
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
We conducted a genome-wide association study of blood DNA methylation and smoking, attempted replication of previously discovered associations, and assessed the reversibility of smoking-associated methylation changes. DNA methylation was measured in baseline peripheral blood samples for 5,044 participants in the Melbourne Collaborative Cohort Study. For 1,032 participants, these measures were repeated using blood samples collected at follow-up, a median of 11 years later. A cross-sectional analysis of the association between smoking and DNA methylation and a longitudinal analysis of changes in smoking status and changes in DNA methylation were conducted. We used our cross-sectional analysis to replicate previously reported associations for current (N = 3,327) and former (N = 172) smoking. A comprehensive smoking index accounting for the biological half-life of smoking compounds and several aspects of smoking history was constructed to assess the reversibility of smoking-induced methylation changes. This measure of lifetime exposure to smoking allowed us to detect more associations than comparing current with never smokers. We identified 4,496 cross-sectional associations at P < 10−7, including 3,296 annotated to 1,326 genes that were not previously implicated in smoking-associated DNA methylation changes at this significance threshold. We replicated the majority of previously reported associations (P < 10−7) for current and former smokers. In our data, we observed for former smokers a substantial degree of return to the methylation levels of never smokers, compared with current smokers (median: 74%, IQR = 63-86%), corresponding to small values (median: 2.75, IQR = 1.5–5.25) for the half-life parameter of the comprehensive smoking index. Longitudinal analyses identified 368 sites at which methylation changed upon smoking cessation. Our study demonstrates the usefulness of the comprehensive smoking index to detect associations between smoking and DNA methylation at CpGs across the genome, replicates the vast majority of previously reported associations, and quantifies the reversibility of smoking-induced methylation changes.
Introduction
Several studies have examined the association between exposure to tobacco smoke and DNA methylation levels in blood [Citation1–Citation13]. A systematic review identified methylation at 1,460 CpG sites to be associated with smoking [Citation14], and a recent large-scale meta-analysis identified 2,623 CpGs with P < 10−7[Citation12]. These associations were identified comparing current with never smokers, and not all were replicated using independent data. Additionally, there is substantial variability by study in the strength of associations, which may be due to characteristics of the cohorts such as age or ethnicity, or methodological issues such as the variables used for adjustment in statistical models or the pipeline used for normalization of the DNA methylation data.
Most of these studies also reported differences in methylation for former smokers compared with never and current smokers, indicating a degree of reversibility of smoking-associated methylation changes. Few studies have examined reversibility patterns beyond assessing the effect of time since quitting [Citation5,Citation9,Citation10,Citation12]. Zeilinger and colleagues concluded that for 36 of 187 CpGs at which DNA methylation was associated with smoking, there was a linear association with time since quitting in former smokers, but the pattern of reversibility was only assessed graphically [Citation9]. Guida and colleagues assessed reversibility in a study based on 745 women and identified two clusters of smoking-associated methylation at CpG sites according to whether methylation reverted back to the level of never smokers within 35 years of quitting [Citation5]. The assessment of reversibility made by Joehanes and colleagues was based on 2,374 participants and concluded that for the majority of the 2,568 CpGs they examined (those with FDR-adjusted P < 0.05 in the comparison of former vs. never smokers) methylation levels returned to those of never smokers within five years of smoking cessation, and for only 36 CpGs did they observe no tendency of a return to the methylation levels of never smokers 30 years after they had quit [Citation12]. Consistent findings were reported by Wilson and colleagues, who made use of repeated methylation measures taken seven years apart to identify methylation at CpG sites that varied longitudinally with changes in smoking status [Citation10]. They also observed differential methylation in former smokers who had quit more than 40 years before methylation measurement, compared to never smokers. Assessing what smoking-associated methylation changes are transient or long-lasting may have important implications for biological understanding and clinical practice [Citation15].
Lifetime exposure to smoking relevant to DNA methylation can be modelled as a function of the smoking history of an individual, including the number of cigarettes smoked, the age at starting smoking and the duration of smoking, and the biological half-life of smoking compounds, expressed as a half-life parameter, i.e. half the time taken for smoking compounds to cease to be active. The resulting comprehensive smoking index (CSI) was shown to substantially improve the prediction of smoking-related disease compared with simpler smoking assessment models [Citation16–Citation18]. A prominent feature of the CSI is that it includes a parameter for biological half-life, representing the rate at which the activity of smoking compounds declines, and is therefore the parameter of interest when assessing reversibility.
