
ABSTRACT
Alu repeats constitute a major fraction of human genome and for a small subset of them a role in gene regulation has been described. The number of studies focused on the functional characterization of particular Alu elements is very limited. Most Alu elements are DNA methylated and then assumed to lie in repressed chromatin domains. We hypothesize that Alu elements with low or variable DNA methylation are candidates for a functional role. In a genome-wide study in normal and cancer tissues, we pinpointed an Alu repeat (AluSq2) with differential methylation located upstream of the promoter region of the DIEXF gene. DIEXF encodes a highly conserved factor essential for the development of zebrafish digestive tract. To characterize the contribution of the Alu element to the regulation of DIEXF we analysed the epigenetic landscapes of the gene promoter and flanking regions in different cell types and cancers. Alternate epigenetic profiles (DNA methylation and histone modifications) of the AluSq2 element were associated with DIEXF transcript diversity as well as protein levels, while the epigenetic profile of the CpG island associated with the DIEXF promoter remained unchanged. These results suggest that AluSq2 might directly contribute to the regulation of DIEXF transcription and protein expression. Moreover, AluSq2 was DNA hypomethylated in different cancer types, pointing out its putative contribution to DIEXF alteration in cancer and its potential as tumoural biomarker.
Introduction
About half of the human genome is made of transposable elements (TEs), which include short interspersed elements (SINEs), long interspersed elements (LINEs) and long terminal repeat (LTR) elements [Citation1,Citation2]. TEs were originally regarded as ‘junk’ DNA. However, numerous studies have demonstrated that some of them have evolved from parasitic sequences into functional genomic elements [Citation3–Citation6]. Among TEs, Alu elements, which are restricted to primates, constitute the most successful SINE family with more than one million copies per haploid human genome [Citation7]. Interestingly, their distribution throughout the genome is non-random as they tend to accumulate in gene-rich regions [Citation1,Citation2]. In recent years, an increasing body of evidence have pointed out the potential of Alu elements to function as regulators of gene expression [Citation3–Citation6]. The influence of Alu elements in gene expression and chromatin structure can be exerted through different mechanisms, including the presence of transcription factor binding sites (TFBSs) [Citation8] and enhancer-like histone modifications [Citation9]. Moreover, the high density of methylated CpG in Alu elements appears to favour nucleosome occupancy, having a direct impact in the positioning of neighbouring nucleosomes [Citation10,Citation11]. Most Alu elements are highly DNA-methylated [Citation12–Citation14] and associated with repressive histone modifications [Citation15], which probably confines their overall potential contribution to gene regulation.
Alterations in DNA methylation are a hallmark of cancer and include both hyper- and hypomethylations. Whereas hypermethylations are mostly focal and associated with CpG islands, hypomethylation often involves extensive blocks of the genome largely affecting repetitive elements [Citation16–Citation19]. The correlation between global loss of DNA methylation and chromosomal alterations in different cancer types has been well established [Citation20–Citation24]. Although some studies have clearly pointed out DNA hypomethylation as an inductor of genomic instability [Citation20–Citation22], the mechanisms and the local impact of Alu DNA hypomethylation to cancer cell’s phenotype and behaviour have not been elucidated.
The application of AUMA (Amplification of Unmethylated Alu’s) to a series of matched normal and tumour colon samples, identified a large number of hypomethylated Alu elements associated with cancer [Citation25]. One of the hypomethylated Alu elements consisted of an AluSq2 located within the promoter region of digestive organ expansion factor homolog (DIEXF) gene. DIEXF (current HGNC symbol: UTP25) is a highly conserved nucleolar protein component of the small ribosomal subunit and participates in the processing of pre-rRNA [Citation26,Citation27]. DIEXF plays an essential role in the development of the digestive organs in zebrafish [Citation28] and mouse [Citation29] and some of its reported functions include regulation of TP53 expression [Citation28,Citation30,Citation31] and TGF beta signalling [Citation32]. More recently, it has been shown that DIEXF is overexpressed in human neuroblastoma and possibly involved in apoptosis evasion [Citation33]. In summary, although there are compelling results supporting a crucial role of DIEXF in development as well as a possible implication in cancer, the mechanisms of action of this protein and its regulation remain to be elucidated.
Here, we report that an Alu repeat element (AluSq2) located within the promoter region of DIEXF displays a tissue/cell type-specific DNA methylation pattern which is consistent with the transcriptional and protein profiles of DIEXF in different cell types. Furthermore, the results suggest that AluSq2 methylation might play an important role in DIEXF expression by regulating the chromatin landscape and determining the transcription start site (TSS). In addition, we have found recurrent DNA hypomethylation of the AluSq2 element in different cancer types, pointing out its potential contribution to DIEXF deregulation in cancer.
Results
The AluSq2 element within the promoter region of DIEXF gene displays tissue/cell type-dependent DNA methylation
Unmethylated Alu sequences can be identified by application of the AUMA technique [Citation25]. One of the recurrently hypomethylated Alu we found in colorectal cancer (Aj2c1) corresponded to a member of the AluSq2 subfamily and was located upstream of the DIEXF gene promoter. Bisulphite sequencing of the complete AluSq2 element in a subset of colorectal normal and tumour tissues confirmed the differential DNA methylation of the SmaI site reported by AUMA ()), and also revealed a polarized DNA methylation profile, with lower methylation levels in the side proximal to the neighbouring DIEXF gene promoter (p-AluSq2). In colorectal tumours, there was a loss of methylation that reached the SmaI site and in some cases spanned the whole Alu element ()). Interestingly, different normal tissues exhibited alternative boundaries in the DNA methylation profile ()).
