
ABSTRACT
We previously identified sequence-dependent allele-specific methylation (sd-ASM) in adult human peripheral blood leukocytes, in which ASM occurs in cis depending on adjacent polymorphic sequences. A number of groups have identified sd-ASM sites in the human and mouse genomes, illustrating the prevalence of sd-ASM in mammalian genomes. In addition, sd-ASM can lead to sequence-dependent allele-specific expression of neighbouring genes. Imprinted genes also often exhibit parent-of-origin–dependent allele-specific methylation (pd-ASM), which causes parent-of-origin–dependent allele-specific expression. However, whether most of the already known sd-ASM and pd-ASM sites are methylated or hydroxymethylated remains unclear due to technical restrictions. Accordingly, a novel method that enables examination of allelic methylation and hydroxymethylation status and also overcomes the drawbacks of conventional methods is needed. Such a method could also be used to elucidate the mechanisms underlying polymorphism-associated inter-individual differences in disease susceptibility and the mechanism of genomic imprinting. Here, we developed a simple method to determine allelic hydroxymethylation status and identified novel sequence- and parent-of-origin–dependent allele-specific hydroxymethylation sites. Correlation analyses of TF binding sequences and methylation or hydroxymethylation between three mouse strains revealed the involvement of Pax5 in strain-specific methylation and hydroxymethylation in exon 7 of Pdgfrb.
Introduction
Mammalian DNA methylation (5mC) at the 5-position of the cytosine of CpG dinucleotides is catalysed by various DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) [Citation1,Citation2]. DNA methylation regulates spatial and temporal gene expression by suppressing the binding of transcription factors to DNA [Citation3]. In addition, DNA methylation is involved in the inactivation of transposable elements [Citation4], genomic stabilization [Citation5], X chromosome inactivation in females [Citation6], and genomic imprinting [Citation7]. Aberrant DNA methylation is involved in the occurrence of various types of cancer [Citation8], and the disruption of DNA methyltransferases leads to embryonic and postnatal lethality in mice [Citation1,Citation2].
Most cytosines of CpG dinucleotides in mammalian genomes are subject to DNA methylation. Methylated CpG dinucleotides are converted to TpG dinucleotides via deamination of the cytosine bases more frequently than unmethylated CpG dinucleotides [Citation9]. Generally, such CpG suppression is infrequent in CpG-rich regions known as CpG islands (CGIs), as most CGIs are unmethylated or minimally methylated in all developmental stages and adult tissues [Citation10,Citation11]. CGIs generally lie in the promoter regions of house-keeping genes and some tissue-specific genes [Citation12,Citation13]. A fraction of CGIs are subject to parent-of-origin–dependent allele-specific methylation (pd-ASM) in imprinted genes [Citation14,Citation15] or random-mono-allelic methylation in X chromosomes [Citation6].
We previously examined the allelic methylation status of CGIs in chromosomes 11 and 21 using our original method, which is designated HpaII-McrBC PCR (HM-PCR) [Citation16,Citation17]. HM-PCR uses methylation-sensitive and -dependent restriction enzymes to discriminate between four allelic methylation states. As a result, we first identified a sequence-dependent allele-specific methylation (sd-ASM) site in adult human peripheral blood leukocytes, in which ASM occurred in cis depending on adjacent polymorphic sequences [Citation16]. It is known that some sd-ASM can also induce allele-specific expression of neighbouring genes. A number of groups have identified sd-ASM sites in the human and mouse genomes, illustrating the prevalence of sd-ASM in mammalian genomes [Citation18–Citation20]. However, the mechanism underlying sd-ASM remains unclear.
Genome-wide association studies have revealed that most polymorphisms associated with inter-individual differences in disease susceptibility occur in non-coding regions [Citation21], although the underlying mechanism remains unclear. Accordingly, the identification of novel sd-ASM sites could clarify the role of polymorphisms in determining inter-individual differences in disease susceptibility [Citation22]. For example, haplotype-specific methylation was shown to be associated with susceptibility to fat mass and obesity–associated (FTO) type II diabetes and obesity [Citation23].
Ten-eleven translocation (Tet) enzymes catalyse the oxidization of 5mC to 5-hydroxymethylation (5hmC) [Citation24]. 5hmC, an intermediate in the demethylation of 5mC, is sequentially converted to formylcytosine (5fC) and carboxylcytosine (5caC) through oxidization by Tet enzymes [Citation25]. 5fC and 5caC can be excised by the DNA repair protein thymine DNA glycosylase, and restored to unmodified cytosine by base excision repair [Citation26]. 5hmC is not only an intermediate in the demethylation of 5mC but also a stable epigenetic DNA modification associated with transcriptional regulation [Citation26]. 5hmC is involved in both positive and negative regulations of gene expression [Citation27,Citation28]. Several 5hmC-specific binding proteins are reported, suggesting its distinct roles from 5mC in transcriptional regulation [Citation29]. 5hmC distribution is highly tissue-specific and 5hmC is abundant in normal brain tissue and ES cells but depleted in some cancers, including acute myeloid leukaemia and glioma [Citation28,Citation30]. 5hmC is involved in neuronal differentiation, the maintenance and differentiation of ES cells and the occurrence of some cancers such as acute myeloid leukaemia and glioma [Citation24,Citation27,Citation30]. In addition, although 5hmC level is lower than 5mC level in the genome, 5hmC is enriched in transcriptional regulatory elements such as promoters, enhancers, and gene bodies [Citation27,Citation28]. It is therefore important to reveal the tissue and genome distribution of 5hmC.
Methods for bisulphite sequencing and methylation analysis using methylation-sensitive restriction enzymes such as HpaII and HhaI or methylation-dependent restriction enzymes such as McrBC and LpnPI (MS/MD-REMM) can distinguish unmodified cytosine from 5mC and 5hmC but not 5mC from 5hmC [Citation16,Citation31]. However, as bisulphite sequencing and MS/MD-REMM are frequently used to identify ASM sites, whether most previously identified sd-ASM and pd-ASM sites are methylated or hydroxymethylated remains unknown.
A modified bisulphite sequencing method known as TAB-Seq can distinguish 5hmC from 5mC and unmodified cytosine at single-base resolution [Citation32]. Another modified bisulphite sequencing method, oxBS-Seq, is also capable of distinguishing 5mC from 5hmC and unmodified cytosine at single-base resolution [Citation33]. However, the bisulphite PCR is usually more difficult than conventional PCR due to the genomic DNA degradation and the reduction of the DNA sequence complexity, making it harder to produce a specific PCR product [Citation34]. In addition, bisulphite sequencing using these modified methods often requires deep sequencing of bisulphite PCR products to determine the methylation or hydroxymethylation status of alleles within a sample, for example, samples in which a portion of cells are allele-specifically methylated or hydroxymethylated and the others biallelically unmethylated [Citation35]. The majority of sd-ASM occurs in a cell type–specific manner [Citation36] and 5hmC level is low in the genome, indicating the necessity of deep sequencing of bisulphite PCR products to determine the methylation or hydroxymethylation status of alleles. However, deep sequencing of bisulphite PCR products from a small number of amplicons is relatively costly and computer analysis of bisulphite sequencing reads obtained is required for determining the methylation or hydroxymethylation status of alleles. Furthermore, bisulphite treatment degrades genomic DNA into short fragments, which often makes it impossible to design bisulphite primers for longer PCR amplicons containing both a sufficient number of CpG sites and polymorphic sites to distinguish parental alleles [Citation34,Citation37].
