrome is caused by the expansion and hypermethylation of a tandem trinucleotide repeat (CGG) in the 5ʹ untranslated region of the FMR1 gene. The adjacent promoter also becomes hypermethylated and inactive leading to the loss of the FMR1 product, FMRP, an RNA binding protein, in neurons and consequent intellectual disability. Both the repeat and the promoter are part of the same CGI. Using dCas9-Tet1 editing with a GG(CGG)6NGG single guide RNA, DNA demethylation was recently targeted to the CGG repeat with minimal off-target effects in induced pluripotent stem cells (iPSCs) derived from FXS patients [Citation47]. This demethylation spread to the promoter, and there was concomitant reactivation of FMR1 gene expression to give 30–90% of wild-type FMR1 RNA levels, depending on the FXS iPSC line that was used. FMR1 transcription reactivation and cellular phenotype rescue was also observed in neurons derived from the treated iPSCs. However, directing demethylation to differentiated neurons, rather than to the iPSCs, gave only a 30% decrease in mCGG demethylation at the FMR1 5ʹ UTR, probably due to a much lower efficacy of targeted Tet1-initiated demethylation in post-mitotic cells. Although the expanded repeat itself might contribute to FXS, this study provides strong evidence for hypermethylation at the 5ʹ end of FMR1 having a large role in the neurological dysfunction in this cognitive disorder.
Immunological disease and DNA hypermethylation
Genetically programmed alterations in DNA methylation play important roles in the normal functioning of the immune system, such as in T-cell biology [Citation117]. Changes in DNMT and TET enzyme levels are responsible for some of these normal increases and decreases in DNA methylation (and hydroxymethylation) [Citation118,Citation119]. Epigenetic changes help to drive the intricate pathways of postnatal differentiation of immune cells, their versatile and rapid responses to infection, and their induced-memory functions. Not unexpectedly, abnormal changes in DNA methylation are associated with autoimmune and autoinflammatory diseases. These include DNA hypermethylation in blood cells or synovial fibroblasts at certain gene regions in rheumatoid arthritis (RA; RA-related CCR6 and SPTBN1), systemic sclerosis (SSc; SSc-related and Wnt-signal modulating DKK1 and SFRP1), Sjogren’s syndrome (T-cell development-related RUNX1), Graves’ disease (autoimmunity-related TSHR), Type 1 diabetes (T-cell development-related FOXP3), and Crohn’s disease (CD; CD-related FASLG, S100A13 and RIPK3) [Citation18,Citation119,Citation120]. The study of epigenetics in the immune system is simplified by the accessibility of samples from the affected tissue but can be complicated by the heterogeneous and changing cellular composition of blood. However, this issue can be addressed by cell fractionation or by computational methods that usually rely on reference genomes [Citation55]. More research is needed on which DNA hypermethylated regions (and hypomethylated regions) characteristic for these diseases are causal vs. just disease markers.
Atherosclerosis, transdifferentiation, and DNA hypermethylation
Although atherosclerotic plaques are quite different from solid tumours, there are marked similarities between atherosclerosis and carcinogenesis, namely, the participation in both of oxidative stress, inflammation, angiogenesis, apoptosis, and proliferation by multiple clonal expansions to give oligoclonal progeny cells [Citation121,Citation122]. Especially interesting are the changes in cellular differentiation that accompany both atherosclerosis and cancer formation (for aberrant cell differentiation in cancer, see the section on cancer stem cells above). In atherosclerosis, monocytes infiltrate the intima of the aortic wall and differentiate into macrophages. Smooth muscle cells (SMCs) can dedifferentiate and lose all or part of their normal contractile phenotype to give a constellation of states (synthetic SMC phenotype) that allows them to actively proliferate through clonal expansion, synthesize increased amounts of certain extra-cellular proteins, lose their ability to contract, and thereby facilitate atherosclerotic changes in the aortic arch [Citation123]. Some of the synthetic SMCs migrate into the atherosclerotic plaque itself as well as into the neointima of the aorta. In addition, some SMCs in the aorta transdifferentiate to acquire a subset of properties of monocytes and may form lipid-laden foam cells further contributing to atherosclerotic changes in the aortic arch [Citation124].
Using publicly available WGBS profiles of an atherosclerotic aorta, a matching control, and additional control aortas [Citation1,Citation125], we confirmed that DNA hypermethylation predominates over DNA hypomethylation in atherosclerotic aorta [Citation125,Citation126]. DNA hypermethylation was present in a large fraction of the cells from the diseased aorta, could not be explained by atherosclerosis-related monocyte/macrophage infiltration, and was preferentially located in tissue-specific enhancer chromatin regions found in normal aorta [Citation126]. Comparisons of atherosclerosis-associated differentially methylated regions (DMRs) and normal aorta-specific DMRs, monocyte DMRs, and other tissue-specific DMRs and known functions of genes associated with the atherosclerotic DMRs indicate that many SMCs in the atherosclerotic aorta had partially acquired a leukocyte-like epigenetic profile. The observed disease-related DNA hypermethylation might be the result of decreases in TET activity that have been linked to atherosclerosis [Citation127,Citation128] (see section on TET enzymes below) and are associated with de novo methylation of certain normally unmethylated DNA regions [Citation13].
Osteoporosis and aberrant DNA methylation
Differential methylation of promoters of genes has been implicated in the development and regulation of osteoblasts and osteoclasts [Citation129]. Wnt signalling genes, which as mentioned above, figure so prominently in many kinds of cancer [Citation57,Citation80,Citation81], are also important in bone development, and the expression of some of these genes is inversely linked to promoter hypermethylation in the skeletal system [Citation129]. Methylation profiling of blood DNA from osteoporotic women and age-matched controls using a microbead methylation platform identified hypermethylated CpGs and hypomethylated CpGs in diseased individuals vs. controls [Citation43]. A subset of these sites was tested and shown to have their abnormal methylation replicable by bisulfite-based pyrosequencing. Although only total blood cell DNA was examined, some of the methylation marker-associated genes have functionality that could explain their osteoporosis association. Another study implicating alterations in DNA methylation in osteoporosis demonstrated that the lack of mechanical loading of a hind limb in a rat osteoporosis model caused hypermethylation of the promoter of H19 in femoral tissue [Citation130]. H19 is an important imprinted gene for osteogenic differentiation of bone marrow stem cells [Citation131]. After three weeks of hindlimb disuse, there was bone loss, upregulation of DNMT1, and H19 repression (which impacts bone homeostasis-related Erk signalling) accompanying H19 promoter hypermethylation. In vivo administration of siDNMT1 RNA to knockdown DNMT1 levels in bone resulted in significantly enhanced osteogenic function [Citation130]. This study provides an important animal model for understanding, at the level of epigenetic memory, the induction of osteoporosis in immobile individuals.