In this study, we aimed to: i) conduct a genome-wide association study of DNA methylation and exposure to tobacco smoking measured using traditional smoking assessment and CSI [Citation16], the latter allowing a better assessment of the methylation reversibility pattern; ii) replicate previously reported associations, including associations observed in former smokers or by time since quitting; iii) assess the association between changes in DNA methylation and changes in smoking using repeated measures taken a median of 11 years apart.
Results
Altogether, 5,044 participants in the Melbourne Collaborative Cohort Study (MCCS) were included in the cross-sectional analysis; at baseline, their median age was 60.7 years (IQR: 53.9–65.4), 3,408 (68%) were males, and 655 (13%) were current, 2,010 (40%) former, and 2,379 (47%) never smokers (). Participants in the longitudinal analysis were on average younger (median age at baseline: 58.5 years).
Table 1. Characteristics of study participants from the Melbourne Collaborative Cohort Study (MCCS) at baseline (1990–1994) and follow-up (2003–2007).
Genome-wide association study of DNA methylation
Comparison of current, former and never smokers
At P < 10−7, we observed 1,851 differentially methylated CpG sites between current and never smokers, and 156 differentially methylated CpGs between former and never smokers, with 140 overlapping CpGs and 16 found in former smokers only. In total, 917 of the 1,851 CpGs (50%) associated with current smoking had not been reported in previous studies at P < 10−7 (Supplementary Table 1); 1,124 (61%) showed some methylation differences (P < 0.05 and same direction of coefficient) in former smokers. Reversibility coefficients (calculated as the ratio of regression coefficients comparing ‘former’ to ‘current’ smokers and ‘never’ to ‘current’ smokers) indicated that for former smokers, there was a substantial degree of return to the average methylation levels of never smokers (median: 74%, IQR = 63% to 86%).
Comprehensive smoking indices (CSI)
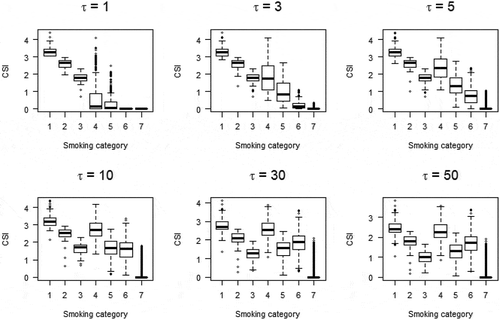
shows the relationship between smoking history and CSI in MCCS participants for six values of τ ranging from 1 to 50. To estimate τ, we first considered plausible values of half-life parameter τ of the CSI based on 3,327 differentially methylated CpGs identified in six previous studies at P < 10−7 (Supplementary Table 2). Estimated τ values were wide-ranging: median: 2.25, IQR: 1 to 5.25 and 3,038 (91%) CpGs had P < 0.05. To further refine the potential for these values to identify new associations, we considered only the 1,277 CpGs for which the previously reported association was replicated in our sample (with the estimated τ) at P < 10−7. For these, the median and 25th and 75th percentile values were 2.75, 1.5 and 5.25 respectively. We thus conducted methylome-wide association studies for each of these three values and identified 3,497 (τ = 2.75), 4,022 (τ = 1.5) and 2,433 (τ = 5.25), respectively, at P < 10−7. From these analyses, 4,496 associations were identified and DNA methylation at these CpGs was classified as smoking-associated in subsequent analyses, including 1,775 overlapping with associations identified using the current and former smoking variables. Of these, 3,296 (73%) had not been reported at P < 10−7 in previous studies, which corresponded to 1,326 genes not previously implicated in smoking-associated DNA methylation changes at this significance threshold. The function of all 2,102 genes identified at P < 10−7 is summarized in Supplementary Table 3. We examined the replication of these associations using the results from Joehanes et al. [Citation12] in which P-values up to 0.019 (FDR-adjusted P < 0.05) were presented for the current vs. never smoking association; we found that 1,189 (36%) of 3,296 were replicated at P < 0.019 with effect estimates in the same direction.
Figure 1. Relationshipa between smoking status and comprehensive smoking indices (equation (1)) for various values of half-life parameter τ.