As 5hmC plays roles both distinct from and equivalent to 5mC in regulating of gene expression depending on conditions [Citation27,Citation28,Citation38], it is necessary to determine whether previously reported sd-ASM and pd-ASM sites are methylated or hydroxymethylated. However, there are only a few reports describing the identification of sequence-dependent allele-specific hydroxymethylation (sd-AShyM) and parent-of-origin–dependent allele-specific hydroxymethylation (pd-AShyM). If sd-AShyM and pd-AShyM are common in the mammalian genome, identification of novel sd-AShyM and pd-AShyM sites could also facilitate elucidation of the mechanisms of polymorphism-associated differences in disease susceptibility and genomic imprinting, respectively.
However, a method that enables the determination of allelic methylation and hydroxymethylation status without bisulphite treatment and deep sequencing is needed in order to identify novel sd-AShyM and pd-AShyM sites. Here, we introduce a modified method for HM-PCR that not only enables the determination of the methylation and hydroxymethylation status of alleles without the need for bisulphite PCR and deep sequencing but also permits scanning of the allelic methylation and hydroxymethylation status of longer amplicons compared to modified bisulphite sequencing methods.
Results
Principle for evaluation of allelic methylation and hydroxymethylation status–dependent PCR
We previously reported the HM-PCR assay for determining allelic methylation status. The HM-PCR assay can distinguish complete, null, composite, and incomplete allelic methylation status according to PCR amplification or lack thereof of each restriction enzyme–digested DNA [Citation16]. However, the HM-PCR assay cannot determine allelic hydroxymethylation status. To overcome this drawback, we modified the HM-PCR assay to develop a novel method designated allelic methylation and hydroxymethylation status–dependent PCR (AMhyMsd-PCR).
In AMhyMsd-PCR, genomic DNA is digested with either a methylation-sensitive restriction enzyme (e.g., HpaII or HhaI), a methylation-dependent restriction enzyme (e.g., McrBC or LpnPI), or a hydroxymethylation-dependent restriction enzyme (e.g., PvuRts1I) () [Citation39]. In addition, genomic DNA is treated with T4-BGT and subsequently digested with a glucosylated 5-hydroxymethylcytosine-sensitive restriction enzyme (e.g., MspI or TaqαI) () [Citation40]. MspI or TaqαI digestion following T4-BGT treatment is hereafter referred to as T4-BGT+MspI or T4-BGT+TaqαI digestion, respectively. Amplicons bearing the recognition site for each restriction enzyme are then PCR-amplified in the respective digested DNAs.
Table 1. Restriction enzymes used for AMhyMsd-PCR.
The right panel in shows PCR amplification patterns of eight representative allelic methylation and hydroxymethylation states in AMhyMsd-PCR. As shown in , AMHyMsd-PCR can distinguish eight allelic methylation and hydroxymethylation statuses according to PCR amplification patterns from each restriction enzyme–digested DNA.
Figure 1. Principle of AMhyMsd-PCR.
The two parallel lines in each rounded rectangle show genomic amplicons from parental alleles. The two parallel lines in each rounded rectangle indicate PCR amplicons from parental alleles. The opened, solid, and shaded circles on the lines indicate unmethylated CCGG (HpaII and T4-BGT+MspI recognition sequences), methylated CmCGG (LpnPI and T4-BGT+MspI recognition sequences), and hydroxymethylated ChmCGG (LpnPI recognition sequences), respectively. The opened, solid, and shaded squares on the lines indicate unmethylated CN11-12/N9-10G, methylated mCN11-12/N9-10G, and hydroxymethylated hmCN11-12/N9-10G (PvuRts1l recognition sequences), respectively. Complete, no, and incomplete digestion of a target amplicon with each restriction enzyme are indicated by +, –, and +/–, respectively. The right panel shows the gel electrophoresis pattern of PCR products obtained from genomic amplicons digested with the indicated restriction enzymes.
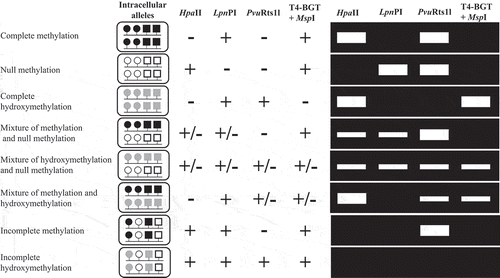
If PCR products from a target amplicon are obtained only from HpaII-, PvuRts1I-, or T4-BGT+MspI–digested genomic DNAs, this indicates that the target amplicon is composed of methylated and hydroxymethylated alleles (designated ‘mixture of methylation and hydroxymethylation’). Allele-specific demethylation and coexistence of both biallelically methylated and hydroxymethylated copies are included in ‘mixture of methylation and hydroxymethylation’. When PCR products from a target amplicon are obtained from all of the restriction enzyme–digested genomic DNAs, this indicates that both hydroxymethylated and unmethylated alleles are present in a target amplicon (designated ‘mixture of hydroxymethylation and null methylation’). Allele-specific hydroxymethylation (AShyM) and the presence of both biallelically hydroxymethylated and unmethylated copies are included in ‘mixture of hydroxymethylation and null methylation’. However, AMHyMsd-PCR cannot distinguish AShyM status from coexistence of both biallelically hydroxymethylated and unmethylated copies in a genomic region. In this case, an amplicon containing polymorphic sites to discriminate parental alleles should be amplified by AMHyMsd-PCR in heterozygotes for the polymorphisms, and direct sequencing of the resulting PCR products is necessary for determining AShyM.
Although more complex allelic methylation and hydroxymethylation statuses, such as if there are both 5mC and 5hmC on the same allele, are also possible, they are not considered in . Note that AMHyMsd-PCR cannot distinguish the more complex allelic methylation and hydroxymethylation statuses from the eight representative allelic methylation and hydroxymethylation states in . Therefore, each PCR amplification pattern obtained from AMHyMsd-PCR could show allelic methylation or hydroxymethylation status other than the eight representative allelic methylation and hydroxymethylation states.
Validation of AMhyMsd-PCR by application to a CGI with pd-ASM on impact
Mouse Impact is a paternally expressed imprinted gene that bears a CGI with maternal ASM in its first intron [Citation41,Citation42]. The CGI is shown as GR #10 in , and GR #10 exhibits a length polymorphism (181 bp) between JF1 and B6 mice, with GR #10 from the B6 strain being longer than that from JF1 mice. We can thus easily distinguish the two alleles for GR #10 in F1 hybrids between B6 and JF1 mice [Citation16,Citation42]. However, whether the pd-ASM on GR #10 is an ASM or an AShyM cannot be determined.
Table 2. Allelic methylation and hydroxymethylation status of target genome regions identified using AMhyMsd-PCR.