Figure 1. Genomic landscape and DNA methylation profile of the DIEXF gene promoter. (a) DNA methylation profile of the AluSq2 element in normal colon and colon cancer samples. Each dot along the line represents an unmethylated (empty), methylated (full black), or partially methylated (grey) CpG dinucleotide (DNA methylation ranges are indicated). Arrowhead indicates the location of the SmaI site reported by AUMA. The Alu side proximal to DIEXF gene (p-AluSq2, coinciding with the free left Alu monomer -FLAM) and the Alu side distal to DIEXF gene (d-AluSq2, coinciding with the free right Alu monomer -FRAM) are indicated. (b) DNA methylation profile of the AluSq2 element in different normal tissues. (c) UCSC genome browser view of the DIEXF gene promoter region. Annotated CpG islands and repetitive elements are indicated. Bisulphite sequencing of the annotated genomic elements in seven normal (embryonic stem cells, induced pluripotent stem cells, fibroblasts and keratinocytes) and colon cancer cell lines. DNA methylation representation was generated using the Methylation plotter application [Citation76].
![Figure 1. Genomic landscape and DNA methylation profile of the DIEXF gene promoter. (a) DNA methylation profile of the AluSq2 element in normal colon and colon cancer samples. Each dot along the line represents an unmethylated (empty), methylated (full black), or partially methylated (grey) CpG dinucleotide (DNA methylation ranges are indicated). Arrowhead indicates the location of the SmaI site reported by AUMA. The Alu side proximal to DIEXF gene (p-AluSq2, coinciding with the free left Alu monomer -FLAM) and the Alu side distal to DIEXF gene (d-AluSq2, coinciding with the free right Alu monomer -FRAM) are indicated. (b) DNA methylation profile of the AluSq2 element in different normal tissues. (c) UCSC genome browser view of the DIEXF gene promoter region. Annotated CpG islands and repetitive elements are indicated. Bisulphite sequencing of the annotated genomic elements in seven normal (embryonic stem cells, induced pluripotent stem cells, fibroblasts and keratinocytes) and colon cancer cell lines. DNA methylation representation was generated using the Methylation plotter application [Citation76].](/cms/asset/1142cb20-6749-4d74-b499-d23457dd85f4/kepi_a_1722398_f0001_oc.jpg)
Next, we analysed the DNA methylation of the AluSq2 element and flanking sequences in a set of cell lines from different tissues ()). Interestingly, in all the samples, the neighbouring region encompassing the CpG island appeared fully unmethylated and this status also embraced the closest downstream AluSx element located in the first intron of the DIEXF gene. The rest of the region was heavily methylated. The DNA methylation of the region was analysed in other normal and cancer tissues using data from ENCODE (Infinium HumanMethylation450 BeadChip) (ENCODE Project Consortium 2012) and the Epigenomics Roadmap (http://www.roadmapepigenomics.org/) (Figure S1), confirming the reported profiles. In summary, the DIEXF promoter is invariably unmethylated, but it displays a tissue and cancer-related upstream boundary located inside the AluSq2 element. Based on the well-stablished role of DNA methylation in gene regulation [Citation34–Citation37], we hypothesize that the mobile DNA methylation boundary observed inside this element in different tissues and cancer cells reflects differential regulation of the associated gene. A direct implication of this association is the putative contribution of the AluSq2 element in the regulation of the DIEXF gene expression.
Association of AluSq2 DNA methylation with DIEXF transcriptional and protein profiles
To test the hypothesis that the variable DNA methylation of the AluSq2 element was associated with DIEXF gene expression we first analysed the DIEXF transcripts, that included three main isoforms according to gene annotation databases ()). Two transcripts comprise the complete coding region including the 12 exons, but differ in the untranslated regions (UTRs), especially the 3ʹUTR that spans either 766 bp or 6118 bp and results in mRNAs of 3.1 kb and 8.5 kb, respectively. The third transcript comprises exons 7–9 and includes a non-overlapping 3ʹUTR of 2149 bp what makes a total length of 2.9 kb ()).
Figure 2. DIEXF gene transcriptional characterization. (a) DIEXF transcripts according to UCSC genes, RefSeq Gene, Ensembl Gene Predictions and Vega Protein-Coding Annotations databases. (b) Genomic location of northern blot probes and RT-qPCR amplicons used to characterize the expression profiles. (c) Northern blot analysis of DIEXF transcripts in SW480 cell line with seven probes covering different DIEXF transcripts. The molecular weight of the three described transcripts is shown on the right. The arrowhead indicates the new isoform. (d) Northern blot analysis of DIEXF expression using probes B (located in the common region of all transcripts) and E (specific of the long 8.5-kb transcript with an extended 3ʹ-UTR) in different cell lines. PPIA (Cyclophilin A) was used as the mRNA loading control. The methylation status of the AluSq2 region proximal to DIEXF (p-AluSq2) for each sample is indicated in a box next to its name using a greyscale (black: full methylation; grey: partially methylated; white: no methylation; see for more details). (e) Relative expression of DIEXF analysed by RT-qPCR in different cell lines using 3 different primer pairs (Exons 8–9 and 12-3ʹUTRa covering different DIEXF transcripts and 3ʹUTRa specific of the long 8.5-kb transcript). The methylation status of p-AluSq2 for each sample is indicated in a box next to its name using a greyscale (black: full methylation; grey: partially methylated; white: no methylation; see for more details). Expression levels were normalized using two reference genes (PPIA and PSMC4) and represented relative to HCT116 cells.