Causes of disease-associated DNA hypermethylation: the biochemical actors
Changes in DNA methyltransferases
One of the mechanisms for establishing disease-related DNA hypermethylation is a pathogenic increase in DNMT activity. This increase could involve the de novo DNA methyltransferases, DNMT3A and DNMT3B, whose main substrates are symmetrically unmethylated CpG dyads (unmethylated at 5ʹ-CpG-3ʹ/3ʹ-GpC-5ʹ), or the maintenance DNMT, DNMT1, which is mostly responsible for replication-linked DNA methylation of hemi-methylated subregions of DNA (5ʹ-5mCpG-3ʹ/3ʹ-GpC-5ʹ) but can also participate in de novo methylation [Citation132–Citation134].
Increases in DNMT activity may arise as follows:
Increases in the amounts of the DNMT1, DNMT3A or DNMT3B mRNAs because of elevated transcription of their genes or decreased miRNA-induced degradation or translation inhibition of the DNMT RNAs [Citation135];
Disruption of normal cell-cycle regulation of DNMT gene expression [Citation136];
Generation of aberrant or unusual mRNA isoforms from DNMT genes [Citation134,Citation137];
Increases in the activity of proteins with which they partner for their DNA methylation activity (e.g., DNMT3L or UHRF1 [Citation132,Citation138,Citation139]);
Decreases in degradation of DNMTs by changes in their post-translational modification [Citation132,Citation140].
Disease-related increases in DNMT activity accompanying DNA hypermethylation have been reported most often for cancer but occasionally for other types of diseases, such as non-neoplastic brain or vascular disease [Citation141,Citation142]. Because DNMT enzymes have preferences for methylating certain kinds of CpG-containing chromatin regions or DNA sequences (sequences flanking CpGs) and because de novo methylation often requires recruitment by sequence-specific DNA-binding proteins, increases in DNMT activity do not lead to uniform increases in CpG methylation throughout the genome [Citation132,Citation143].
Changes in TET (DNA hydroxymethylating) enzymes
Maintaining 5mC levels in DNA requires removing adventitious gains in DNA methylation. Many studies indicate that the main pathways for this homeostatic DNA demethylation in humans involve the conversion of 5mC to 5hmC as the first step [Citation13,Citation144]. This hydroxymethylation is followed by a kind of epigenetic repair that can occur either upon DNA replication or in its absence [Citation2,Citation12]. Therefore, although 5hmC can be a stable base in DNA with its own distinct influence on transcription control [Citation4,Citation145,Citation146], a major role for this modified genomic base is acting as an intermediate in DNA demethylation. TET1, TET2 and TET3 are 2-oxoglutarate-dependent dioxygenases that catalyze the formation of 5hmC from 5mC and further oxidize 5hmC. TET2-IDAX complexes, TET1, and TET3 bind preferentially to high concentrations of unmethylated CpGs in CGIs or in long methylation canyons via their CXXC Zn finger DNA-binding domains [Citation147]. This preference for mostly unmethylated, CpG-rich regions by TET enzymes allows them to serve as guardians for keeping such regions essentially free of 5mC. This can help explain why CpG island-promoter methylation increases upon TET enzyme depletion [Citation13]. It is probably partly through this mechanism that TET genes can function as tumour suppressor genes [Citation81,Citation82]. Decreases in TET enzyme activity are seen not only in cancer but also in certain non-neoplastic diseases, e.g., the loss of TET2 enzyme activity in the intimal layer of aorta in atherosclerosis [Citation128].
Aberrant DNA hypermethylation may arise by repression of the TET pathway for DNA demethylation as follows:
Mutation of TET genes, especially TET2, e.g., in ~45% of examined chronic myelomonocytic leukemias [Citation12];
TET1 CGI-promoter hypermethylation in primary cancers; this abnormal methylation is associated with repression of TET1 and is implicated in decreased genome-wide removal of aberrant de novo DNA hypermethylation [Citation82];
Post-transcriptional changes in TET proteins, e.g., by miRNA targeting [Citation148];
Gain-of-function mutations in IDH1 or IDH2 isocitrate dehydrogenase interfering with the α-ketoglutarate cofactor for TET-catalyzed hydroxymethylation of DNA [Citation149]; these mutations can lead to extensive gains in DNA methylation in cancer [Citation60].
Remarkably, in acute myelogenous leukaemias containing both DNA hypermethylation-provoking IDH2 mutations and DNA hypomethylating DNMT3A mutations, these two types of mutations seem to functionally oppose one another to give lower overall rates of DNA methylation change than seen in leukaemias with only one of the two genes mutated [Citation85].
Changes in chromatin structure
DNA methylation and chromatin structure influence each other in various ways [Citation1,Citation102,Citation133,Citation150] either by one driving the other or by acting jointly. The following is a summary of some of the ways that DNA methylation is influenced by chromatin structure.
DNMTs can be recruited to DNA sequences by the following chromatin proteins or chromatin modifications:
Certain histone modifying enzymes or complexes [Citation133], such as PRMT5 [Citation151];
Chromatin regions enriched in certain histone modifications, e.g., DNMT3B recruitment by H3K36me3 in actively transcribed gene bodies [Citation134];
The DNA replication machinery during maintenance replication, including UHRF1 recruiting DNMT1 to newly replicated, hemi-methylated DNA regions [Citation134];
Proteins that bind selectively to and are part of certain types of chromatin, e.g., heterochromatin protein HP1 [Citation133].
Selective recruitment of DNA modifying enzymes by transcription factors
Although the binding of some TFs can help protect against cancer-related DNA hypermethylation at CGIs [Citation37], a variety of other TFs by themselves or in conjunction with histone modification enzymes recruit DNMTs to DNA [Citation102,Citation133,Citation152]. These include P53, E2F6 and the methylated DNA-preferring MBD2 [Citation133,Citation153]. In addition, certain ncRNAs can direct some of the de novo DNA methylation (and DNA demethylation) in cancer to specific DNA sequences or chromatin locations [Citation154].
Physiological and environmental factors contributing to disease-associated DNA hypermethylation
As discussed above in the section on osteoporosis, DNA methylation is altered not only in a tissue-specific and cell type-specific manner but also with certain physiological changes, although to a much lesser extent. Obesity is known to be associated with changes in DNA methylation as well as with a proclivity to develop certain diseases, notably, type 2 diabetes [Citation155]. There is evidence that the intrauterine environment, physical inactivity and diet can affect both obesity and DNA methylation [Citation156]. Moreover, using Mendelian randomization analysis (which compares GWAS and EWAS data) of methylomes from blood and GWAS data for obesity as well as an adipose DNA methylation database, Wahl et al. [Citation155] concluded that the associations of blood or adipose DNA hypermethylation with obesity in large populations of individuals are primarily the consequence of obesity and not its cause. Among the many deleterious environmental changes linked to DNA hypermethylation and/or hypomethylation are smoking and prolonged morphine use, including at genes implicated in cancer (for smoking) or neural function (for morphine use) as seen in a meta-analysis of DNA methylation in human blood and buccal samples [Citation157] or in a rat model [Citation158]. Similarly, the relevance to cancer of many smoking-associated hypermethylated CpGs in the deduced epithelial compartment of buccal swab samples was demonstrated by comparison of methylomes from buccal cells (from smokers or non-smokers) and lung cancer [Citation45].