Smoking categories:1: Current smoker; 20 cigarettes or more per day; 2: Current smoker; between 10 and 20 cigarettes per day; 3: Current smoker; less than 10 cigarettes per day; 4: Former smokers for less than 15 years; 20 cigarettes or more per day; 5: Former smokers for less than 15 years; less than 20 cigarettes per day; 6: Former smokers for more than 15 years; 20 cigarettes or more per day; 5: Former smokers for more than 15 years; less than 20 cigarettes per day;
Sensitivity analyses conducted without adjustment for alcohol consumption or BMI, or both, showed very similar results (Supplementary Table 4). Results using the pack-year variable were only partly consistent with those obtained using the CSI. The EWAS of the (log) pack-years of smoking identified a total of 930 CpGs at which methylation was associated with smoking (P < 10−7) of which 889 overlapped with the 4,496 associations identified at P < 10−7 with CSIs (Supplementary Table 3).
Interaction analyses
Using the Bonferroni correction for multiple testing (P = 0.05/4,496 = 1.1x10−5) and the CSI with τ = 1.5, we observed a weaker association for DNA methylation in women at a CpG not annotated to a gene, and a weaker association for participants with higher BMI at five CpGs, including two in AHRR (Supplementary Table 5). Significant interaction was also observed by country of birth at AHRR, at which UK-born participants had less pronounced methylation changes and at an unannotated region, where weaker associations were observed for participants born in the UK and Italy. No significant interaction with smoking status was observed at this significance threshold by age, alcohol consumption, or future case status.
Replication of previously reported associations
We examined the replication in the MCCS of 3,327 associations between current smoking and whole-blood DNA methylation previously reported at P < 10−7 in any of the six studies considered. We replicated, with coefficients consistent in direction, 2,795 (84%) at P < 0.05 and 934 (28%) at P < 10−7 using the current vs. never comparison. These numbers were 2,946 (89%) and 1,200 (36%), respectively, when considering any of the CSIs with τ = 1.5, τ = 2.75 or τ = 5.25 (, Supplementary Table 2). Of the 2,500 associations that had been reported in one study only, we replicated 1,983 (79%) at P < 0.05 using the current smoking variable; and 97% of associations that had been reported in two or more studies (). Finally, using our current vs former smoking comparison, we examined the replication of associations reported using an FDR-adjusted P < 0.05 to account for multiple testing in two of the six previous studies. The study by Besingi et al. [Citation2] reported 54 associations with P > 10−7. Of these, we replicated 45 (83%) at P < 0.05, and 42 (78%) using the Bonferroni correction for multiple testing (P < 9x10−4). The study by Joehanes et al. reported 16,138 associations with P > 10−7 [Citation12]. Of these, we replicated 9,337 (58%) at P < 0.05, and only 1,363 (8%) using the Bonferroni correction for multiple testing (P < 3x10−6). A total of 2,392 CpGs were not identified at any statistical significance thresholds by the 6 studies we compared our results with.
Table 2. Replication of previously reported associations (P < 10−7 in six large studies) using the ‘Current vs Never’ smoker comparison, and for 3 comprehensive smoking indices (τ = 1.5, τ = 2.75 or τ = 5.25).
We then examined the replication of associations identified for former compared with never smoking previously reported in any of four large studies. Of the 146 associations that had been reported at P < 10−7 in one study only, we replicated 129 (88%) at P < 0.05 and 60 (41%) at P < 10−7 using the former smoking variable. All associations that had been reported two or more times were replicated at P < 0.05 using the MCCS data (, Supplementary Table 6).
Reversibility of associations
Estimated τ values for the 4,496 associations were wide-ranging (Supplementary Table 7) but 90% were less than 6, with median [IQR] of 1.75 [1.25–3], consistent with and the 3,327 previously reported associations. The median τ was equal to 2 for CpGs that were differentially methylated in current or former smokers, compared with never smokers. shows the relationship between estimated values of τ and: i) reversibility coefficients; this analysis showed greater values of τ for CpGs at which methylation levels in former smokers were similar to those of current smokers, ii) the strength of association observed in current compared with never smokers; this analysis showed slightly greater τ values for most strongly differentially methylated CpGs in the cross-sectional EWAS, and iii) the strength of association observed in the longitudinal analysis, showing that CpGs at which methylation changed more strongly longitudinally did not have smaller half-life parameter estimates in the cross-sectional analysis.