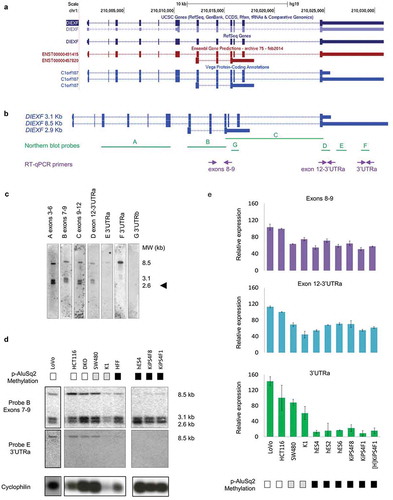
To characterize the DIEXF expression we performed northern blots of SW480 colon cancer cell line with probes reporting different transcripts or combinations of transcripts ()). Results showed no expression of transcript 2.9 kb, as revealed by probe G (3ʹUTRb) ()). Alternatively, an unexpected novel transcript revealed by probes A (exons 3–6), B (exons 7–9), C (exons 9–12) and D (short 3ʹUTR in exon 12) and with a size of about 2.6 kb was found. E and F probes (spanning the long 3ʹUTR) identified the expected transcript 8.5 kb ()).
Interestingly, northern blot of different cell lines revealed an association between the p-AluSq2 methylation and the expression of the 8.5-kb transcript. This is, cells with low DNA methylation of p-AluSq2 expressed high levels of the long transcript (HCT116, DKO, SW480, K1), while those cells with full methylation of the p-AluSq2 showed low or null levels (hES4, KiPS4F8, KiPS4F1, and HFF) ()). Conversely, the shorter transcripts were expressed in all samples independently of p-AluSq2 methylation. These results were confirmed by RT-qPCR using different primer sets ( and Figure S2). Altogether these findings pointed out an association between low DNA methylation of the p-AluSq2 element and expression of the DIEXF 8.5-kb transcript.
Next, we wondered whether differences in the AluSq2 DNA methylation were associated with changes in the DIEXF protein content. Western blot of cell fractions confirmed the nuclear localization of DIEXF ()), as previously described [Citation30]. Interestingly, only colon cancer cell lines expressing the long 8.5-kb transcript and with low or null DNA methylation in the p-AluSq2 showed high levels of DIEXF protein, while normal fibroblasts and human embryonic stem cells with full DNA methylation of the p-AluSq2 did not express DIEXF protein ()).
Figure 3. DIEXF protein and mRNA isoforms expression. (a) DIEXF protein expression measured by western blot from cytoplasmic and nuclear fractions in SW480 and HCT116 cell lines using tubulin as a loading cytoplasmic control and TATA as a loading nuclear control. (b) DIEXF protein expression measured by western blot in total extract of different cell lines using tubulin as a loading control. The p-AluSq2 DNA methylation level of each sample is represented in boxes using a greyscale (black: full methylation; grey: partially methylated; white: no methylation; see for more details). (c) Genomic location of the siRNAs used to silence DIEXF targeting different transcripts (siDIEXF#1, siDIEXF#2 and siDIEXF#3). (d) RT-qPCR of DIEXF gene in HCT116 using different sets of primers, at 24, 48, and 72 h after transfection of siRNAs. Expression levels were normalized using two reference genes (PPIA and PSMC4) and expressed relative to Mock. (e) Western blot of DIEXF protein in HCT116 cells at 24, 48, and 72 h after transfection of siRNAs. HCT116 plus Lipofectamine 2000 (Mock) and HCT116 transfected with a non-targeting siRNA (siCtrl) were used as controls. The band that corresponds to DIEXF protein is indicated with an arrowhead.
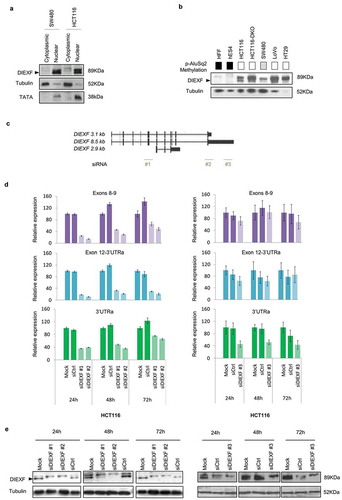
To further determine the contribution of the long 3ʹUTR to the presence of DIEXF protein, we treated HCT116, SW480 and HCT116-DKO cell lines with three different siRNAs targeting either the coding region shared by all transcripts (siDIEXF #1 and #2) and the 3ʹUTR of the long 8.5-kb transcript (siDIEXF #3) ()). All siRNAs induced reduction of both DIEXF mRNA and protein levels (,e) and Figure S3), which confirms the critical involvement of the long 3ʹUTR to produce the protein and supports the hypothesis that the long 8.5-kb transcript is the main one encoding for the protein.
Fine characterization of DIEXF UTRs
To better understand the transcriptional profile of DIEXF we applied the 5ʹ and 3ʹ-rapid amplification of cDNA ends method (5ʹ/3ʹ RACE) to characterize the 5ʹ and 3ʹ UTRs ()) in cell lines with different DNA methylation profiles of the AluSq2 element. Cloning and sequencing of the amplicons associated with the 5ʹ end showed that the new transcript not only shared exons 3 to 12, as revealed by northern blots (), but also exons 1 and 2 (). According to different gene annotation databases (RefSeq, Ensemble, Vega), there were discrepancies regarding the DIEXF TSS. Interestingly, RACE identified dispersed TSSs located within a 100 bp region upstream the ATG codon. The most frequent TSS in cells with full methylation of the p-AluSq2 and not-expressing the 8.5-kb long DIEXF transcript (hES4 and HFF cell lines) was located at −92 bp of the ATG codon ( and Figure S4). Conversely, SW480 and HCT116 cells harbouring low or null methylation of the p-AluSq2 and expressing the 8.5-kb long transcript displayed heterogeneous profiles with a prevalent TSS at −34 bp in SW480 cells, while the HCT116 cell line showed a profile similar to those observed in DNA-methylated cells.