We validated the AMhyMsd-PCR method by applying it to GR #10 with maternal ASM. As shown in ), PCR products of 1261 bp and 1442 bp were obtained from genomic DNAs from brain tissues of adult JF1 and B6 mice, respectively. Both the JF1 and B6 alleles were PCR-amplified in undigested DNAs from brain tissues of (JF1 × B6) F1 (designated JF/B6 F1) or (B6 × JF1) F1 (designated B6/JF F1) hybrids (7‒12 weeks of age). As GR #10 is maternally methylated and HpaII cannot digest methylated or hydroxymethylated DNAs, only the maternally derived JF1 and B6 alleles were amplified from the HpaII-digested DNAs in the JF1/B6 and B6/JF F1 hybrids, respectively ()). On the other hand, only the paternal B6 and JF1 alleles were amplified from the LpnPI-digested DNAs, in which methylated or hydroxymethylated alleles were digested, from JF1/B6 and B6/JF F1 mice, respectively ()). These results demonstrated maternal ASM or AShyM of GR #10, consistent with previous observations [Citation42].
Figure 2. (Continued).
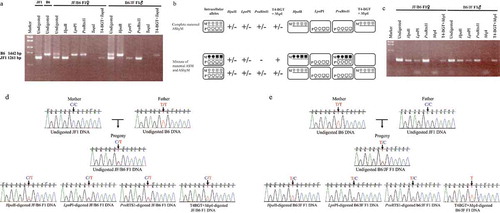
Figure 2. Proof-of-principle for AMhyMsd-PCR.
JF1, B6, JF/B6 F1, and B6/JF F1 indicate JF1, B6NJ, and reciprocal F1 hybrids of JF and B6NJ mice, respectively. (a) Application of AMhyMsd-PCR to GR #10 with maternal ASM of Impact. AMhyMsd-PCR was applied to the maternally methylated CGI, GR #10, located in the first intron of Impact. Undigested, HpaII, LpnPI, PvuRTS1l, TaqαI, and T4-BGT+TaqαI indicate the PCR products from undigested, HpaII-, LpnPI-, PvuRTS1l-, TaqαI-, and T4-BGT+TaqαI-digested DNAs, respectively. The PCR products from undigested, HpaII-, LpnPI-, PvuRTS1l-, TaqαI-, and T4-BGT+TaqαI-digested DNAs from JF1, B6, JF/B6 F1, and B6/JF F1 mice were electrophoresed, stained with ethidium bromide, and visualized by UV illumination. (b) AMhyMsd-PCR can distinguish complete AShyM from ‘mixture of ASM and AShyM’. The ‘M’ and ‘P’ in each rounded rectangle indicate maternal and paternal alleles, respectively. The right panel shows parental alleles exempt from restriction enzyme digestion of the amplicon. (c) Application of AMhyMsd-PCR for GR #9, the Nhlrc1 promoter region with sd-AShyM. Undigested, HpaII, LpnPI, PvuRTS1l, MspI, and T4-BGT+MspI indicate the PCR products from undigested, HpaII-, LpnPI-, PvuRTS1l-, MspI- and T4-BGT+MspI-digested DNAs, respectively. The PCR products from undigested, HpaII-, LpnPI-, PvuRTS1l-, MspI-, or T4-BGT+MspI-digested DNAs from JF/B6 F1 or B6/JF F1 mice were electrophoresed and stained with ethidium bromide. (d) Direct sequencing of AMhyMsd-PCR products from GR #9 in a JF/B6 F1 hybrid. The vertical arrowhead shows the SNP site (rs48369422: C/T). (e) Direct sequencing of AMhyMsd-PCR products from GR #9 in a B6/JF F1 hybrid. The vertical arrowhead shows the SNP site (rs48369422: C/T). (f) Bisulphite sequencing of GR #9 in JF/B6 and B6/JF F1 hybrids. Each row of circles indicates a clone of bisulphite PCR products. Open and closed circles show unmethylated and methylated CpG sites, respectively. The CpG sites within MspI recognition sequences (CCGG) in the AMhyMsd-PCR amplicon are indicated by vertical arrowheads. The SNP site (rs48369422: C/T) is indicated by a vertical arrowhead. (g) Quantitative PCR targeting GR #9 in undigested and each restriction enzyme-digested DNAs from a B6/JF F1 hybrid. Undigestion, HpaII, MspI, and T4-BGT+MspI indicate enzyme-resistant DNA levels of GR #9 in undigested, HpaII-, MspI- and T4-BGT+MspI-digested DNAs, respectively.

In addition, PvuRTS1 digests hydroxymethylated DNAs but not unmethylated or methylated DNAs. Only the paternal B6 allele was amplified from the PvuRTS1-digested DNA from a JF1/B6 F1 hybrid mouse, whereas only the paternal JF1 allele was amplified from the PvuRTS1-digested DNA from a B6/JF F1 hybrid mouse ()). In contrast to PvuRTS1 digestion, T4-BGT+TaqαI digestion led to the cleavage of unmethylated and methylated DNAs but not hydroxymethylated DNAs. PCR amplification occurred only from the maternal JF1 or B6 allele in the T4-BGT+TaqαI-digested DNAs from JF1/B6 F1 or B6/JF F1 hybrids, respectively ()). These results demonstrate maternal AShyM of GR #10 in mouse whole brain tissues.
Accordingly, AMhyMsd-PCR could discriminate null methylation (paternal alleles in the F1 hybrids), complete hydroxymethylation (maternal alleles in the F1 hybrids), and ‘mixture of hydroxymethylation and null methylation’ (both alleles in the F1 hybrids), demonstrating of the accuracy of the AMhyMsd-PCR method.
A previous study reported that ASM in imprinted differentially methylated regions (DMRs) often involves both methylation and hydroxymethylation (designated ‘mixture of ASM and AShyM’) [Citation43]. Relevant to this finding, AMhyMsd-PCR clearly demonstrated that GR #10 is subject to complete maternal AShyM in brain tissues in which the maternal allele is only hydroxymethylated and the paternal allele unmethylated. In AMhyMsd-PCR, the maternal allele of a genomic region with complete maternal AShyM is completely digested with LpnPI or PvuRTS1 but not HpaII or T4-BGT+MspI, allowing PCR amplification only from maternal alleles in HpaII- and T4-BGT+MspI–digested DNAs ()). In contrast, the paternal allele of the genomic region is completely digested with HpaII or T4-BGT+MspI but not LpnPI or PvuRTS1, allowing PCR amplification only from paternal alleles in LpnPI- or PvuRTS1-digested DNAs. On the other hand, if the genomic region shows ‘mixture of maternal ASM and AShyM’, PCR amplification from maternal, paternal, biallelic, or maternal alleles will be obtained from HpaII-, LpnPI-, PvuRTS1-, or T4-BGT+MspI–digested DNAs, respectively ()). For this reason, AMhyMsd-PCR can discriminate complete AShyM from ‘mixture of ASM and AShyM’.
Validation of AMhyMsd-PCR by application to a CGI with sd-AShyM on Nhlrc1
Kawasaki et al. reported that the promoter of the NHL repeat-containing 1 (Nhlrc1) gene is subject to B6-specific AShyM in the cerebrums of JF/B6 and B6/JF F1 mice [Citation44]. We therefore examined whether AMhyMsd-PCR could be used to determine if GR #9 in the promoter region of Nhlrc1 is subject to B6-specific AShyM in the brain of JF/B6 and B6/JF F1 mice ().