Stochastic & age-related factors favouring DNA hypermethylation and disease
There is some heterogeneity in DNA methylation patterns within the same cell type even among replicate cell clones at individual CpGs, and this heterogeneity appears to be partly stochastic [Citation159]. Related to this variation is the discovery that the best biochemical correlate of ageing in humans is the progressive accumulation of DNA hypermethylation at certain CpGs and hypomethylation at others in a partially tissue-independent manner [Citation160]. Moreover, superimposed on this DNA methylation clock of chronological age is a health-predictor function of DNA methylation at age-related epigenetically unstable CpG sites. DNA methylation in blood at these methylation/age CpGs has been reported to be an even better predictor of all-cause mortality than chronological age [Citation161].
This methylation clock is associated with diet and obesity, smoking, alcohol use, prolonged psychological stress, social class, an increased risk of breast cancer, and physical and mental fitness, but not with the number of cell divisions [Citation160,Citation162]. Using a DNA methylation clock, Horvath et al. [Citation163] found that cancers display significant age-acceleration and that ESC and iPSC exhibit apparent ages close to zero [Citation160]! Furthermore, Hutchinson Gilford Progeria Syndrome (HGPS) patients, who experience an early, drastic, and eventually fatal acceleration of ageing, display increased molecular ageing in their skin fibroblast cultures (patients < 10 yo, p = 0.02; all patients, p = 0.06). This was demonstrated by monitoring methylation at 391 CpGs in various parts of the genome in HGPS vs. control fibroblast cultures.
McKinney et al. [Citation164] analyzed changes in DNA methylation in postmortem orbital frontal cortex from individuals of different ages and demonstrated that age-associated DNA hypermethylation was more prevalent than hypomethylation. These age-related epigenetic changes were preferentially found at risk genes for schizophrenia, Alzheimer’s disease and major depressive disorder. They occurred more often in genes that exhibited age-associated changes in expression. CpGs at least 2 kb distant from CGIs were the favoured sites for these methylation changes.
A study by Tao et al. [Citation46] of DNA hypermethylation in colon adenocarcinomas at CIMP-associated CGIs provided evidence that ageing-related CIMP-type hypermethylation contributes to carcinogenesis. They generated a mouse colon-derived organoid model with a BrafV600E mutant proto-oncogene that could be experimentally activated. Braf is a MAPK signalling gene whose up-regulatory mutation can lead to either cell proliferation or, in pre-malignant lesions, to tumour-suppressing cell senescence (oncogene-induced senescence) [Citation165]. Tao et al. found that colon organoids expressing BrafV600E display a dysplastic phenotype when cultured for 2–3 months after BrafV600E activation. Only after several more months of culturing is a frankly malignant phenotype acquired by the organoids, including elevated expression of stem cell-associated genes and cancer-like autonomous activation of Wnt signalling. The long-term culture of the organoids was not consistently associated with the generation of key cancer-driver mutations but was invariably linked to progressive CIMP-type CGI-promoter hypermethylation whose gene targets partly overlapped those seen in human colon adenocarcinomas. There was also significant overlap of genes that became promoter-hypermethylated upon long-term culture of the BrafV600E-activated colon organoids and those that are hypermethylated in aged normal colon organoids and aged mouse colon. Using CRISPR mediation on two-month-old BrafV600E-expressing colon organoids, Tao et al. concurrently inactivated four cell-cycle or Wnt regulator genes (Cdkn2a, Cdx2, Sfrp4, and Sox17) that are subject to CIMP in vivo in proximal colon adenocarcinomas. Knockout of these four genes led to tumour-like growth after only a short incubation of the organoid subsequent to BrafV600Eactivation, thereby apparently accelerating tumorigenesis. Of these four genes, only three, Cdkn2a, Sfrp4, and Sox17, were subject to promoter hypermethylation in the BrafV600E-activated organoids. Cdx2 was progressively repressed in the organoids without promoter hypermethylation although it displays CIMP-associated hypermethylation in proximal colon adenocarcinomas. Overall, the findings from this study are consistent with the authors’ hypothesis of the biological importance of CIMP at promoters that are repressed in normal cells by PcG chromatin marks. Cancer-associated and age-related DNA hypermethylation at CGI promoters may result in more stable inactivation of some key tumour suppressor genes than is obtained by their PcG repression in non-tumour cells. In the case of BrafV600E-driven carcinogenesis, a small subset of the genes with cancer-hypermethylated CGI promoters (rather than the very many bystander-methylated genes) might allow the pre-cancerous cells to avoid BrafV600E-induced cell senescence during tumour formation.
Conclusions
There is very much evidence indicating causal roles for a subset of disease-linked alterations in DNA methylation. These findings are consistent with disease-associated acceleration of the DNA methylation-based molecular clock in cancer and HGPS (a premature ageing syndrome) and with parallels between ageing-linked changes in DNA methylation and many ageing-associated diseases. New insights into how altered DNA methylation affects or just monitors disease need to be translated into more clinical applications. First, many more diagnostic or prognostic assays should be developed for clinical use to detect aberrant, disease-related DNA methylation in either the target tissue, when it is accessible, or blood, sputum, buccal cells, or feces samples, when they are informative surrogates for the target tissue or contain DNA released from the target tissue. These assays should have the necessary specificity, sensitivity, precision and ease of handling. Secondly, despite the difficulties of in vivo molecular targeting using genetic technology and the concerns about off-target effects, there should be more research on the development of DNA sequence-specific epigenetic treatments, as modeled in a recent study in which part of the phenotype of the FXS mental retardation syndrome was reversed in cultured cells using CRISPR-mediated targeted demethylation [Citation47].
In the case of cancer treatment, personalized therapy is gaining recognition as being of the utmost importance because of the high variability in the resistance of cancers of a given type to many standard treatment protocols. This resistance is due, in part, to epigenetic heterogeneity between cancer subtypes and within malignant lesions. Cancer stem cells, which are critical to carcinogenesis and metastasis, often have different sensitivities to a given cancer treatment than do the cells in the bulk of the tumour probably due to differences in their epigenetics and genetics [Citation59,Citation80,Citation106,Citation166]. Challenging future clinical applications of epigenetics to cancer are the inclusion of CSC markers as well as bulk cancer markers in diagnostic and treatment-decision assays and the development of treatments aimed at CSCs (as well as bulk cancer cells) while sparing normal stem cells.