Figure 4. DIEXF 3ʹ and 5ʹ ends characterization. (a) Genomic location of 5ʹ- and 3ʹ-RACE primers. We designed 2 primers to characterize 5ʹ ends (3R and 7R) and 2 other primers to characterize 3ʹ ends (3'UTRaF and 12F). PCR products of 5ʹ-RACE (b) and of 3ʹ-RACE (c) in SW480, HFF, and hES4 cells. Black arrows show the 8.5-kb and 3.1-kb transcripts; red arrows show the novel transcripts; the asterisk indicates the fragment that could not be sequenced.
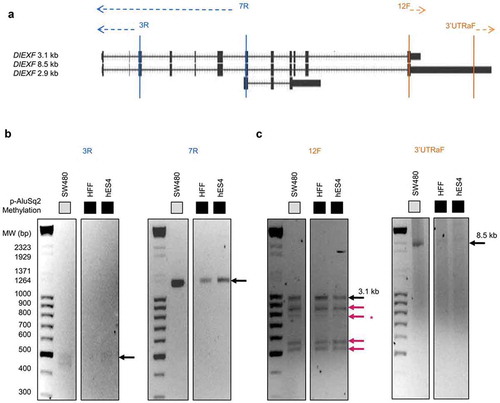
The analysis by RACE of the 3ʹ UTR allowed us to identify new alternative 3ʹends (), red arrows). After cloning and sequencing the associated amplicons (except one because of technical problems), we confirmed the already annotated 3ʹends (corresponding to the canonical 8.5-kb and 3.1-kb transcripts) and found new transcripts with similar molecular weight but containing different shorter 3ʹUTRs, in consistence with the results obtained by northern blot (). The new transcripts were present in all the analysed cell lines independently of the methylation state of p-AluSq2 ()). Altogether, RACE results support the existence of multiple transcripts in consistence with the northern blot analyses and point out the long 8.5-kb transcript as the main isoform with translational capacity.
Chromatin histone modification landscape of the DIEXF promoter
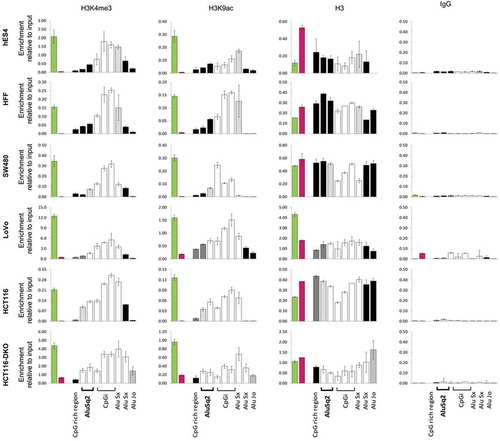
Next, we analysed the chromatin landscape of the DIEXF promoter region including the AluSq2 element in six cell lines with different DNA methylation profiles. We applied chromatin immunoprecipitation (ChIP) to analyse histone modifications associated with open/active chromatin (H3K4me3 and H3K9ac) and closed/repressed chromatin (H3K27me3 and H3K9me3). The region was negative for repressed chromatin histone marks (Figure S5), but an enrichment of the active histone marks was found in all the analysed cell lines (). Interestingly, the extent of the active chromatin domain window was clearly associated with the DNA methylation profile of the AluSq2 element, being wider when AluSq2 was totally unmethylated. It is of note that the spreading of active chromatin (H3K4me3 and H3K9ac as well as DNA hypomethylation) also affected other Alu repeats downstream of the CpG island and located in the first intron of the gene ().
Figure 5. Enrichment in histone modifications associated with active chromatin along the DIEXF promoter region, including Alu repeats, in cell lines with different DNA methylation profile. ChIP assays were performed with antibodies against 3meK4H3, AcK9H3, H3 and non-specific antibody (IgG). Different genomic elements within DIEXF promoter region (see )) were analysed by qPCR. GAPDH (green bar) and 16CEN (red bar) were used as positive and negative control, respectively. Results are reported as enrichment of immunoprecipitated DNA relative to the input. The DNA methylation levels of each region are depicted using a greyscale (black: full methylation; grey: partially methylated; white; no methylation; see for more details).
AluSq2 methylation is recurrently altered in cancer
To assess the extent of AluSq2 hypomethylation in colorectal cancer, we analysed a series of 40 patients by bisulphite sequencing. Classification of samples according to their DNA methylation profile revealed two main clusters (): a first group with a majority of tumours (29 tumours and 6 normal tissues) in which the p-AluSq2 was unmethylated, while the second group, integrated by most normal tissues and 13 tumours, displayed heavier methylation of AluSq2, although the distal-proximal decreasing DNA methylation gradient was maintained. To check whether the altered methylation pattern of AluSq2 was specific of colorectal cancer, we examined a variety of other cancers. Interestingly, AluSq2 hypomethylation also occurred in breast, lung (both squamous and adenocarcinoma), prostate, thyroid tumours with different extents although less frequently than in colorectal cancer (Figure S6).
Figure 6. DNA methylation profile of the AluSq2 element in 40 patients with colorectal cancer. DNA samples were analysed by bisulphite sequencing. The methylation level of each CpG (annotated according to its position in the Alu sequence) is displayed using a greyscale in which black corresponds to full methylation and white to full unmethylation. The order of the CpGs has been maintained according to their position in the genome. Samples have been clustered using the ClustVis tool [Citation81] applying Ward clustering and Euclidian distances. Normal (N, green) and tumour (T, orange).