AMhyMsd-PCR leads to amplification of all restriction enzyme–digested DNAs from whole-brain tissues of adult JF/B6 and B6/JF F1 hybrids (7‒12 weeks of age), except for the MspI–digested DNA used as a negative control ()). We next performed direct sequencing of the PCR products to determine the allelic methylation and hydroxymethylation status. To discriminate JF1 and B6 alleles on GR #9 in JF/B6 and B6/JF F1 hybrids, a C/T SNP (rs48369422) was used, with the C and T alleles obtained from JF1 and B6 mice, respectively (). The amplicon bearing the C/T SNP site also contains CpG sites 1 and 7 embedded in the CCGG motif of the HpaII, LpnPI and MspI recognition sequences, in which methylation status was investigated by bisulphite sequencing ()).
Direct sequencing of the PCR products from undigested JF/B6 and B6/JF F1 DNAs displayed double peaks of C and T at the C/T SNP site (). A relatively higher T peak in comparison to that of C was obtained from HpaII- and T4-BGT+MspI–digested JF/B6 F1 DNAs, but double peaks of T and C were obtained from LpnPI- and PvuRTS1-digested JF/B6 F1 DNAs ()). This result was consistent with that of the EnIGMA method [Citation44] but not that of bisulphite sequencing ()). As qPCR and bisulphite sequencing revealed low and variable 5mC and 5hmC levels at HpaII/MspI recognition sites in GR #9 (), this inconsistency between the results obtained from different methodologies may result from the lack of deep sequencing process in bisulphite sequencing, indicating the necessity of deep sequencing of bisulphite PCR products to determine the allelic methylation and hydroxymethylation status of GR #9. Collectively, these results demonstrate the presence of GR #9 with B6-AShyM, biallelic hydroxymethylation, and biallelic null methylation in subpopulations of brain tissue of JF/B6 F1 hybrid mice. In contrast, a higher T peak in comparison to that of C was obtained from HpaII-digested B6/JF F1 DNA, but only a single T peak was obtained from T4-BGT+MspI–digested B6/JF F1 DNA ()). Furthermore, double peaks of T and C were obtained from LpnPI- or PvuRTS1-digested B6/JF F1 DNAs ()). This result was consistent with those of the EnIGMA method and hairpin TAB sequencing [Citation44] but not that of bisulphite sequencing ()). This result may also result from low and variable 5mC and 5hmC levels at HpaII/MspI recognition sites in GR #9 ()). Collectively, these results suggest that GR #9 is subject to B6-AShyM and biallelic null methylation in subpopulations of brain tissue of B6/JF F1 hybrid mice.
Application of AMhyMsd-PCR to analysis of DMRs between two inbred mouse strains
Schilling et al. screened for DMRs between macrophages from B6 and BALB/c mice using methyl-CpG immunoprecipitation and locus-wide tiling arrays, identifying over 400 DMR candidates [Citation20]. They also validated differences in methylation status between the strains in a validation set of DMR candidates and reported that the majority (13/18) were differentially methylated between macrophages from the two inbred strains of mice using MALDI-TOF MS analysis of bisulphite-treated DNAs. They also found that 5 of the 13 validated DMRs were subject to sd-ASM in F1 hybrids between B6 and BALB/c mice. We therefore expected that the 13 confirmed DMRs are candidates subject to sd-AShyM in JF/B6 and B6/JF F1 hybrids.
With reference to the B6 and JF1 genome databases, we chose eight genomic regions among the 13 confirmed DMRs as candidates to apply AMhyMsd-PCR. These regions contained at least one polymorphic site between JF1 and B6 mice and at least two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites. In contrast, more than two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites but not at least one polymorphic site between JF1 and B6 mice were contained in another confirmed DMR (GR #8 in ). However, a DNA region adjacent to GR #8 contained at least one polymorphic site between JF1 and B6 mice and more than two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites. We therefore selected the DNA region adjacent to GR #8 as a candidate to apply AMhyMsd-PCR. Although two genomic regions (GR #6 and #7 in ) in a validation set of the DMR candidates exhibited BALB/c- and B6-specific methylated insertions and were not the confirmed DMRs, we analysed these two candidate template regions by AMhyMsd-PCR as controls for the confirmed DMRs.
To confirm the polymorphic and HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites within the 11 abovementioned candidate template regions in JF1 and B6 mice, the genomic regions were PCR-amplified from genomic DNA from JF1 and B6 mice. The PCR products obtained were directly sequenced using a PCR primer or cloned into the pCR4-TOPO vector and sequenced. As a result, we confirmed the presence of at least one polymorphic site and more than two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites in 10 of 11 candidate template regions (except GR #7) in JF1 and B6 mice. GR #7 contains at least two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites but not at least one polymorphic site in JF1 and B6 mice. For this reason, we could not discriminate parental alleles in GR #7, but we included GR #7 among the template regions for AMhyMsd-PCR as controls for the confirmed DMRs, as mentioned above.
We ultimately applied AMhyMsd-PCR to a total of 10 target regions (GR #1, #2, #3, #4, #5, #6, #7, #8, #11, #12), as shown in . The genome sequences for the 10 target regions in JF1 and C57BL/6NJ mice are listed in Supplemental Table S1. Furthermore, because the normal adult brain genome is enriched in 5hmC compared to genomes from other adult tissues, we used genomic DNA of normal adult whole brain from F1 hybrids (7‒12 weeks of age) of JF1 and B6 mice for AMhyMsd-PCR.
The results of AMhyMsd-PCR and bisulphite sequencing for the ten target regions are shown in and Supplemental Figures S1‒9. If not distinguishing between methylation and hydroxymethylation, the results of AMhyMsd-PCR were consistent with those of bisulphite sequencing for all 10 target regions ( and Supplemental Figures S1‒9). These results indicate that AMhyMsd-PCR is suitable for determining allelic methylation status.
Figure 3. (Continued).
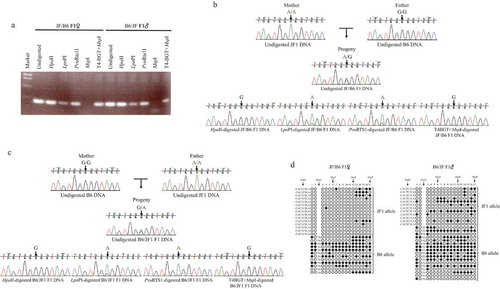
Figure 3. GR #11 of Pdgfrb exon 7 exhibits sd-AShyM in the brain of adult JF/B6 and B6/JF F1 hybrids.
JF1, B6, JF/B6 F1, and B6/JF F1 indicate JF1, B6NJ, and reciprocal F1 hybrids of JF and B6NJ mice, respectively. (a) AMhyMsd-PCR of GR #11 in exon 7 of Pdgfrb. Undigested, HpaII, LpnPI, PvuRTS1l, MspI, and T4-BGT+MspI indicate the PCR products from undigested, HpaII-, LpnPI-, PvuRTS1l-, MspI-, and T4-BGT+MspI-digested DNAs, respectively. The PCR products from undigested, HpaII-, LpnPI-, PvuRTS1l-, MspI-, or T4-BGT+MspI-digested DNAs from JF/B6 or B6/JF F1 hybrids were electrophoresed, stained with ethidium bromide, and visualized by UV illumination. (b) Direct sequencing of AMhyMsd-PCR products from GR #11 in a JF/B6 F1 hybrid. The vertical arrowhead shows the A/G SNP site (chr18:61,225,744). (c) Direct sequencing of AMhyMsd-PCR products from GR #11 in a B6/JF F1 hybrid. The vertical arrowhead shows the A/G SNP site (chr18:61,225,744). (d) Bisulphite sequencing of GR #11 in the brain of adult JF/B6 and B6/JF F1 hybrids. The CpG sites within MspI recognition sequences (CCGG) in the AMhyMsd-PCR amplicon are indicated by vertical arrowheads. The indel sites and insertion sequences are also indicated by vertical arrowheads. (e) Bisulphite sequencing of GR #11 in the brain of adult JF and B6 mice. The insertion sequences are indicated by a vertical arrowhead. (f) Multiple sequence alignment of GR #11 from three mouse strains. The asterisk shows an identical base among these mouse strains.