Figure 2. Estimated values of half-life parameter τ according to other features of smoking-associated CpG sitesa.
a Values of τ above 40 (N = 44 CpGs) were omitted for graphical presentation
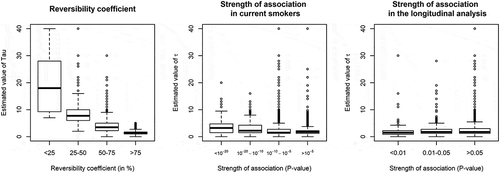
We then examined the distribution of τ values according to the reversibility patterns observed in three previous studies. First, Guida et al [Citation5]. grouped differentially methylated CpGs into persistent (N = 149) or reversible (N = 602) clusters. We found weak evidence (Wilcoxon rank-sum test one-sided P = 0.03) that τ values were greater in the persistent cluster (median τ (IQR): 3.75 [1.75–5.25]) compared with the reversible cluster (2.75 [1.75–5.00]). Second, Joehanes et al [Citation12]. identified 36 CpGs at which methylation levels did not return to never-smoker levels 30 years after smoking cessation: for these CpGs, we found τ values that were greater than for other differentially methylated CpGs (6.25 [3.25–13], one-sided P < 0.001). Third, in Wilson et al [Citation10]., 15 CpGs were differentially methylated in participants who had quit smoking for 40 years or more: for these 15 CpGs, we found weak evidence (one-sided P = 0.05) of greater τ values (3.75 [2.5–5.25] than for other differentially methylated CpGs.
We further examined the 4,496 cross-sectional associations for longitudinal associations using repeated methylation measures and smoking information collected a median of 11 years apart. After adjustment for baseline smoking status (CSI with τ = 1.5), the results were, comparing with smokers at both time points, 368 differentially methylated CpGs (P < 0.05 and same direction of association as in cross-sectional analyses) in participants who had quit from baseline to follow-up, 280 differentially methylated CpGs in former smokers at baseline and 262 in never smokers. The results without adjustment for baseline M-value were qualitatively similar, albeit identifying fewer longitudinal associations (Supplementary Table 8a).
When no adjustment for baseline smoking status was made, compared with participants who were smokers at both time points, 432 CpGs were differentially methylated (P < 0.05 and same direction of association as in cross-sectional analyses) in participants who had quit between baseline and follow-up, 1,233 differentially methylated CpGs in former smokers at baseline, and 1,495 in never smokers; (Supplementary Table 8b). Using the results with adjustment for baseline smoking status and baseline DNA methylation, we found no evidence that most strongly differentially methylated CpGs in current-to-former compared with current smokers at both time points had lower τ values ().
Discussion
Our study identified several thousand differentially methylated CpG sites with respect to smoking; 3,296 CpGs with P < 10−7 that had not been reported at this statistical significance threshold before were discovered in our cross-sectional EWAS and 1,189 (36%) of these were replicated using the results from a previous large study [Citation12].
Our literature review might have missed some previously discovered smoking-associated methylation measures at CpGs, but we likely included the majority of them. Additionally, for former smoking, we replicated a substantial proportion (90%) of previously reported associations and identified many differentially methylated CpGs at P < 10−7. It should be noted that three of the six studies [Citation2,Citation12,Citation19] we compared our results with accounted for multiple testing using an FDR-adjusted P < 0.05 and identified a substantially larger number of associations, but these would likely be of smaller magnitude, hence possibly less replicable and biologically relevant. This is consistent with the relatively lower replication rate observed for CpGs discovered in the study by Joehanes et al [Citation12]. and a simulation study that estimated an optimal multiple testing correction threshold for the HM450 assay to be 2.4x10−7 [Citation19].