![Figure 6. DNA methylation profile of the AluSq2 element in 40 patients with colorectal cancer. DNA samples were analysed by bisulphite sequencing. The methylation level of each CpG (annotated according to its position in the Alu sequence) is displayed using a greyscale in which black corresponds to full methylation and white to full unmethylation. The order of the CpGs has been maintained according to their position in the genome. Samples have been clustered using the ClustVis tool [Citation81] applying Ward clustering and Euclidian distances. Normal (N, green) and tumour (T, orange).](/cms/asset/087c9675-c388-4189-81f8-02f15d5c8f58/kepi_a_1722398_f0006_oc.jpg)
Discussion
Most studies analysing DNA methylation of Alu repeats in cancer have performed bulk estimates and used these values as surrogate measurements of the genome’s global DNA methylation [Citation38–Citation47]. The number of studies focused on the identification and functional characterization of particular Alu elements with differential methylation in cancer is very limited [Citation12,Citation25,Citation48], probably due to the intrinsic difficulties in analysing these regions and unambiguously interpreting the results.
It has been widely postulated that Alu elements play a structural and epigenetic role with a direct impact in gene expression regulation [Citation8,Citation12,Citation49–Citation57], including the delimitation of active chromatin boundaries and definition of genome compartments [Citation12,Citation58–Citation60].
Here we have focused our analysis in an Alu displaying a polarized DNA methylation profile with a sliding barrier characteristic of a dynamic chromatin boundary. This profile is not typical among Alu repeats [Citation12,Citation48]. Indeed, most Alu elements show a homogeneous methylation pattern [Citation12,Citation61]. Interestingly the DNA methylation profile along the AluSq2 appeared to be associated with the expression of the neighbouring gene DIEXF ( and ).
After an exhaustive characterization of DIEXF transcriptional profile in different cell models, we identified three novel transcripts in addition to the ones already reported in gene annotation databases, which makes up a total of six isoforms. Although this is not an uncommon event, as most genes are associated with a repertoire of alternative transcripts that can differ either in protein-coding regions and/or UTRs and give rise to different protein isoforms [Citation62,Citation63], the mechanisms underlying the generation of these multiple mRNA variants and the functional benefits of such diversity are still unclear.
Interestingly, all DIEXF transcripts, except one, shared the same exons and they only differed in the 5ʹ and 3ʹ UTRs. The DIEXF gene presents five polyadenylation sites, all of them transcriptionally active. Importantly, only the 8.5-kb transcript harbouring the longest 3ʹUTR appeared to be protein coding. This observation contrasts with previous studies revealing that mRNAs with shorter 3ʹUTRs tend to be more stable and produce more protein as they have less microRNA target sites [Citation64].
Regarding the 5ʹ UTR, multiple TSSs were found, something that is especially common in genes with a promoter CpG island [Citation65–Citation67]. Interestingly, a high proportion (35% to 55%) of the identified TSS in hES4 and HFF cells harbouring methylated AluSq2 and non-expressing the coding transcript (8.5-kb) were located at −92 bp from the ATG, whereas in SW480 and HCT116 cells harbouring low or null methylation of p-AluSq2 and expressing the long coding transcript this figure was lower (3% and 30% of all the clones, respectively).
The mechanism connecting the methylation profile of the AluSq2 element and the choice of TSS remains to be elucidated. The use of these multiple TSSs could be regulated epigenetically, either by DNA methylation, by histone modifications or by chromatin remodelling [Citation68]. The role of epigenetic modifications in gene regulation is well established [Citation69–Citation71]. By analysis of epigenetic and transcriptomic data from the Roadmap Epigenomics (http://www.roadmapepigenomics.org), we observe that the landscape of the DIEXF gene and the associated promoter, including the AluSq2 repeat, shows a tissue-specific pattern (Figure S7(a)). Tissues with high DIEXF expression exhibit a broad promoter state that spans the CpG island and spreads over the neighbouring upstream and downstream Alu repeats (Figure S7(a)). This profile is clearly consistent with DNA methylation profiles of normal and cancer cells (, , S6 and S7B). We hypothesize that the epigenetic status of AluSq2 element could determine the preferential TSS through chromatin remodelling and nucleosome positioning of the promoter region of DIEXF gene. The AluSq2, and especially the proximal side to DIEXF gene (p-AluSq2), is rich in predicted binding sites for multiple transcription factors including Sp1 (involved in development) and SRF (involved in cell cycle, apoptosis, differentiation and growth) (Figure S7(c)). The expansion of the open chromatin would facilitate the binding of transcription factors, playing a role in the usage of different TSSs according to the cell type or the biological context. The usage of TFBSs in Alu repeats to model chromatin states, including insulators, is well known [Citation8,Citation72]. In line with these results, a recent study reported that the differential methylation of an Alu element located in the promoter region of the MIEN1 gene could participate in the regulation of the associated gene, probably through the mediation of USF1/USF2 binding [Citation73]. Further functional studies are being performed to elucidate the specific mechanisms by which AluSq2 might exert these effects.
In colorectal cancer, we observed that about two thirds of the tumours show a sliding of the methylation barrier inwards the AluSq2 element. Our current analysis has been limited to a short series of patients with different cancer types, precluding insights into the clinicopathological correlates of AluSq2 hypomethylation. Future studies in larger series should address the potential utility of this alteration as a biomarker and its association with features of the tumour and disease’s outcome.
In conclusion, Alu and other retroelements are kept in repressed chromatin states in the human genome. We have identified an Alu element upstream of the DIEXF gene CpG island promoter displaying a sliding DNA methylation profile and associated with active chromatin histone modifications. The epigenetic characterization of the whole promoter region, together with the transcriptional and protein profiles of DIEXF and the distribution of predicted TFBSs, points out this specific Alu element as a principal determinant of expression regulation. The DNA methylation profile of this Alu element appears to be altered in colorectal and other cancer types. The significance of this alteration remains to be elucidated.