Four of the 10 target regions (GR #2, #3, #11, #12) exhibited ‘mixture of hydroxymethylation and null methylation’ in AMhyMsd-PCR in which PCR products are obtained from HpaII/HhaI-, LpnPI-, PvuRTS1-, and T4-BGT+MspI/TaqαI-digested DNAs (). We therefore examined whether GR #2, #3, #11, and #12 exhibiting ‘mixture of hydroxymethylation and null methylation’ are subject to sd-AShyM by direct sequencing of the AMhyMsd-PCR products from restriction enzyme–digested DNAs. To discriminate parental JF1 and B6 alleles on GR #2, #3, #11, #12 in the F1 hybrids, we analysed C/T (chr4:46,614,000 for NCBI37/mm9), A/G (rs30171093), A/G (chr18:61,225,744 for NCBI37/mm9), and C/A (chrX:120,217,176 for NCBI37/mm9) SNPs, respectively.
An amplicon bearing the A/G SNP site on GR #11 in exon 7 of Pdgfrb was amplified by AMhyMsd-PCR using genomic DNA from JF/B6 and B6/JF F1 hybrids, with the A allele from the JF1 strain and the G allele from the B6 strain (). The amplicon also contained CpG sites 12 and 17 embedded in the CCGG motif of HpaII, LpnPI, and MspI recognition sequences ()). Direct sequencing of the PCR products from undigested JF/B6 and B6/JF F1 DNAs displayed double peaks of A and G at the A/G SNP site (). A higher G peak in comparison to that of A was obtained from HpaII- and T4-BGT+MspI-digested JF/B6 and B6/JF F1 DNAs, but a higher A peak in comparison to that of G was obtained from LpnPI- and PvuRTS1-digested JF/B6 and B6/JF F1 DNAs (). In addition, bisulphite sequencing showed that GR #11 is more methylated/hydroxymethylated in the B6 allele than the JF1 allele in the brain of adult JF/B6 and B6/JF F1 hybrids and methylated or hydroxymethylated to a greater extent in B6 mice than JF1 mice (). These results indicate that GR #11 exhibits nearly complete sequence-dependent B6-AShyM, with the B6 allele almost completely hydroxymethylated and JF1 allele almost fully unmethylated in the brain of adult JF/B6 and B6/JF F1 hybrids. Schilling et al. reported that GR #11 also exhibits sequence-dependent B6-ASM in normal adult testes from F1 hybrids of B6 and BALB/c mice. These results indicate that cis-regulatory sequences found only in B6 (B6-specific cis-regulatory sequences) or BALB/c and JF1 (BALB/c- and JF1-specific cis-regulatory sequences) mice promote sd-ASM or sd-AShyM of GR #11. Although GR #11 from B6 and BALB/c mice shares the same sequence at almost all the positions without asterisks in the multiple alignment of ), there are BALB/c- and JF1-specific insertion sequences (BALB/c: CATTCTCA; JF1: CACTCTCA) or B6-specific deletions only at base 198. Therefore, BALB/c- and JF1-specific insertions or B6-specific deletions may result in sequence-dependent B6-AShyM of GR #11. A search of transcription factor (TF) binding motifs using the transfac database [Citation45] revealed that PAX5 binding sequences (gcaggatgCATTGgtgtctgggcataac) are within the B6-specific deletion site but not the BALB/c- and JF1-specific insertion sites ( and )). The presence or absence of PAX5 binding sequences in GR #11 may result in B6-specific sd-ASM or sd-AShyM of GR #11 in these mouse strains.
Table 3. TF binding sequences associated with strain-specific methylation or hydroxymethylation in target genome regions.
In contrast, direct sequencing of AMhyMsd-PCR products and bisulphite sequencing showed that GR #2 in the promoter region of Coro2a is subject to sequence-dependent JF1-AShyM in the brain of normal adult JF/B6 and B6/JF F1 hybrids (Supplemental Figure S1(a‒d)). However, in contrast to GR #11, direct sequencing of PCR products from LpnPI- and PvuRTS1-digested JF/B6 and B6/JF F1 DNAs revealed double peaks of C and T at the C/T SNP site (Figure S1(b,c). These results suggest that GR #2 is subject to JF1-AShyM in some cells but biallelically unmethylated in others, as the brain tissues of the F1 hybrids are composed of two types of cells. GR #2 exhibits sequence-dependent BALB/c-ASM in bone marrow–derived macrophages from F1 hybrids between B6 and BALB/c mice [Citation20]. These results indicate that B6-specific or BALB/c- and JF1-specific cis-regulatory sequences are responsible for sd-ASM and sd-AShyM of GR #2. A search of TF binding motifs revealed that the PAX5 (tcccctgccacactgCTTTGgccctggc at base 500) and SZF11 (ctgctttggCCCTGg at base 512) binding sequences are present in B6 mice but not BALB/c and JF1 mice ( and Supplemental Figure S1(e)). The presence or absence of these TF binding sequences in GR #2 could result in sequence-dependent BALB/c-ASM or JF1-AShyM of GR #2 in these three mouse strains.
Our data revealed that GR #12 in the promoter region of Cldn34c1 is subject to sequence-dependent JF1-AShyM in the brain of adult JF/B6 and B6/JF F1 hybrids by direct sequencing of AMhyMsd-PCR products and bisulphite sequencing (Supplemental Figure S2(a‒e)). In addition, similar to GR #2, direct sequencing of PCR products from LpnPI- or PvuRTS1-digested JF/B6 or B6/JF F1 DNAs displayed double peaks of C and A at the C/A SNP site (Supplemental Figure S2(b) and C). These results also suggest that GR #12 is subject to JF1-AShyM in some cells but totally unmethylated in others, as brain tissues of the F1 hybrids consist of different types of cells. GR #12 exhibits BALB/c-specific methylation in bone marrow–derived macrophages and normal adult spleens from B6 and BALB/c mice [Citation20]. These results indicate that BALB/c-specific methylation or sequence-dependent JF1-AShyM of GR #12 is associated with B6-specific cis-regulatory sequences or BALB/c- and JF1-specific cis-regulatory sequences. There are many TF binding sequences in GR #12 that are found only in mouse strains exhibiting methylation or hydroxymethylation and that are also found only in mouse strains exhibiting null methylation ( and Supplemental Figure S2(e)). Therefore, we could not narrow down the TF binding sequences responsible for strain-specific methylation or hydroxymethylation of GR #12, unlike GR #11. However, these mouse strain-specific TF binding sequences correlated with methylation or hydroxymethylation status may promote strain-specific methylation or hydroxymethylation in GR #12.