Acknowledgments
This research was supported in part by grants from the National Institutes of Health (UL1TR001417, NS04885, R01AR069055, and U19AG055373) and the Louisiana Cancer Center.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015 Feb 19;518(7539):317–330. PubMed PMID: 25693563;.
- Pang AP, Sugai C, Maunakea AK. High-throughput sequencing offers new insights into 5-hydroxymethylcytosine. Biomol Concepts. 2016 Jun 1;7(3):169–178. PubMed PMID: 27356236. DOI:10.1515/bmc-2016-0011.
- Koh CWQ, Goh YT, Toh JDW, et al. Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on the mitochondrial genome. Nucleic Acids Res. 2018 Dec 14;46(22):11659–11670. PubMed PMID: 30412255.
- Ehrlich M, Ehrlich KC. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics. 2014;6(6):563–566. PubMed PMID: 25531248; PubMed Central PMCID: PMCPMC4321890. DOI:10.2217/epi.14.48.
- Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013 Aug 9;341(6146):1237905. PubMed PMID: 23828890; PubMed Central PMCID: PMC3785061.
- Li L, Gao Y, Wu Q, et al. New guidelines for DNA methylome studies regarding 5-hydroxymethylcytosine for understanding transcriptional regulation. Genome Res. 2019 Apr;29(4):543–553. PubMed PMID: 30782641.
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21(35):5400–5413.
- Nestor C, Ruzov A, Meehan R, et al. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques. 2010 Apr;48(4):317–319. PubMed PMID: 20569209.
- Ponnaluri VK, Ehrlich KC, Zhang G, et al. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics. 2016 February;Dec:1–16. PubMed PMID: 27911668.
- Nestor CE, Ottaviano R, Reddington J, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012 Mar;22(3):467–477. PubMed PMID: 22106369.
- Wen L, Li X, Yan L, et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014 Mar 4;15(3):R49. PubMed PMID: 24594098.
- Koivunen P, Laukka T. The TET enzymes. Cell Mol Life Sci. 2018 Apr;75(8):1339–1348. PubMed PMID: 29184981.
- Wiehle L, Raddatz G, Musch T, et al. Tet1 and Tet2 protect DNA methylation canyons against hypermethylation. Mol Cell Biol. 2016 Nov 23;36(3):452–461. PubMed PMID: 26598602.
- Hon GC, Hawkins RD, Caballero OL, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012 Feb;22(2):246–258. PubMed PMID: 22156296.
- Ehrlich M, Lacey M. DNA hypomethylation and hemimethylation in cancer. Adv Exp Med Biol. 2013;754:31–56. PubMed PMID: 22956495.
- Xiong L, Wu F, Wu Q, et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat Commun. 2019 Jan 18;10(1):335. PubMed PMID: 30659195;.
- Gangisetty O, Cabrera MA, Murugan S. Impact of epigenetics in aging and age related neurodegenerative diseases. Front Biosci (Landmark Ed). 2018 Mar 1;23:1445–1464. PubMed PMID: 29293444.
- Mazzone R, Zwergel C, Artico M, et al. The emerging role of epigenetics in human autoimmune disorders. Clin Epigenetics. 2019 February 26;11(1):34. DOI:10.1186/s13148-019-0632-2.
- Varley KE, Gertz J, Bowling KM, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013 Jan 16;23(3):555–567. PubMed PMID: 23325432.
- Tsumagari K, Baribault C, Terragni J, et al. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics. 2013;8(3):317–332.
- Kawai T, Richards JS, Shimada M. The cell type-specific expression of Lhcgr in mouse ovarian cells: evidence for a DNA-demethylation-dependent mechanism. Endocrinology. 2018 May 1;159(5):2062–2074. PubMed PMID: 29579175.
- Bartholdy B, Lajugie J, Yan Z, et al. Mechanisms of establishment and functional significance of DNA demethylation during erythroid differentiation. Blood Adv. 2018 Aug 14;2(15):1833–1852. PubMed PMID: 30061308.
- Boothby MR, Hodges E, Thomas JW. Molecular regulation of peripheral B cells and their progeny in immunity. Genes Dev. 2019 Jan 1;33(1–2):26–48. PubMed PMID: 30602439.
- Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007 Apr;39(4):457–466. PubMed PMID: 17334365.
- Neri F, Rapelli S, Krepelova A, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017 Mar 02;543(7643):72–77. PubMed PMID: 28225755.
- Maunakea AK, Chepelev I, Cui K, et al. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013 Aug 13;23(11):1256–1269. PubMed PMID: 23938295.
- Ehrlich KC, Paterson HL, Lacey M, et al. DNA hypomethylation in intragenic and intergenic enhancer chromatin of muscle-specific genes usually correlates with their expression. Yale J Biol Med. 2016;89(4):441–455.
- Edgar RD, Jones MJ, Meaney MJ, et al. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl Psychiatry. 2017 Aug 1;7(8):e1187. PubMed PMID: 28763057; PubMed Central PMCID: PMCPMC5611738.
- Easwaran H, Johnstone SE, Van Neste L, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012 May;22(5):837–849. PubMed PMID: 22391556.
- Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013 Feb 28;152(5):1146–1159. PubMed PMID: 23434322.
- Mahe EA, Madigou T, Salbert G. Reading cytosine modifications within chromatin. Transcription. 2018;9(4):240–247. PubMed PMID: 29406813; PubMed Central PMCID: PMCPMC6104689. DOI:10.1080/21541264.2017.1406435.
- Collings CK, Anderson JN. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenetics Chromatin. 2017;10:18. PubMed PMID: 28413449; PubMed Central PMCID: PMCPMC5387343. DOI:10.1186/s13072-017-0125-5.
- Baribault C, Ehrlich KC, Ponnaluri VKC, et al. Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription. Epigenetics. 2018 Mar;2(Epub ahead of print):1–15. PubMed PMID: 29498561. DOI:10.1080/15592294.2018.1445900.
- Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011 Nov 3;479(7371):74–79. PubMed PMID: 21964334.
- Chandra S, Baribault C, Lacey M, et al. Myogenic differential methylation: diverse associations with chromatin structure. Biology (Basel). 2014;3(2):426–451. PubMed PMID: 24949935; PubMed Central PMCID: PMC4085616. DOI:10.3390/biology3020426.
- Wang H, Maurano MT, Qu H, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012 Sep;22(9):1680–1688. PubMed PMID: 22955980.
- Berman BP, Weisenberger DJ, Aman JF, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2011 Nov 27;44(1):40–46. PubMed PMID: 22120008; PubMed Central PMCID: PMCPMC4309644.
- Ehrlich KC, Lacey M, Ehrlich M. Tissue-specific epigenetics of atherosclerosis-related ANGPT and ANGPTL genes. Epigenomics. 2019 Feb;11(2):169–186. PubMed PMID: 30688091.