We assessed associations using a comprehensive smoking index representing lifetime exposure to smoking relevant to DNA methylation which included several aspects of a person’s smoking history as well as, for each methylation site, the estimated biological half-life of smoking compounds. This modelling strategy has several limitations, including our assumptions that there was no lag-time between smoking exposure and changes in DNA methylation, and that the number of cigarettes smoked contributed equally to methylation changes throughout the lifetime. Another limitation is that because the CSI was log-transformed, the interpretation of the parameter τ was no longer that of a biological half-life, i.e. the time required for a biological substance to reduce to half its initial value [Citation16]. Specific to our study, this means that τ is not interpretable as half the time by which methylation levels of former smokers would return to the level of never smokers. Our values can nevertheless be used to rank CpGs by their rate of reversibility. We observed (by definition) a clear correspondence between the values of τ and the reversibility coefficients we calculated, suggesting that our analysis provides a more complete picture of how smoking-associated methylation changes vary over time. We also found that half-life parameter estimates were relatively similar across varying strength of evidence of association between smoking and DNA methylation, i.e. that more strongly differentially methylated CpGs did not show quicker return of methylation levels to normal. Although our half-life parameter estimates require formal validation in external studies, there was some degree of consistency between the reversibility patterns observed in our study and others [Citation5,Citation10,Citation12]. The main strength of the CSI is that it captures in a single variable several aspects of a smoking history that individually contribute to differential methylation, hence resulting in a more accurate measure of the effects of smoking (illustrated by e.g. >4,000 CpGs identified with τ = 1.5, which was substantially more that with the current smoking variable). Finally, the reversibility coefficients calculated in this study were substantially lower than those observed in our previous analysis of alcohol consumption [Citation20], which suggests that smoking-associated methylation marks might be more frequent but less persistent compared to alcohol-associated methylation changes.
Our longitudinal analysis had less precision due to fewer participants with relatively small variation in smoking status over a decade in this age range, and there was no clear correspondence with reversibility patterns observed from the cross-sectional data as CpG sites with smaller half-life parameter estimates were not associated with more statistically significant changes in DNA methylation longitudinally. It nevertheless identified many CpGs at which methylation levels returned towards normal in participants who had quit at follow-up compared with those still currently smoking, but these findings need to be replicated. The use of several models with or with adjustment for baseline DNA methylation and smoking indicated that confounding was likely to play a strong role in these analyses; this may be due in part to the considerable strength of association between smoking and DNA methylation or to potential ‘horse-racing’ or regression to the mean [Citation21] introduced via adjustment.
Another limitation of our study is the potential for residual confounding, especially by white blood cell type composition, which is strongly associated with smoking and DNA methylation. Cell composition was estimated with the widely used Houseman algorithm [Citation22,Citation23] and we did not assess sensitivity to the method used for deriving cell composition [Citation24]. Additionally, we reported in a previous study that many differentially methylated CpGs with respect to alcohol drinking are also associated with smoking, so it may be difficult to tease out the individual effects or joint influences on many of these CpGs across the genome [Citation20]. Finally, we included participants who later developed cancer, which could give rise to collider bias given the strong association of smoking with cancer risk [Citation25], but, by assessing effect modification by case-control status, we found no evidence of such bias in our setting. The findings from this analysis suggested that associations in controls and future cancer cases were of similar magnitude.
To conclude, our study provides evidence that several thousand associations between smoking and DNA methylation at CpGs exist across the genome that had not been discovered or replicated before. Smoking-associated methylation changes appeared largely reversible after smoking cessation. We also proposed a way to quantify the reversibility of methylation changes due to smoking by using a comprehensive smoking index that accounts for both the biological half-life of smoking compounds and several aspects of smoking history that are relevant to DNA methylation.
Materials and methods
Study participants
Between 1990 and 1994 (baseline), 41,513 participants were recruited to the Melbourne Collaborative Cohort study (MCCS). The majority (99%) were aged 40 to 69 years and 41% were men. Southern European migrants were oversampled to extend the range of lifestyle factors and genetic variation [Citation26] Participants were contacted again between 2003 and 2007 (follow-up). Blood samples were taken at baseline and follow-up from 99% and 64% of participants, respectively. Baseline samples were stored as dried blood spots on Guthrie cards for the majority (73%), as mononuclear cell samples for 25% and as buffy coat samples for 2% of the participants. Follow-up samples were stored as buffy coat samples and dried blood spots on Guthrie cards. All participants provided written informed consent and the study protocols were approved by the Cancer Council Victoria’s Human Research Ethics Committee.