Direct sequencing of AMhyMsd-PCR products revealed that adult brain tissues of JF/B6 and B6/JF F1 hybrids can be divided into two populations, one in which GR #3 in the first intron of Asb4 is biallelically hydroxymethylated, and another in which it is biallelically unmethylated (data not shown). Bisulphite sequencing also suggested the co-occurrence of both biallelic methylation or hydroxymethylation and biallelic unmethylation of GR #3 in the brain of adult JF/B6 F1 hybrid mice (Supplemental Figure S3(a)). In addition, Schilling et al. reported that GR #3 exhibits sequence-dependent BALB/c-ASM in bone marrow–derived macrophages from F1 hybrids of B6 and BALB/c mice. These results indicate that GR #3 may be subject to null methylation in bone marrow–derived macrophages from B6 mice but hydroxymethylation and null methylation in the brain from adult B6 and JF1 mice.
Discussion
Modified bisulphite sequencing methods enable the examination of 5hmC levels of alleles at the single-base resolution but often require deep sequencing of bisulphite PCR products to determine the allelic methylation and hydroxymethylation status. In addition, as the modified methods degrade genomic DNA into short fragments, it is often impossible to design primers for long amplicons that bear a sufficient number of CpG and polymorphic sites to allow for allelic discrimination. HM-PCR is based on MS/MD-REMM and can detect sd-ASM or pd-ASM without bisulphite treatment and deep sequencing by using both methylation-sensitive and dependent restriction enzymes compared with bisulphite sequencing method. However, HM-PCR cannot distinguish hydroxymethylated alleles from methylated alleles.
Here we developed AMhyMsd-PCR, a modified method for HM-PCR that can distinguish hydroxymethylated alleles from methylated alleles. In addition, compared with modified bisulphite sequencing methods, AMhyMsd-PCR does not degrade genomic DNA into short fragments and does not require deep sequencing of bisulphite PCR products for determining allelic methylation and hydroxymethylation status. Indeed, although GR #10 is too enriched in long tandem repeat sequences to design bisulphite primers containing both CpG sites in the long tandem repeat sequences and the polymorphic indel within an amplicon, AMhyMsd-PCR enabled us to identify pd-AShyM of GR #10 using primers for the long amplicon. In addition, using AMhyMsd-PCR, we could clearly detect sd-AShyM of GR #9 and GR #12, even though only a small subset of brain tissues exhibit AShyM ( and Supplemental Figure 2(b–e)). In this case, modified bisulphite sequencing methods would require deep sequencing to identify sd-AShyM of GR #9 and GR #12.
One drawback of AMhyMsd-PCR, however, is the possibility of false-positive and -negative PCR amplification from the enzyme-digested genome due to incomplete and excessive digestion by restriction enzymes, respectively. It has been reported that the digestion efficiency of MspI is dramatically reduced with symmetrical hydroxymethylation of its recognition sequences and partially reduced with hemi-hydroxymethylation of its recognition sequences, whereas there is relatively no decrease in the cleavage efficiency of TaqαI with symmetrical hydroxymethylation of its recognition sequences [Citation46]. Indeed, we encountered complete digestion of some unmethylated genomic regions by addition of excessive LpnPI or PvuRTS1 followed by no PCR amplification and incomplete digestion of hydroxymethylated GR #7 and GR #10 by MspI followed by PCR amplification (data not shown). Databases integrating WGBS data across a variety of species provide an interactive browser for visualization of methylation status in each genomic region [Citation47,Citation48]. Therefore, it appears necessary to optimize the amount of restriction enzymes and choose optimal restriction enzymes for AMhyMsd-PCR, referring to these databases as a reference or using the internal control regions where methylation or hydroxymethylation statuses are known. In addition, there are also some limitations to AMhyMsd-PCR. One is that in order to conduct the allele-specific analysis, there needs to be at least one allele-specific polymorphism in relatively close proximity to the cytosines being interrogated. The second limitation is that it is difficult to determine the methylation status at specific CpG dinucleotides within an amplicon if more than one methylation state is present on a single allele. For example, if there are multiple MspI/HpaII sites within the target sequence and at least one is unmethylated, no product will be produced when treated with HpaII, and it is not possible to determine whether one or more than one of the HpaII site(s) is/are unmethylated. This leads to an underestimation of the amount of methylation that is actually present. The third limitation is that AMhyMsd-PCR provides limited insight into the methylation status across the locus. In other words, AMhyMsd-PCR can examine allelic methylation and hydroxymethylation status only at restriction enzyme recognition sites. This indicates that AMhyMsd-PCR cannot determine allelic methylation and hydroxymethylation status in genomic regions with no restriction enzyme recognition sites and allelic methylation and hydroxymethylation status at single-base resolution.
We examined the allelic methylation and hydroxymethylation status of a total of 12 genomic regions, including GR #9 and GR #10, used for validation experiments by AMhyMsd-PCR to find one novel genomic region with pd-AShyM, GR #10, and three novel genomic regions with sd-AShyM, GR #2, GR #11, and GR #12.
Enrichment of 5hmC of the methylated allele at specific imprinted DMRs is observed in both the human placenta and brain [Citation43]. It was also reported that ASM in imprinted DMRs often involves both methylation and hydroxymethylation (i.e., ‘mixture of ASM and AShyM’) [Citation43]. Impact bears GR #10 with maternal ASM in the first intron that would contribute to paternal allele-specific expression [Citation41,Citation42]. AMhyMsd-PCR revealed that GR #10 in the first intron of Impact is subject to complete maternal AShyM in the whole brain, with the paternal allele completely unmethylated and the maternal allele completely hydroxymethylated. In other words, AMhyMsd-PCR enabled us to discriminate complete AShyM from ‘mixture of ASM and AShyM’ using both hydroxymethylation-sensitive and -dependent restriction enzymes ()). Therefore, maternal allele-specific repression of Impact in the adult brain would be regulated not by maternal ASM but maternal AShyM of GR #10.
Although the mechanism underlying sd-ASM remains unclear, sd-ASM may result from disruption of trans-acting TF binding to cis-acting sequences [Citation22,Citation49]. GR #9 showed only a single T peak in T4-BGT+MspI–digested B6/JF F1 DNA but a relatively higher T peak in comparison to that of C in T4-BGT+MspI–digested JF/B6 F1 DNA (). Furthermore, the results of bisulphite sequencing of GR#9 were different between JF/B6 and B6/JF F1 hybrids ()). These inconsistencies between JF/B6 and B6/JF F1 hybrids suggest that sd-AShyM is also promoted by not only cis-acting sequences but also trans-acting TF binding to the cis-acting sequences.
Schilling et al. and we showed that GR #11 bears sequence-dependent B6-ASM and B6-AShyM in the testes of adult F1 hybrids of B6 and BALB/c mice and in brain tissues of adult F1 hybrids between B6 and JF1 mice, respectively. It was validated by qRT-PCR analysis that Pdgfrb associated with GR #11 in the exon 7 is more highly expressed in bone marrow–derived macrophages of BALB/c mice than in those of B6 mice, suggesting that B6-ASM of GR #11 contributes to reduced expression of Pdgfrb in B6 mice compared to BALB/c mice [Citation20]. Sequence-dependent B6-AShyM of GR #11 may thus also lead to higher expression of Pdgfrb in JF1 alleles than in B6 alleles in the brain of JF/B6 or B6/JF F1 hybrids. In addition, we revealed that PAX5 binding sequences occur only in GR #11 of B6 mice and not in GR #11 of BALB/c and JF1 mice. These data suggest that the PAX5 binding sequence is associated with sequence-dependent B6-ASM or B6-AShyM of GR #11 in these mouse strains. Indeed, Pax5 is involved in ASM and histone modification in the 3’ regulatory region of the Immunoglobulin Heavy-Chain Locus [Citation50]. As many TFs are known to regulate methylation or demethylation in their binding regions, our approach described above could also reveal other novel cis regulatory sequences responsible for sd-ASM and sd-AShyM [Citation51].