- Chandra S, Terragni J, Zhang G, et al. Tissue-specific epigenetics in gene neighborhoods: myogenic transcription factor genes. Hum Mol Genet. 2015 Jun 3;24(16):4660–4673. PubMed PMID: 26041816.
- Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol. 2001 May 14;153(4):773–784. PubMed PMID: 11352938; PubMed Central PMCID: PMCPMC2192370. DOI:10.1083/jcb.153.4.773.
- Thomas M, Knoblich N, Wallisch A, et al. Increased BDNF methylation in saliva, but not blood, of patients with borderline personality disorder. Clin Epigenetics. 2018 Aug 22;10(1):109. PubMed PMID: 30134995.
- Story Jovanova O, Nedeljkovic I, Spieler D, et al. DNA methylation signatures of depressive symptoms in middle-aged and elderly persons: meta-analysis of multiethnic epigenome-wide studies. JAMA Psychiatry. 2018 Sep 1;75(9):949–959. PubMed PMID: 29998287; PubMed Central PMCID: PMCPMC6142917.
- Cheishvili D, Parashar S, Mahmood N, et al. Identification of an epigenetic signature of osteoporosis in blood DNA of postmenopausal women. J Bone Miner Res. 2018 Nov;33(11):1980–1989. PubMed PMID: 29924424.
- Zhang X, Moen EL, Liu C, et al. Linking the genetic architecture of cytosine modifications with human complex traits. Hum Mol Genet. 2014 Nov 15;23(22):5893–5905. PubMed PMID: 24943591; PubMed Central PMCID: PMCPMC4204771.
- Zheng SC, Breeze CE, Beck S, et al. Identification of differentially methylated cell types in epigenome-wide association studies. Nat Methods. 2018 Dec;15(12):1059–1066. 10.1038/s41592-018-0213-x. PubMed PMID: 30504870; PubMed Central PMCID: PMCPMC6277016.
- Tao Y, Kang B, Petkovich DA, et al. Aging-like spontaneous epigenetic silencing facilitates Wnt activation, stemness, and Braf(V600E)-induced tumorigenesis. Cancer Cell. 2019 Feb 11;35(2):315–328 e6. PubMed PMID: 30753828.
- Liu XS, Wu H, Krzisch M, et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell. 2018 Feb 22;172(5):979–992 e6. PubMed PMID: 29456084.
- Rauscher GH, Kresovich JK, Poulin M, et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer. 2015 Oct 29;15:816. PubMed PMID: 26510686.
- Kim M, Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017 Apr 28;49(4):e322. PubMed PMID: 28450738. DOI:10.1038/emm.2017.10.
- Oudinet C, Braikia FZ, Dauba A, et al. Developmental regulation of DNA cytosine methylation at the immunoglobulin heavy chain constant locus. PLoS Genet. 2019 Feb;15(2):e1007930. PubMed PMID: 30779742.
- Koch A, Joosten SC, Feng Z, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018 Jul;15(7):459–466. 10.1038/s41571-018-0004-4. PubMed PMID: 29666440.
- Kraan CM, Godler DE, Amor DJ. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev Med Child Neurol. 2019 Feb;61(2):121–127. PubMed PMID: 30084485.
- Moller M, Strand SH, Mundbjerg K, et al. Heterogeneous patterns of DNA methylation-based field effects in histologically normal prostate tissue from cancer patients. Sci Rep. 2017 Jan 13;7:40636. PubMed PMID: 28084441.
- Giannopoulou L, Kasimir-Bauer S, Lianidou ES. Liquid biopsy in ovarian cancer: recent advances on circulating tumor cells and circulating tumor DNA. Clin Chem Lab Med. 2018 Jan 26;56(2):186–197. PubMed PMID: 28753534.
- Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018 Nov 29;9(1):5068. PubMed PMID: 30498206.
- Aref-Eshghi E, Rodenhiser DI, Schenkel LC, et al. Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. Am J Hum Genet. 2018 Jan 4;102(1):156–174. PubMed PMID: 29304373; PubMed Central PMCID: PMCPMC5777983.
- Weisenberger DJ, Liang G, Lenz HJ. DNA methylation aberrancies delineate clinically distinct subsets of colorectal cancer and provide novel targets for epigenetic therapies. Oncogene. 2018 Feb 1;37(5):566–577. PubMed PMID: 28991233.
- Ferreira HJ, Esteller M. CpG islands in cancer: heads, tails, and sides. Methods Mol Biol. 2018;1766:49–80. PubMed PMID: 29605847. DOI:10.1007/978-1-4939-7768-0_4.
- Pangeni RP, Zhang Z, Alvarez AA, et al. Genome-wide methylomic and transcriptomic analyses identify subtype-specific epigenetic signatures commonly dysregulated in glioma stem cells and glioblastoma. Epigenetics. 2018;13(4):432–448. PubMed PMID: 29927689.
- Nebbioso A, Tambaro FP, Dell’Aversana C, et al. Cancer epigenetics: moving forward. PLoS Genet. 2018 Jun;14(6):e1007362. PubMed PMID: 29879107; PubMed Central PMCID: PMCPMC5991666.
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev. 2004 Dec;4(12):988–993. PubMed PMID: 15573120.
- Semaan A, van Ellen A, Meller S, et al. SEPT9 and SHOX2 DNA methylation status and its utility in the diagnosis of colonic adenomas and colorectal adenocarcinomas. Clin Epigenetics. 2016;8:100. PubMed PMID: 27660666; PubMed Central PMCID: PMCPMC5028994. DOI:10.1186/s13148-016-0267-5.
- Yamashita K, Hosoda K, Nishizawa N, et al. Epigenetic biomarkers of promoter DNA methylation in the new era of cancer treatment. Cancer Sci. 2018 Dec;109(12):3695–3706. PubMed PMID: 30264476.
- Su J, Huang YH, Cui X, et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018 Aug 10;19(1):108. 10.1186/s13059-018-1492-3. PubMed PMID: 30097071; PubMed Central PMCID: PMCPMC6085761.
- Su YH, Kim AK, Jain S. Liquid biopsies for hepatocellular carcinoma. Transl Res. 2018 Nov;201:84–97. PubMed PMID: 30056068.
- Vrba L, Futscher BW. A suite of DNA methylation markers that can detect most common human cancers. Epigenetics. 2018;13(1):61–72. PubMed PMID: 29212414.
- Jurgens B, Schmitz-Drager BJ, Schulz WA. Hypomethylation of L1 LINE sequences prevailing in human urothelial carcinoma. Cancer Res. 1996 Dec 15;56(24):5698–5703. PubMed PMID: 8971178.
- Narayan A, Ji W, Zhang X-Y, et al. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int J Cancer. 1998;77:833–838.
- Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011 Jun 26;43(8):768–775. PubMed PMID: 21706001.