The present study sample comprised MCCS participants selected for inclusion in one of seven previously conducted nested-case control studies of DNA methylation of colorectal, gastric, kidney, lung, and prostate cancer, B-cell lymphoma, and urothelial cell carcinoma (UCC) [Citation27–Citation32]. All participants were cancer-free at blood draw. Controls were matched to incident cases of prostate, colorectal, gastric, lung or kidney cancer, UCC or mature B-cell neoplasms on sex, year of birth, country of birth, baseline sample type and smoking status (the latter for the lung cancer study only). In the UCC case-control study, 303 participants had their blood sample collected at follow-up (2004–2007); we excluded these participants from our cross-sectional analyses because their questionnaire data and storage time were different from those with blood collected at baseline (1990–1994). We also excluded cases from the lung and UCC studies to avoid bias due to the strong association between smoking and these cancers [Citation25]. Methylation data for baseline blood samples (baseline study) were available from a total of 2,777 controls and 2,267 cases after quality control and exclusions. Additionally, methylation measures (Guthrie cards) were repeated at follow-up (2004–2007) for a subset of 1,100 of the controls who also had their baseline sample collected on a Guthrie card, of which 1,088 were available after quality control.
Description of the smoking variables is presented in . Participants with missing data for smoking variables were excluded from the analysis, as were those who had never smoked cigarettes but had smoked cigars or pipes. Missing data for confounders (<1% for BMI or alcohol drinking) were imputed using the median of the distribution.
Methods relating to DNA extraction, and DNA methylation processing and quality are presented in Supplementary Material.
Previously reported associations
Previous studies were identified using the keywords (‘smoking’ and ‘blood’ and ‘methylation’), which returned 416 articles in PubMed (31 July 2018). We retained from this search the six EWAS of smoking and blood DNA methylation we judged were the most important on this topic [Citation2,Citation5,Citation9,Citation10,Citation12,Citation19]. Other studies were identified but not selected due to small sample size, not identifying associations other than those reported in these six studies, or due to methodological issues, such as not adjusting for potential confounders of the association [Citation1,Citation3,Citation4,Citation6–Citation8,Citation11,Citation33–Citation37]. The six studies retained identified 3,327 associations with a P-value less than 10−7, 2500 (75%) in one study only, 438 (13%) in two, and 389 (12%) in three or more studies. Of the six studies, four also reported differentially methylated CpGs for former compared with never smokers [Citation5,Citation9,Citation12,Citation19], identifying 172 associations (at P < 10−7), including 146 in only one study.
Comprehensive smoking index (CSI)
We constructed a CSI following the recommendations of Leffondré and colleagues [Citation16]. The half-life parameter τ needs to be specified. Whereas in the case of lung cancer [Citation16], τ was estimated as the values maximizing the likelihood of models of association with lung cancer risk, τ was estimated in our study as the values maximizing the likelihood of models of association with DNA methylation. We observed better model fits (data not shown) when using the log-transformed version of the CSI: ln(CSI)+1, referred to as simply ‘CSI’, and we assumed no lag-time between exposure to smoking and changes in DNA methylation [Citation16]. The CSI was defined in our study as:
where T is duration of smoking in years, tsc the time since smoking cessation in years, N the average number of cigarettes smoked per day and τ the half-life parameter. We estimated τ from the data as follows: (i) by visual inspection of CSI values obtained for various τ values (), we concluded that for smaller values of τ, the CSI was both sensitive and more consistent with assumed biological activity by smoking history; (ii) for a CpG of interest, we fitted the same model for every CSI with τ value within the grid: {0.001; 0.005; 0.01; 0.025; 0.05; 0.1 to 1 by increment of 0.1; 1 to 10 by increment of 0.25; 10 to 30 by increment of 1; and 30 to 100 by increment of 10}; (iii) the estimated τ that maximized model fit [Citation16], based on the restricted maximum likelihood from a linear mixed model (see following section).
Genome-wide association study of DNA methylation (EWAS)
We assessed cross-sectional associations (baseline data) for methylation at each individual CpG by regressing DNA methylation M-values on smoking status using linear mixed-effects regression models, using the function lmer from the R package lme4 [Citation38]. Models were adjusted by fitting fixed effects for baseline values of age (continuous), country of birth (Australia/New-Zealand, Italy, Greece, United Kingdom/Malta), sex, alcohol drinking in the previous week (continuous, in grams/day), BMI (≤25 kg/m2, >25 to ≤30, >30 to ≤35, >35), sample type (peripheral blood mononuclear cells, dried blood spots, buffy coats) and estimated white blood cell composition (percentage of CD4 + T cells, CD8 + T cells, B cells, NK cells, monocytes and granulocytes, estimated using the Houseman algorithm [Citation22,Citation23]), and random effects for study, plate, and chip. Although we did not remove outliers for the methylation or smoking variables from our dataset, we tested the influence of these using robust regression models and results were similar (not shown).