In this study, only GR #7 exhibited ‘mixture of methylation and hydroxymethylation’ in AMhyMsd-PCR analysis of the brain of adult F1 hybrids of B6 and JF1 mice. However, as GR #7 contains no polymorphic sites in JF1 and B6 mice, we could not discriminate parental alleles and determine the hydroxymethylation status of alleles in GR #7. Intriguingly, it has been reported that some imprinted DMRs are subject to allele-specific demethylation (i.e., ‘mixture of methylation and hydroxymethylation’ in ) in which one allele is methylated and the other hydroxymethylated. Hydroxymethylation at these DMRs is enriched in transcribed alleles of associated imprinted genes [Citation43,Citation52]. The hydroxymethylated allele in this allele-specific demethylation often involves both methylation and hydroxymethylation (designated ‘mixture of allele-specific demethylation and biallelic methylation’) [Citation43,Citation52]. Here, it is theoretically possible that AMhyMsd-PCR discriminates complete allele-specific demethylation, in which one allele is completely methylated and the other completely hydroxymethylated, from ‘mixture of allele-specific demethylation and biallelic methylation’ in a manner similar to ). Thus, AMhyMsd-PCR could be used to reveal novel imprinted and sequence-dependent allele-specific demethylation in the future.
Materials and methods
Animals
This study was approved under the guidelines of the Animal Care and Use Committee of Kanazawa University (No. AP-143251). Mice (Mus musculus molossinus; Japanese Fancy Mouse 1; JF1/Ms; 9‒11 weeks of age) were purchased from the National Institute of Genetics (Mishima, Japan). A material transfer agreement (No. S2014-002) for the use of JF1 mice was concluded between Kanazawa University and the National Institute of Genetics. C57BL/6NJ mice (Mus musculus domesticus; B6; 7 weeks of age) were purchased from SLC Japan (Shizuoka, Japan). The reciprocal F1 hybrid mice, (B6 × JF) F1 (B6/JF F1) and (JF × B6) F1 (JF/B6 F1), were bred in the Animal Centre at the Institute of Medical Science, Kanazawa University.
Sequencing of target genomic regions in JF1 and B6 mice
Genomic positions for a validation set of DMRs between B6 and BALB/c mice reported by Schilling et al. were obtained [Citation20]. With reference to the B6 and JF1 genome databases [Citation53], PCR primer pairs for these genomic regions were designed to bear at least one candidate polymorphic site between JF1 and B6 and more than two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites within the amplicons. Genomic DNA from brain tissues of JF1 and B6 mice (7‒12 weeks of age) was extracted using a standard phenol-chloroform method. The genomic regions were PCR-amplified from genomic DNA from JF1 and B6 mice. The PCR products obtained were directly sequenced using a PCR primer or cloned into the pCR4-TOPO vector (Thermo Fisher Scientific) and sequenced.
Allelic methylation and hydroxymethylation status-dependent PCR
Genomic DNA from whole-brain tissues of adult B6, JF1, and their reciprocal F1 hybrid mice (7‒12 weeks of age) was extracted by a standard phenol-chloroform method, incubated in 1 M NaCl overnight at room temperature, purified by phenol-chloroform and ethanol precipitation, and dissolved in TE buffer (10 mM Tris-HCl (pH 8.0), 1 mM EDTA). The purified mouse genomic DNA (1 μg) was digested with 50 units of HpaII/HhaI (TaKaRa) overnight at 37°C; 0.2 units of LpnPI (New England Biolabs) overnight at 37°C; or 0.6 units of PvuRts1I (Nippon Gene) overnight at 30°C in 50 μL of the recommended buffer for each enzyme.
Aside from the treatments described above, purified mouse genomic DNA (1 μg) was treated with 2.5 units of T4 phage β-glucosyltransferase (T4-BGT) (New England Biolabs) overnight at 37°C in 50 μL of appropriate buffer supplemented with 0.04 mM UDP-glucose. In addition, as a negative control, purified mouse genomic DNA (1 μg) was treated in the same manner except without the addition of T4-BGT. For GR #2, #5 and #9 (shown in ), 300 units of MspI (New England Biolabs) were added to each sample treated with T4-BGT and the untreated sample and incubated overnight at 37°C. For GR #7 and #10 (shown in ), 50‒100 units of TaqαI (New England Biolabs) were added to each sample treated with T4-BGT and the untreated sample and incubated overnight at 65°C. For GR #1, #3, #4, #6, #8, #11 and #12 (shown in ), genomic DNAs treated and untreated with T4-BGT were purified using Agencourt AMpure XP beads (Beckman Coulter) and then digested with 50 units of MspI (TaKaRa) overnight at 37°C in 50 μL of the recommended buffer. The digested DNAs were recovered by ethanol precipitation, and dissolved in TE buffer.
Subsequently, 50 ng of genomic DNA digested with each enzyme was used as a template for PCR. PCR primer pairs were designed to bear at least one polymorphic site between JF1 and B6 mice and more than two HpaII/HhaI, LpnPI, PvuRTS1, and MspI/TaqαI recognition sites within the amplicons. PCR for each amplicon was conducted in 10 μL of the recommended buffer supplemented with PCR enhancer (Invitrogen) using the primer pairs shown in supplemental Table S1. The amplified products were electrophoresed on a 1–2% agarose gel, stained with ethidium bromide, and visualized by UV illumination.
Real-time PCR
The enzyme-resistant DNAs present in undigested and each restriction enzyme-digested samples were quantified by SYBR Green qPCR. The SYBR Green qPCR was performed using TB Green® Premix Ex Taq II (TaKaRa) in Step One Real-time PCR system (Applied Biosystems). No template controls were used as negative controls. The qPCR for each sample was run in duplicate. The enzyme-resistant DNA levels in undigested and each restriction enzyme-digested samples were normalized against that of a DNA region in the exon 3 of histamine H4 receptor (Hrh4) not containing HpaII/MspI recognition sequences. Serial dilutions of the control genome were performed to produce standard-curve for each primer pairs. Each measured DNA level relative to Hrh4 was normalized to the undigested DNA. The primers used for the Hrh4 region were: Forward, 5’-TGTCTGTTCACAATTGTCCTTTCAAC-3’ and Reverse, 5’-TGGTTGCTTTGTCACACAAAGTATCT-3’, yielding a PCR product of 180 bp. The qPCR for GR #9 in the Nhlrc1 promoter region was performed using primers 5’-GACGGCGACATCCAGTGGGTACTTC-3’ (forward) and 5’-GACTCTCACCTGCTACCACGCCTTC-3’ (reverse) to amplify a fragment of 216 bp. The primers used for GR #12 in the Cldn34c1 promoter region were: Forward, 5’-AGGGGGGAATGGGCGAGCGGT-3’ and Reverse, 5’-GGAAAGGCCCGCTGGGAAAGGT-3’, yielding a PCR product of 191 bp.