- Nishiyama R, Qi L, Lacey M, et al. Both hypomethylation and hypermethylation in a 0.2-kb region of a DNA repeat in cancer. Molec Cancer Res. 2005;3:617–626.
- Merlo A, Herman JG, Mao L, et al. 5ʹ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1(7):686–692.
- Nguyen C, Liang G, Nguyen TT, et al. Susceptibility of nonpromoter CpG islands to de novo methylation in normal and neoplastic cells. J Natl Cancer Inst. 2001;93(19):1465–1472. PubMed PMID: 11584062.
- Ehrlich M, Jiang G, Fiala ES, et al. Hypomethylation and hypermethylation in Wilms tumors. Oncogene. 2002;21:6694–6702.
- Bae MG, Kim JY, Choi JK. Frequent hypermethylation of orphan CpG islands with enhancer activity in cancer. BMC Med Genomics. 2016 Aug 12;9 Suppl 1:38. 10.1186/s12920-016-0198-1. PubMed PMID: 27534853; PubMed Central PMCID: PMCPMC4989897.
- Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007 Feb;39(2):232–236. PubMed PMID: 17200670.
- Hinoue T, Weisenberger DJ, Lange CP, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012 Feb;22(2):271–282. PubMed PMID: 21659424.
- Court F, Arnaud P. An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research. Oncotarget. 2017 Jan 17;8(3):4110–4124. PubMed PMID: 27926531; PubMed Central PMCID: PMCPMC5354816. DOI:10.18632/oncotarget.13746.
- Zhang Y, Charlton J, Karnik R, et al. Targets and genomic constraints of ectopic Dnmt3b expression. Elife. 2018 Nov 23;7. DOI:10.7554/eLife.40757. PubMed PMID: 30468428; PubMed Central PMCID: PMCPMC6251628.
- Verma N, Pan H, Dore LC, et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat Genet. 2018 Jan;50(1):83–95. 10.1038/s41588-017-0002-y. PubMed PMID: 29203910; PubMed Central PMCID: PMCPMC5742051.
- Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017 Feb 1;16(1):29. PubMed PMID: 28148257.
- Neri F, Dettori D, Incarnato D, et al. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene. 2015 Aug 6;34(32):4168–4176. PubMed PMID: 25362856.
- Li L, Li C, Mao H, et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci Rep. 2016 May 26;6:26591. PubMed PMID: 27225590.
- Song Q, Decato B, Hong EE, et al. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS One. 2013;8(12):e81148. PubMed PMID: 24324667; PubMed Central PMCID: PMC3855694. DOI:10.1371/journal.pone.0081148.
- Kent WJ, Sugnet CW, Furey TS, et al. UCSC Genome Browser. 2016 [cited 2016 June 12]. Available from: http://ucsc.genome.edu
- Glass JL, Hassane D, Wouters BJ, et al. Epigenetic Identity in AML depends on disruption of nonpromoter regulatory elements and Is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov. 2017 Aug;7(8):868–883. PubMed PMID: 28408400; PubMed Central PMCID: PMCPMC5540802.
- Wang Y, Hao DP, Li JJ, et al. Genome-wide methylome and chromatin interactome identify abnormal enhancer to be risk factor of breast cancer. Oncotarget. 2017 Jul 4;8(27):44705–44719. PubMed PMID: 28621677; PubMed Central PMCID: PMCPMC5546512.
- Brinkman AB, Gu H, Bartels SJ, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012 Jun;22(6):1128–1138. PubMed PMID: 22466170; PubMed Central PMCID: PMCPMC3371717.
- Statham AL, Robinson MD, Song JZ, et al. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res. 2012 Jun;22(6):1120–1127. PubMed PMID: 22466171; PubMed Central PMCID: PMCPMC3371705.
- Modur V, Singh N, Mohanty V, et al. Defective transcription elongation in a subset of cancers confers immunotherapy resistance. Nat Commun. 2018 Oct 23;9(1):4410. 10.1038/s41467-018-06810-0. PubMed PMID: 30353012; PubMed Central PMCID: PMCPMC6199328.
- Arechederra M, Daian F, Yim A, et al. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat Commun. 2018 Aug 8;9(1):3164. 10.1038/s41467-018-05550-5. PubMed PMID: 30089774; PubMed Central PMCID: PMCPMC6082886.
- Couture F, Sabbagh R, Kwiatkowska A, et al. PACE4 undergoes an oncogenic alternative splicing switch in cancer. Cancer Res. 2017 Dec 15;77(24):6863–6879. PubMed PMID: 28993410.
- Gama-Sosa MA, Slagel VA, Trewyn RW, et al. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–6894.
- Ehrlich M. The controversial denouement of vertebrate DNA methylation research. Biochemistry (Mosc). 2005 May;70(5):568–575. PubMed PMID: 15948710.
- Malta TM, Sokolov A, Gentles AJ, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018 Apr 5;173(2):338–354 e15. PubMed PMID: 29625051.
- Poli V, Fagnocchi L, Zippo A. Tumorigenic cell reprogramming and cancer plasticity: interplay between signaling, microenvironment, and epigenetics. Stem Cells Int. 2018;2018:4598195. PubMed PMID: 29853913; PubMed Central PMCID: PMCPMC5954911. DOI:10.1155/2018/4598195.
- Valle S, Martin-Hijano L, Alcala S, et al. The ever-evolving concept of the cancer stem cell in pancreatic cancer. Cancers (Basel). 2018 Jan 26;10(2). PubMed PMID: 29373514; PubMed Central PMCID: PMCPMC5836065. DOI:10.3390/cancers10020033.
- Shah M, Cardenas R, Wang B, et al. HOXC8 regulates self-renewal, differentiation and transformation of breast cancer stem cells. Mol Cancer 2017 Feb;16;16(1):38. 10.1186/s12943-017-0605-z. PubMed PMID: 28202042; PubMed Central PMCID: PMCPMC5312582.
- Wang Y, Shang Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp Cell Res. 2013 Jan 15;319(2):160–169. PubMed PMID: 22935683. DOI:10.1016/j.yexcr.2012.07.019.
- Zhou D, Alver BM, Li S, et al. Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biol. 2018 Mar 27;19(1):43. 10.1186/s13059-018-1420-6. PubMed PMID: 29587824; PubMed Central PMCID: PMCPMC5872397.
- Roscigno G, Quintavalle C, Donnarumma E, et al. MiR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget. 2015 Jan 5;7(1):580–592. PubMed PMID: 26556862; PubMed Central PMCID: PMCPMC4808019.
- Li S, Han Z, Zhao N, et al. Inhibition of DNMT suppresses the stemness of colorectal cancer cells through down-regulating Wnt signaling pathway. Cell Signal. 2018 Jul;47:79–87. PubMed PMID: 29601907.
- Lorincz MC, Schubeler D. Evidence for converging DNA methylation pathways in placenta and cancer. Dev Cell. 2017 Nov 6;43(3):257–258. PubMed PMID: 29112847.