We hypothesized that older participants, women, participants drinking more alcohol, those with lower BMI, as well as those who developed cancer or born outside Australia, could be more sensitive to tobacco smoke, hence tested heterogeneity in the association between smoking and methylation by age (continuous), sex, alcohol intake in the previous week (continuous), BMI (continuous) and future case status was assessed using likelihood ratio tests for interaction. These were performed for differentially methylated CpG sites identified in the EWAS (N = 4,496) and the Bonferroni correction was applied to account for multiple testing (P < 0.05/4,496 = 1.1x10−5).
Sensitivity analyses were conducted to assess the robustness of our findings by: i) fitting the same models without adjustment for BMI, or alcohol consumption, or both, and ii) fitting the same models using the pack-year variable (log-transformed) instead of CSI.
We estimated τ for the 3,327 CpGs previously reported to be associated with smoking. We assumed that the median, and the 25th and 75th percentile of the distribution of τ were the values most likely to detect novel associations between smoking and DNA methylation. We thus ran cross-sectional EWAS analyses for: i) current compared with never smoking, ii) former compared with never smoking; and iii) CSI (continuous variable) with τ = 1.5, τ = 2.75, and τ = 5.25. Given the substantial correlation between these tests, we did not correct further for multiple testing and used a threshold of P < 10−7 to identify associations for any of the three cross-sectional EWAS [Citation39–Citation41]. For all associations with P < 10−7 in our cross-sectional EWAS we estimated the half-life τ that provided the best model fit for the CSI, as described previously. We also calculated a ‘reversibility coefficient’, expressed as a percentage and defined as the regression coefficient comparing ‘former’ to ‘current’ smokers divided by the coefficient comparing ‘never’ to ‘current’ smokers, as done previously [Citation20].
Longitudinal analysis
Linear mixed effects regression models were used to assess the relationship between change in smoking status and change in methylation for individual differentially methylated CpGs in our cross-sectional EWAS (P < 10−7). In a first model, we used the following longitudinal smoking patterns: current (at baseline)-current (at follow-up), current-former, former-former, and never-never. Study was included as a random effect and the following variables were included as fixed effects: sex, country of birth (four categories), baseline age (continuous), baseline alcohol intake (continuous), baseline BMI (continuous), baseline cell composition (as defined previously), change in age, BMI and alcohol intake (all continuous), the difference between baseline and follow-up composition for each cell type (continuous), baseline smoking (expressed using a CSI with τ = 1.5 because it identified the greatest number of associations in the cross-sectional EWAS) and the baseline methylation M-value of the CpG. As adjustment for baseline methylation in analyses of change in methylation may lead to bias in some circumstances [Citation21], we conducted a sensitivity analysis using models without adjustment for baseline M-value. We also carried the analysis not adjusting for baseline smoking status.
All statistical analyses were performed using the statistical software R (version 3.4.4).
Supplemental Material
Download MS Excel (9.2 MB)Supplemental Material
Download MS Word (31.2 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Allione A, Marcon F, Fiorito G, et al. Novel epigenetic changes unveiled by monozygotic twins discordant for smoking habits. PLoS One. 2015;10:e0128265.
- Besingi W, Johansson A. Smoke-related DNA methylation changes in the etiology of human disease. Hum Mol Genet. 2014;23:2290–2297.
- Dogan MV, Shields B, Cutrona C, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151.
- Elliott HR, Tillin T, McArdle WL, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6:4.
- Guida F, Sandanger TM, Castagne R, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24:2349–2359.
- Harlid S, Xu Z, Panduri V, et al. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the Sister Study. Environ Health Perspect. 2014;122:673–678.
- Shenker NS, Polidoro S, van Veldhoven K, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22:843–851.
- Tsaprouni LG, Yang TP, Bell J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9:1382–1396.
- Zeilinger S, Kuhnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8:e63812.
- Wilson R, Wahl S, Pfeiffer L, et al. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics. 2017;18:805.
- Breitling LP, Yang R, Korn B, et al. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–457.