Bisulphite sequencing
Mouse genomic DNA (10 μg) from brain tissues was treated using an EpiSight Bisulphite Conversion kit (Wako). One-tenth of the bisulphite-treated DNA was used for PCR in a 10-μL of the recommended buffer containing 0.5 U of Ex-Taq DNA polymerase (TaKaRa) and 2.5 pmols of each primer. Primer sequences are described in Supplemental Table S1. The amplified products were subsequently cloned into the pCR4-TOPO vector (Thermo Fisher Scientific) and sequenced.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Supplemental Material
Download MS Word (16.7 KB)Supplemental Material
Download MS Excel (18.7 KB)Supplemental Material
Download MS Power Point (1.5 MB)Acknowledgments
This work was supported by Japan Society for the Promotion of Science KAKENHI under Grant Numbers 24510266 and 15K06894 and Shibuya Science Culture and Sports Foundation. We thank Daisuke Fukushima in School of Electrical and Computer Engineering, Kanazawa University for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Related Research Data
References
- Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257.
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926.
- Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466.
- Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99.
- Chen RZ, Pettersson U, Beard C, et al. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93.
- Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Annu Rev Genet. 1997;31:571–610.
- Kaneda M, Okano M, Hata K, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903.
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428.
- Simmen MW. Genome-scale relationships between cytosine methylation and dinucleotide abundances in animals. Genomics. 2008;92:33–40.
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220.
- Rakyan VK, Hildmann T, Novik KL, et al. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the human epigenome project. PLoS Biol. 2004;2:e405.
- Illingworth RS, Bird AP. CpG islands - ‘A rough guide guide’. FEBS Lett. 2009;583:1713–1720.
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282.
- Wasson JA, Birol O, Katz DJ. A resource for the allele-specific analysis of DNA methylation at multiple genomically imprinted loci in mice. G3 (Bethesda). 2017;8:91–103.
- Wutz A, Smrzka OW, Schweifer N, et al. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature. 1997;389:745–749.
- Yamada Y, Watanabe H, Miura F, et al. A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 21q A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 21q. Genome Res. 2004;14:247–266.
- Yamada Y, Shirakawa T, Taylor TD, et al. A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 11q: comparison with chromosome 21q. DNA Seq. 2006;17:300–306.
- Kerkel K, Spadola A, Yuan E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–908.
- Xie W, Barr CL, Kim A, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831.
- Schilling E, Chartouni C, Rehli M. Allele-specific DNA methylation in mouse strains is mainly determined by cis-acting sequences. Genome Res. 2009;19:2028–2035.
- Edwards SL, Beesley J, French JD, et al. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–797.
- Do C, Lang CF, Lin J, et al. Mechanisms and disease associations of haplotype-dependent allele-specific DNA methylation. Am J Hum Genet. 2016;98:934–955.
- Bell CG, Finer S, Lindgren CM, et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One. 2010;5:e14040.
- Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES cell self-renewal, and ICM specification. Nature. 2010;466:1129–1133.
- Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303.
- Hardwick JS, Lane AN, Brown T. Epigenetic modifications of cytosine: biophysical properties, regulation, and function in mammalian DNA. Bioessays. 2018;40:1700199.
- Pfeifer GP, Kadam S, Jin SG. 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics Chromatin. 2013;6:10.
- Hahn MA, Szabó PE, Pfeifer GP. 5-Hydroxymethylcytosine: a stable or transient DNA modification? Genomics. 2014;104:314–323.
- Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159.
- Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015;7:9.
- Cohen-Karni D, Xu D, Apone L, et al. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc Natl Acad Sci U S A. 2011;108:11040–11045.
- Yu M, Hon GC, Szulwach KE, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380.
- Booth MJ, Branco MR, Ficz G, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937.
- Kondratyev N, Golov A, Alfimova M, et al. Prediction of smoking by multiplex bisulfite PCR with long amplicons considering allele-specific effects on DNA methylation. Clin Epigenetics. 2018;10:130.
- Arai Y, Fukukawa H, Atozi T, et al. Ultra-deep bisulfite sequencing to detect specific DNA methylation patterns of minor cell types in heterogeneous cell populations: an example of the pituitary tissue. PLoS One. 2016;11:e0146498.
- Meaburn EL, Schalkwyk LC, Mill J. Allele-specific methylation in the human genome: implications for genetic studies of complex disease. Epigenetics. 2010;5:578–582.
- Liu Y, Siegmund KD, Laird PW, et al. Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13:R61.
- Mellén M, Ayata P, Dewell S, et al. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430.
- Sun Z, Dai N, Borgaro JG, et al. A sensitive approach to map genome-wide 5-Hydroxymethylcytosine and 5-Formylcytosine at single-base resolution. Mol Cell. 2015;57:750–761.
- Song CX, Yu M, Dai Q, et al. Detection of 5-hydroxymethylcytosine in a combined glycosylation restriction analysis (CGRA) using restriction enzyme TaqαI. Bioorg Med Chem Lett. 2011;21:5075–5077.
- Hagiwara Y, Hirai M, Nishiyama K, et al. Screening for imprinted genes by allelic message display: identification of a paternally expressed gene impact on mouse chromosome 18. Proc Natl Acad Sci U S A. 1997;94:9249–9254.
- Okamura K, Hagiwara-Takeuchi Y, Li T, et al. Comparative genome analysis of the mouse imprinted gene Impact and its nonimprinted human homolog IMPACT: toward the structural basis for species-specific imprinting. Genome Res. 2000;10:1878–1889.
- Hernandez Mora JR, Sanchez-Delgado M, Petazzi P, et al. Profiling of oxBS-450K 5-hydroxymethylcytosine in human placenta and brain reveals enrichment at imprinted loci. Epigenetics. 2018;13:182–191.
- Kawasaki Y, Kuroda Y, Suetake I, et al. A Novel method for the simultaneous identification of methylcytosine and hydroxymethylcytosine at a single base resolution. Nucleic Acids Res. 2017;45:e24.
- Wingender E, Chen X, Hehl R, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319.
- Ichiyanagi K. Inhibition of MspI cleavage activity by hydroxymethylation of the CpG site a concern for DNA modification studies using restriction endonucleases. Epigenetics. 2012;7:131–136.
- Song Q, Decato B, Hong EE, et al. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS One. 2013;8:e81148.
- Li R, Liang F, Li M, et al. MethBank 3.0: a database of DNA methylomes across a variety of species. Nucleic Acids Res. 2017;46:D288–D295.
- Onuchic V, Lurie E, Carrero I, et al. Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science. 2018;28:361.
- Giambra V, Volpi S, Emelyanov AV, et al. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3’ regulatory region of the immunoglobulin heavy chain locus. Mol Cell Biol. 2008;28:6123–6133.
- Suzuki T, Maeda S, Furuhata E, et al. A screening system to identify transcription factors that induce binding site-directed DNA demethylation. Epigenetics Chromatin. 2017;10:60.
- Wood AJ, Bourc’his D, Bestor TH, et al. Allele-specific demethylation at an imprinted mammalian promoter. Nucleic Acids Res. 2007;35:7031–7039.
- Takada T, Ebata T, Noguchi H, et al. The ancestor of extant Japanese fancy mice contributed to the mosaic genomes of classical inbred strains. Genome Res. 2013;23:1329–1338.