- Ehrlich M, Gama-Sosa M, Huang L-H, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982;10:2709–2721.
- Smith ZD, Shi J, Gu H, et al. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017 Sep 28;549(7673):543–547. PubMed PMID: 28959968.
- De Rubis G, Krishnan SR, Bebawy M. Circulating tumor DNA - Current state of play and future perspectives. Pharmacol Res. 2018 Oct;136:35–44. PubMed PMID: 30142423.
- Howard CM, Valluri J, Alberico A, et al. Analysis of chemopredictive assay for targeting cancer stem cells in glioblastoma patients. Transl Oncol. 2017 Apr;10(2):241–254. PubMed PMID: 28199863; PubMed Central PMCID: PMCPMC5310181.
- Lister R, Mukamel EA. Turning over DNA methylation in the mind. Front Neurosci. 2015;9:252. PubMed PMID: 26283895.
- Davies MN, Volta M, Pidsley R, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012 Jun 15;13(6):R43. PubMed PMID: 22703893; PubMed Central PMCID: PMCPMC3446315.
- Cui D, Xu X. DNA methyltransferases, DNA methylation, and age-associated cognitive function. Int J Mol Sci. 2018 Apr 28;19(5). PubMed PMID: 29710796.
- The_GTEx_Consortium. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015 May 8;348(6235):648–660. PubMed PMID: 25954001; PubMed Central PMCID: PMCPMC4547484. DOI:10.1126/science.1262110.
- Herculano-Houzel S, Lent R. Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J Neurosci. 2005 Mar 9;25(10):2518–2521. PubMed PMID: 15758160. DOI:10.1523/JNEUROSCI.4526-04.2005.
- Semick SA, Bharadwaj RA, Collado-Torres L, et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer’s disease. Acta Neuropathol. 2019 Feb 2. 10.1007/s00401-019-01966-5. PubMed PMID: 30712078.
- Sanchez-Mut JV, Heyn H, Vidal E, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry. 2016 Jan 19;6:e718. PubMed PMID: 26784972.
- Oliveira AM. DNA methylation: a permissive mark in memory formation and maintenance. Learn Mem. 2016 Oct;23(10):587–593. PubMed PMID: 27634149.
- Hannon E, Spiers H, Viana J, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2016 Jan;19(1):48–54. PubMed PMID: 26619357.
- Williams-Karnesky RL, Sandau US, Lusardi TA, et al. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest. 2013 Aug;123(8):3552–3563. PubMed PMID: 23863710.
- Schmidl C, Delacher M, Huehn J, et al. Epigenetic mechanisms regulating T-cell responses. J Allergy Clin Immunol. 2018 Sep;142(3):728–743. PubMed PMID: 30195378.
- Nair N, Wilson AG, Barton A. DNA methylation as a marker of response in rheumatoid arthritis. Pharmacogenomics. 2018 Sep;18(14):1323–1332. PubMed PMID: 28836487.
- Alvarez-Errico D, Vento-Tormo R, Ballestar E. Genetic and epigenetic determinants in autoinflammatory diseases. Front Immunol. 2017;8:318. PubMed PMID: 28382039.
- de la Rica L, Urquiza JM, Gomez-Cabrero D, et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun. 2013 Mar;41:6–16. PubMed PMID: 23306098.
- Tapia-Vieyra JV, Delgado-Coello B, Mas-Oliva J. Atherosclerosis and cancer; A resemblance with far-reaching implications. Arch Med Res. 2017 Jan;48(1):12–26. PubMed PMID: 28577865. DOI:10.1016/j.arcmed.2017.03.005.
- Chappell J, Harman JL, Narasimhan VM, et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016 Dec 9;119(12):1313–1323. PubMed PMID: 27682618.
- Allahverdian S, Chaabane C, Boukais K, et al. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018 Mar 15;114(4):540–550. PubMed PMID: 29385543; PubMed Central PMCID: PMCPMC5852505.
- Dubland JA, Francis GA. So Much Cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol. 2016 Apr;27(2):155–161. PubMed PMID: 26836481. DOI:10.1097/MOL.0000000000000279.
- Zaina S, Heyn H, Carmona FJ, et al. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014 Oct;7(5):692–700. PubMed PMID: 25091541.
- Lacey M, Baribault C, Ehrlich KC, et al. Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers. Atherosclerosis. 2019 Nov;27(280):183–191. PubMed PMID: 30529831. DOI:10.1016/j.atherosclerosis.2018.11.031.
- Liu R, Jin Y, Tang WH, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013 Oct 29;128(18):2047–2057. PubMed PMID: 24077167; PubMed Central PMCID: PMC3899790.
- Peng J, Yang Q, Li AF, et al. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE-/- mice. Oncotarget. 2016 Nov 22;7(47):76423–76436. PubMed PMID: 27821816; PubMed Central PMCID: PMCPMC5363520.
- Husain A, Jeffries MA. Epigenetics and bone remodeling. Curr Osteoporos Rep. 2017 Oct;15(5):450–458. 10.1007/s11914-017-0391-y. PubMed PMID: 28808893; PubMed Central PMCID: PMCPMC5710824.
- Li B, Zhao J, Ma JX, et al. Overexpression of DNMT1 leads to hypermethylation of H19 promoter and inhibition of Erk signaling pathway in disuse osteoporosis. Bone. 2018 Jun;111:82–91. PubMed PMID: 29555308.
- Zhou P, Li Y, Di R, et al. H19 and Foxc2 synergistically promotes osteogenic differentiation of BMSCs via Wnt-beta-catenin pathway. J Cell Physiol. 2019 Jan 11. PubMed PMID: 30633332. DOI:10.1002/jcp.28060.
- Gowher H, Jeltsch A. Mammalian DNA methyltransferases: new discoveries and open questions. Biochem Soc Trans. 2018 Oct 19;46(5):1191–1202. PubMed PMID: 30154093.
- Hervouet E, Peixoto P, Delage-Mourroux R, et al. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin Epigenetics. 2018;10:17. PubMed PMID: 29449903.
- Gujar H, Weisenberger DJ, Liang G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes (Basel). 2019 Feb 23;10(2). PubMed PMID: 30813436. DOI:10.3390/genes10020172.
- Wu H, Zhang W, Wu Z, et al. miR-29c-3p regulates DNMT3B and LATS1 methylation to inhibit tumor progression in hepatocellular carcinoma. Cell Death Dis. 2019 Jan 18;10(2):48. PubMed PMID: 30718452.
- Robertson KD, Keyomarsi K, Gonzales FA, et al. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000 May 15;28(10):2108–2113. PubMed PMID: 10773079; PubMed Central PMCID: PMCPMC105379.
- Siddiqui S, White MW, Schroeder AM, et al. Aberrant DNMT3B7 expression correlates to tissue type, stage, and survival across cancers. PLoS One. 2018;13(8):e0201522. PubMed PMID: 30071066.