- Joehanes R, Just AC, Marioni RE, et al. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet. 2016;9:436–447.
- Lohoff FW, Sorcher JL, Rosen AD, et al. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol Psychiatry. 2018;23:1–11.
- Gao X, Jia M, Zhang Y, et al. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7:113.
- van der Harst P, de Windt LJ, Chambers JC. Translational perspective on epigenetics in cardiovascular disease. J Am Coll Cardiol. 2017;70:590–606.
- Leffondre K, Abrahamowicz M, Xiao Y, et al. Modelling smoking history using a comprehensive smoking index: application to lung cancer. Stat Med. 2006;25:4132–4146.
- Dietrich T, Hoffmann K. A comprehensive index for the modeling of smoking history in periodontal research. J Dent Res. 2004;83:859–863.
- Hoffmann K, Bergmann MM. Re: “Modeling smoking history: a comparison of different approaches”. Am J Epidemiol. 2003;158:393; author reply 393–394.
- Ambatipudi S, Cuenin C, Hernandez-Vargas H, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618.
- Dugué PA, Wilson R, Lehne B, et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: analysis of cross-sectional and longitudinal data. BioRxiv. 2018.
- Glymour MM, Weuve J, Berkman LF, et al. When is baseline adjustment useful in analyses of change? An example with education and cognitive change. Am J Epidemiol. 2005;162:267–278.
- Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
- McGregor K, Bernatsky S, Colmegna I, et al. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 2016;17:84.
- Cole SR, Platt RW, Schisterman EF, et al. Illustrating bias due to conditioning on a collider. Int J Epidemiol. 2010;39:417–420.
- Milne RL, Fletcher AS, MacInnis RJ, et al. Cohort profile: the melbourne collaborative cohort study (Health 2020). Int J Epidemiol. 2017.
- Baglietto L, Ponzi E, Haycock P, et al. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int J Cancer. 2017;140:50–61.
- Dugué PA, Brinkman MT, Milne RL, et al. Genome-wide measures of DNA methylation in peripheral blood and the risk of urothelial cell carcinoma: a prospective nested case-control study. Br J Cancer. 2016.
- Dugué PA, English DR, MacInnis RJ, et al. Reliability of DNA methylation measures from dried blood spots and mononuclear cells using the HumanMethylation450k BeadArray. Sci Rep. 2016;6:30317.
- FitzGerald LM, Naeem H, Makalic E, et al. Genome-wide measures of peripheral blood dna methylation and prostate cancer risk in a prospective nested case-control study. Prostate. 2017;77:471–478.
- Wong Doo N, Makalic E, Joo JE, et al. Global measures of peripheral blood-derived DNA methylation as a risk factor in the development of mature B-cell neoplasms. Epigenomics. 2016; 8:55-66.
- Dugué PA, Bassett JK, Joo JE, et al. DNA methylation-based biological aging and cancer risk and survival: pooled analysis of seven prospective studies. Int J Cancer. 2018;142:1611–1619.
- Beach SR, Dogan MV, Lei MK, et al. Methylomic aging as a window onto the influence of lifestyle: tobacco and alcohol use alter the rate of biological aging. J Am Geriatr Soc. 2015;63:2519–2525.
- Chen LM, Nergard JC, Ni L, et al. Long-term exposure to cigarette smoke extract induces hypomethylation at the RUNX3 and IGF2-H19 loci in immortalized human urothelial cells. PLoS One. 2013;8:e65513.
- Wan ES, Qiu W, Baccarelli A, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;21:3073–3082.
- Zaghlool SB, Al-Shafai M, Al Muftah WA, et al. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenetics. 2015;7:6.
- Zhang Y, Schottker B, Florath I, et al. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ Health Perspect. 2016;124:67–74.
- Bates D, Mächler M, Bolker B, et al. Fitting linear mixed-effects models using lme4. arXiv preprint arXiv:1406.5823. 2014.
- Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86.
- Geurts YM, Dugue PA, Joo JE, et al. Novel associations between blood DNA methylation and body mass index in middle-aged and older adults. Int J Obes (Lond). 2018;42:887–896.
- Chamberlain JA, Dugue PA, Bassett JK, et al. Dietary intake of one-carbon metabolism nutrients and DNA methylation in peripheral blood. Am J Clin Nutr. 2018;108:611–621.