Materials and methods
Tissues, cell lines, and sample processing
Colorectal tumours and their paired non-adjacent normal colonic mucosa as well as the corresponding whole blood DNA were obtained from the Hospital Universitari de Bellvitge (Barcelona, Spain). Normal and tumoural thyroid tissues were obtained as described [Citation45]. The study protocol was reviewed and approved by the Hospital Germans Trias i Pujol Ethics Committee (EO-11-134/PI-19-092; PI-18-257). Written informed consents were obtained from all participants. Genomic DNA from breast, prostate and lung samples were obtained from the Spanish National DNA Bank (BNADN, Salamanca, Spain). Human colon cancer cell lines were obtained from the American Type Culture Collection (ATCC), except the DNA methyltransferase deficient HCT116-DKO, provided by B. Vogelstein [Citation74]. Cell lines were grown in their corresponding medium, complemented with 10% heat-inactivated FBS, 2mM L-glutamine and 1mM Pyruvate (Gibco) at 37ºC in 5% CO2. Pluripotent cell lines (including human embryonic stem (hES) cells and induced pluripotent stem (iPS) cells), and primary cultures of fibroblasts and keratinocytes were grown as previously described [Citation75]. Cultures were tested and found to be mycoplasma free. Genomic DNA was extracted by standard methods.
Bisulphite sequencing
DNA bisulphite treatment was performed using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer’s instructions. From the transformed DNA, two independent nested PCRs, using primers listed in Supplemental Table 1, were performed and the pooled PCR products were purified (PCR Product JETquick Spin Purification Kit, Genomed GmbH). Purified products were sent for Sanger sequencing (GATC Biotech AG, Germany). The degree of methylation was calculated by comparing the peak height of the cytosine residues with the peak of the thymine residues [C/(C + T)*100] and represented using the Methylation plotter, a web tool for dynamic visualization of DNA methylation data [Citation76]. We considered ranges of DNA methylation, specifically those shown in (0–5%, 6–25%, 27–50%, 51–75%, 76–100%), for each CpG, and calculated the methylation status of the Alu region proximal to DIEXF gene promoter (p-AluSq2; see ) for details) as the mean of the methylation of the 9 CpGs within this region (corresponding to the free left Alu monomer(FLAM) of the AluSq2 [Citation77]).
RNA extraction and reverse transcription – quantitative PCR (RT-qPCR)
Total RNA was isolated using TriPure® Isolation Reagent (Roche Life Science) according to the manufacturer’s protocol, and residual DNA was digested with 1 U of DNAse I (DNA-free™ DNA removal Kit, Ambion, Thermo Fisher Scientific Inc.) per μg of RNA.
First-strand cDNA was synthesized from 500 ng of total RNA using SuperScript® III Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific Inc.). Gene expression was analysed by real-time PCR and quantified by the LightCycler System 480 (Roche Diagnostics) with the LightCycler DNA Master SYBR Green I. The reactions were performed in triplicate. Expression levels were normalized using 2 reference genes and represented relative to HCT116 cells. RT-qPCRs were repeated from 1 to 3 times, depending on the sample. Primers are listed in Supplemental Table 2.
Northern blot
Fifty μg of total RNA per lane were resolved on a denaturing 1.1% agarose gel containing 30% formaldehyde, transferred onto a Hybond-XL nylon membrane (GE Healthcare), and crosslinked at 1200 mJ using a UV crosslinker. Membranes were hybridized with probes random labelled with [γ-32P]-ATP (PerkinElmer) using Ready-to-Go DNA Labelling Beads (dCTP) (GE Healthcare). The radioactive signal was then obtained by autoradiography or using a Typhoon Phosphorimager (GE Healthcare). Northen blots were repeated from 1 to 3 times, depending on the sample. Probes are listed in Supplemental Table 3.
siRNA-mediated DIEXF knockdown
Three small interfering RNAs (siRNAs) were designed using BLOCK-iT™ RNAi Designer web (Invitrogen, Thermo Fisher Scientific) and transfected according to the manufacturer’s instructions using Lipofectamine 2000 (Invitrogen). After transfection, cells were collected and analysed at 24, 48, and 72 h. siDIEXF#1 and siDIEXF#2 are located in the DIEXF ORF region (siRNAs starting at 840 and 2299 bp, respectively), and siDIEXF#3 is located in the DIEXF long 3ʹUTR (siRNA starting at 7118 bp). As control, we used the cells treated with Lipofectamine 2000 (Mock) and the cells transfected with a non-targeting siRNA oligonucleotide (siCtrl) (MISSION siRNA Universal Negative Control #1, Sigma-Aldrich)
(https://www.sigmaaldrich.com/catalog/product/sigma/sic001?lang=es®ion=ES). Experiments were repeated from 1 to 3 times, depending on the cell line. Sequences of siRNAs are listed in Supplemental Table 4.
Western blot
Cytoplasmic extracts were obtained using RIPA buffer [Citation78] and nuclear extracts were performed as described [Citation79]. Samples were resolved by SDS-PAGE and transferred onto PVDF (polyvinylidene fluoride) membranes. The membranes were blocked and incubated with first antibodies: anti-DIEXF (1:250, Ab111508, Abcam), anti-tubulin (1:4000, T5168, Sigma) and anti-TATA binding protein (1:2000, Ab818, Abcam), the last two being used as cytoplasmic and nuclear loading controls, respectively. After washing, the membranes were incubated with the corresponding secondary HRP-antibody (Dako) and visualized using the Lumminata Crescendo Western HRP Substrate (Millipore).
Western blots were repeated from 1 to 3 times, depending on the sample.
Rapid amplification of cDNA ends (RACE)
5′ and 3′ -rapid amplification of cDNA ends (RACE) was carried out using SMARTer RACE cDNA Amplification kit (Clontech) following the manufacturer’s recommendations. The primers used are listed in Supplemental Table 5. PCR products were cloned into pGEM®-T Easy vector (Promega), and used for DNA Sanger sequencing (GATC Biotech AG, Germany).