- Li J, Wang R, Hu X, et al. Activated MEK/ERK pathway drives widespread and coordinated overexpression of UHRF1 and DNMT1 in cancer cells. Sci Rep. 2019 Jan 29;9(1):907. 10.1038/s41598-018-37258-3. PubMed PMID: 30696879; PubMed Central PMCID: PMCPMC6351616.
- Subramaniam D, Thombre R, Dhar A, et al. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:80. PubMed PMID: 24822169; PubMed Central PMCID: PMCPMC4013461. DOI:10.3389/fonc.2014.00080.
- Cheng J, Yang H, Fang J, et al. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat Commun. 2015 May 11;6:7023. PubMed PMID: 25960197.
- Symmank J, Zimmer G. Regulation of neuronal survival by DNA methyltransferases. Neural Regen Res. 2017 Nov;12(11):1768–1775. PubMed PMID: 29239313.
- Jiang YZ, Jimenez JM, Ou K, et al. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res. 2014 Jun 20;115(1):32–43. PubMed PMID: 24755985; PubMed Central PMCID: PMCPMC4065854.
- Emperle M, Rajavelu A, Kunert S, et al. The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids Res. 2018 Apr 6;46(6):3130–3139. PubMed PMID: 29518238.
- Li J, Wu X, Zhou Y, et al. Decoding the dynamic DNA methylation and hydroxymethylation landscapes in endodermal lineage intermediates during pancreatic differentiation of hESC. Nucleic Acids Res. 2018 Apr 6;46(6):2883–2900. PubMed PMID: 29394393.
- Wu X, Li G, Xie R. Decoding the role of TET family dioxygenases in lineage specification. Epigenetics Chromatin. 2018 Oct 5;11(1):58. 10.1186/s13072-018-0228-7. PubMed PMID: 30290828; PubMed Central PMCID: PMCPMC6172806.
- Cakouros D, Hemming S, Gronthos K, et al. Specific functions of TET1 and TET2 in regulating mesenchymal cell lineage determination. Epigenetics Chromatin. 2019 Jan 3;12(1):3. 10.1186/s13072-018-0247-4. PubMed PMID: 30606231; PubMed Central PMCID: PMCPMC6317244.
- Ko M, An J, Bandukwala HS, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013 May 2;497(7447):122–126. PubMed PMID: 23563267.
- Chen Q, Yin D, Zhang Y, et al. MicroRNA-29a induces loss of 5-hydroxymethylcytosine and promotes metastasis of hepatocellular carcinoma through a TET-SOCS1-MMP9 signaling axis. Cell Death Dis. 2017 Jun 29;8(6):e2906. PubMed PMID: 28661477.
- Chen K, Zhang J, Guo Z, et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016 Jan;26(1):103–118. PubMed PMID: 26680004.
- Reddington JP, Perricone SM, Nestor CE, et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 2013 Mar 25;14(3):R25. PubMed PMID: 23531360; PubMed Central PMCID: PMCPMC4053768.
- Zhao Q, Rank G, Tan YT, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009 Mar;16(3):304–311. PubMed PMID: 19234465.
- Schafer A, Mekker B, Mallick M, et al. Impaired DNA demethylation of C/EBP sites causes premature aging. Genes Dev. 2018 Jun 1;32(11–12):742–762. PubMed PMID: 29884649; PubMed Central PMCID: PMCPMC6049513.
- Stirzaker C, Song JZ, Ng W, et al. Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene. 2017 Mar;36(10):1328–1338. PubMed PMID: 27593931.
- Bure I, Geer S, Knopf J, et al. Long noncoding RNA HOTAIR is upregulated in an aggressive subgroup of gastrointestinal stromal tumors (GIST) and mediates the establishment of gene-specific DNA methylation patterns. Genes Chromosomes Cancer. 2018 Nov;57(11):584–597. PubMed PMID: 30248209.
- Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017 Jan 5;541(7635):81–86. PubMed PMID: 28002404; PubMed Central PMCID: PMCPMC5570525.
- Nilsson E, Ling C. DNA methylation links genetics, fetal environment, and an unhealthy lifestyle to the development of type 2 diabetes. Clin Epigenetics. 2017;9:105. PubMed PMID: 29026446; PubMed Central PMCID: PMCPMC5627472. DOI:10.1186/s13148-017-0399-2.
- Ma Y, Li MD. Establishment of a strong link between smoking and cancer pathogenesis through DNA methylation analysis. Sci Rep. 2017 May 12;7(1):1811. 10.1038/s41598-017-01856-4. PubMed PMID: 28500316; PubMed Central PMCID: PMCPMC5431893.
- Barrow TM, Byun HM, Li X, et al. The effect of morphine upon DNA methylation in ten regions of the rat brain. Epigenetics. 2017;12(12):1038–1047. PubMed PMID: 29111854.
- Riggs AD, Xiong Z. Methylation and epigenetic fidelity. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):4–5. PubMed PMID: 14695893; PubMed Central PMCID: PMCPMC314126. DOI:10.1073/pnas.0307781100.
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. PubMed PMID: 24138928; PubMed Central PMCID: PMCPMC4015143. DOI:10.1186/gb-2013-14-10-r115.
- Ecker S, Beck S. The epigenetic clock: a molecular crystal ball for human aging? Aging (Albany NY). 2019 Jan 21;11(2):833–835. PubMed PMID: 30669120.
- Kresovich JK, Xu Z, O’Brien KM, et al. Methylation-based biological age and breast cancer risk. J Natl Cancer Inst. 2019 Feb 22. PubMed PMID: 30794318. DOI:10.1093/jnci/djz020.
- Horvath S, Oshima J, Martin GM, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY). 2018 Jul 26;10(7):1758–1775. PubMed PMID: 30048243; PubMed Central PMCID: PMCPMC6075434.
- McKinney BC, Lin CW, Rahman T, et al. DNA methylation in the human frontal cortex reveals a putative mechanism for age-by-disease interactions. Transl Psychiatry. 2019 Jan 29;9(1):39. 10.1038/s41398-019-0372-2. PubMed PMID: 30696804; PubMed Central PMCID: PMCPMC6351569.
- Moulana FI, Priyani AAH, de Silva MVC, et al. BRAF-oncogene-induced senescence and the role of thyroid-stimulating hormone signaling in the progression of papillary thyroid carcinoma. Horm Cancer. 2018 Feb;9(1):1–11. 10.1007/s12672-017-0315-4. PubMed PMID: 29209896.
- Perusina Lanfranca M, Thompson JK, Bednar F, et al. Metabolism and epigenetics of pancreatic cancer stem cells. Semin Cancer Biol. 2018 Sep 28. PubMed PMID: 30273655. DOI:10.1016/j.semcancer.2018.09.008.