Chromatin immunoprecipitation
As previously described [Citation80], cells were cross-linked with 0.5% formaldehyde and lysed, and chromatin was sonicated to obtain DNA fragments ranging from 200 to 500 bp (Bioruptor, Diagenode). Sonicated lysates were immunoprecipitated using EZ-Magna ChIP Kit (Millipore) following the manufacturer’s protocol. We used ChIP-grade antibodies (1 to 5 μg) specific for H3K4me3 (17–614, Millipore) and H3K9ac (07–352, Millipore) as active marks, H3K9me3 (pAb-ab8898, Diagenode) and H3K27me3 (07–449, Millipore) as inactive marks, H3 (ab1791, Abcam) as total H3, and normal rabbit serum (011-000-120, Jackson Immunoresearch) as negative control. Equal amounts of immunoprecipitated DNA were used for qPCR and normalized to total chromatin input (6% of the total chromatin). Data are represented as the mean ± SD. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a qPCR positive control for active marks and negative control for H3K9me3, while a centromeric region of chromosome 16 (16CEN) was used as a negative control for active marks and positive control for H3K9me3. Dopamine receptor D1 (DRD1) was used as a qPCR positive control for H3K27me3, while Complexin 2 (CPLX2) was used as a negative control. Experiments were repeated from 1 to 3 times, depending on the cell line. Primers are listed in Supplemental Table 6.
Author contributions
BM, SP, and MJB performed experiments and analyzed data. ADV, IM, JC, MAP, and MJ analyzed data. BM, MAP, and MJ conceived the study, designed the experiments, and wrote the manuscript.
Supplemental Material
Download PDF (921.6 KB)Supplemental Material
Download PDF (1.4 MB)Acknowledgments
We thank Antònia Pastor and Mar Muñoz for technical help.
Disclosure statement
MAP is cofounder and equity holder of Aniling, a biotech company with no interests in this paper. MAP lab has received research funding from Celgene. The rest of the authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001:409(6822):860–921.
- Wicker T, Sabot F, Hua-Van A, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–982.
- Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351(6274):aac7247.
- Goke J, Ng HH. CTRL+INSERT: retrotransposons and their contribution to regulation and innovation of the transcriptome. EMBO Rep. 2016;17(8):1131–1144.
- Chen LL, Yang L. ALUternative regulation for gene expression. Trends Cell Biol. 2017;27(7):480–490.
- Tomilin NV. Regulation of mammalian gene expression by retroelements and non-coding tandem repeats. Bioessays. 2008;30(4):338–348.
- Deininger P. Alu elements: know the SINEs. Genome Biol. 2011;12(12):236.
- Polak P, Domany E. Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics. 2006;7:133.
- Su M, Han D, Boyd-Kirkup J, et al. Evolution of Alu elements toward enhancers. Cell Rep. 2014;7(2):376–385.
- Tanaka Y, Yamashita R, Suzuki Y, et al. Effects of Alu elements on global nucleosome positioning in the human genome. BMC Genomics. 2010;11:309.
- Collings CK, Anderson JN. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenetics Chromatin. 2017;10:18.
- Jorda M, Diez-Villanueva A, Mallona I, et al. The epigenetic landscape of Alu repeats delineates the structural and functional genomic architecture of colon cancer cells. Genome Res. 2017;27(1):118–132.
- Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322.
- Xie H, Wang M, Bonaldo Mde F, et al. High-throughput sequence-based epigenomic analysis of Alu repeats in human cerebellum. Nucleic Acids Res. 2009;37(13):4331–4340.
- Kondo Y, Issa JP. Enrichment for histone H3 lysine 9 methylation at Alu repeats in human cells. J Biol Chem. 2003;278(30):27658–27662.
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153.
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068.
- Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–259.
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298.
- Gaudet F, Graeme JG, Eden A, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300(5618):489–492.
- Eden A, Gaudet F, Waghmare A, et al. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300(5618):455.
- Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005;65(19):8635–8639.
- Daskalos A, Nikolaidis G, Xinarianos G, et al. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer. 2009;124(1):81–87.
- Rodriguez J, Frigola J, Vendrell E, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006;66(17):8462–9468.
- Rodriguez J, Vives L, Jorda M, et al. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res. 2008;36(3):770–784.
- Charette JM, Baserga SJ. The DEAD-box RNA helicase-like Utp25 is an SSU processome component. RNA. 2010;16(11):2156–2169.
- Goldfeder MB, Oliveira CC. Utp25p, a nucleolar Saccharomyces cerevisiae protein, interacts with U3 snoRNP subunits and affects processing of the 35S pre-rRNA. Febs J. 2010;277(13):2838–2852.
- Chen J, Ruan H, Ng SM, et al. Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 2005;19(23):2900–2911.
- Aryal NK, Wasylishen AR, Pant V, et al. Loss of digestive organ expansion factor (Diexf) reveals an essential role during murine embryonic development that is independent of p53. Oncotarget. 2017;8(61):103996–104006.
- Tao T, Shi H, Guan Y, et al. Def defines a conserved nucleolar pathway that leads p53 to proteasome-independent degradation. Cell Res. 2013;23(5):620–634.
- Guan Y, Huang D, Chen F, et al. Phosphorylation of def regulates nucleolar p53 turnover and cell cycle progression through def recruitment of calpain3. PLoS Biol. 2016;14(9):e1002555.
- Zhu Z, Chen J, Xiong JW, et al. Haploinsufficiency of def activates p53-dependent TGFbeta signalling and causes scar formation after partial hepatectomy. PLoS One. 2014;9(5):e96576.
- Tao T, Sondalle SB, Shi H, et al. The pre-rRNA processing factor DEF is rate limiting for the pathogenesis of MYCN-driven neuroblastoma. Oncogene. 2017;36(27):3852–3867.
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254.
- Tuesta LM, Zhang Y. Mechanisms of epigenetic memory and addiction. Embo J. 2014;33(10):1091–1103.
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492.
- Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends Biochem Sci. 2011;36(7):381–387.
- Yang AS, Estecio MR, Doshi K, et al. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38.
- Weisenberger DJ, Campan M, Long TI, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33(21):6823–6836.
- Wu HC, Delgado-Cruzata L, Flom JD, et al. Global methylation profiles in DNA from different blood cell types. Epigenetics. 2011;6(1):76–85.
- Yoshida T, Yamashita S, Takamura-Enya T, et al. Alu and Satalpha hypomethylation in Helicobacter pylori-infected gastric mucosae. Int J Cancer. 2011;128(1):33–39.
- Choi IS, Estecio MR, Nagano Y, et al. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod Pathol. 2007;20(7):802–810.
- Jintaridth P, Mutirangura A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics. 2010;41(2):194–200.
- Klein Hesselink EN, Zafon C, Villalmanzo N, et al. Increased global DNA hypomethylation in distant metastatic and dedifferentiated thyroid cancer. J Clin Endocrinol Metab. 2018;103(2):397–406.
- Buj R, Mallona I, Diez-Villanueva A, et al. Quantification of unmethylated Alu (QUAlu): a tool to assess global hypomethylation in routine clinical samples. Oncotarget. 2016;7(9):10536–10546.
- Xiang S, Liu Z, Zhang B, et al. Methylation status of individual CpG sites within Alu elements in the human genome and Alu hypomethylation in gastric carcinomas. BMC Cancer. 2010;10:44.
- Gilson E, Horard B. Comprehensive DNA methylation profiling of human repetitive DNA elements using an MeDIP-on-RepArray assay. Methods Mol Biol. 2012;859:267–291.
- Xie H, Wang M, Bonaldo MD, et al. Epigenomic analysis of Alu repeats in human ependymomas. Proc Natl Acad Sci U S A. 2010;107(15):6952–6957.
- Hellmann-Blumberg U, Hintz MF, Gatewood JM, et al. Developmental differences in methylation of human Alu repeats. Mol Cell Biol. 1993;13(8):4523–4530.
- Rodriguez J, Vives L, Jordà M, et al. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res. 2007;36(3):770–784.
- Brohede J, Rand KN. Evolutionary evidence suggests that CpG island-associated Alus are frequently unmethylated in human germline. Hum Genet. 2006;119(4):457–458.
- Rand KN, Molloy PL. Sensitive measurement of unmethylated repeat DNA sequences by end-specific PCR. Biotechniques. 2010;49(4):xiii–xvii.
- Luo Y, Lu X, Xie H. Dynamic Alu methylation during normal development, aging, and tumorigenesis. Biomed Res Int. 2014;2014:784706.
- Mollet IG, Ben-Dov C, Felicio-Silva D, et al. Unconstrained mining of transcript data reveals increased alternative splicing complexity in the human transcriptome. Nucleic Acids Res. 2010;38(14):4740–4754.
- Bakshi A, Herke S, Batzer MA, et al. DNA methylation variation of human-specific Alu repeats. Epigenetics. 2016;11(2):163–173.
- Brosius J. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene. 1999;238(1):115–134.
- Bouttier M, Laperriere D, Memari B, et al. Alu repeats as transcriptional regulatory platforms in macrophage responses toM. tuberculosis infection. Nucleic Acids Res. 2016;44(22):10571–10587.
- Lunyak VV, Prefontaine GG, Nunez E, et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science. 2007;317(5835):248–251.
- Wang X, Fan J, Liu D, et al. Spreading of Alu methylation to the promoter of the MLH1 gene in gastrointestinal cancer. PLoS One. 2011;6(10):e25913.
- Rollins RA, Haghighi F, Edwards JR, et al. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16(2):157–163.
- Barrera V, Peinado MA. Evaluation of single CpG sites as proxies of CpG island methylation states at the genome scale. Nucleic Acids Res. 2012;40(22):11490–11498.
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108.
- De Klerk E,T, Hoen PA. Alternative mRNA transcription, processing, and translation: insights from RNA sequencing. Trends Genet. 2015;31(3):128–139.
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–684.
- Frith MC, Mori R, Asai K. A mostly traditional approach improves alignment of bisulfite-converted DNA. Nucleic Acids Res. 2012;40(13):e100.
- Carninci P, Sandelin A, Lenhard B, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38(6):626–635.
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022.
- Davuluri RV, Suzuki Y, Sugano S, et al. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008;24(4):167–177.
- Hatchwell E, Greally JM. The potential role of epigenomic dysregulation in complex human disease. Trends Genet. 2007;23(11):588–595.
- Rach EA, Winter DR, Benjamin AM, et al. Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet. 2011;7(1):e1001274.
- Zhang Z, Ma X, Zhang MQ. Bivalent-like chromatin markers are predictive for transcription start site distribution in human. PLoS ONE. 2012;7(6):e38112.
- Molto E, Fernandez A, Montoliu L. Boundaries in vertebrate genomes: different solutions to adequately insulate gene expression domains. Brief Funct Genomic Proteomic. 2009;8(4):283–296.
- Rajendiran S, Gibbs LD, Van Treuren T, et al. MIEN1 is tightly regulated by SINE Alu methylation in its promoter. Oncotarget. 2016;7(40):65307–65319.
- Rhee I, Bachman KE, Park BH, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416(6880):552–556.
- Barrero MJ, Berdasco M, Paramonov I, et al. DNA hypermethylation in somatic cells correlates with higher reprogramming efficiency. Stem Cells. 2012;30(8):1696–1702.
- Mallona I, Diez-Villanueva A, Peinado MA. Methylation plotter: a web tool for dynamic visualization of DNA methylation data. Source Code Biol Med. 2014;9(1):11.
- Ullu E, Tschudi C. Alu sequences are processed 7SL RNA genes. Nature. 1984;312(5990):171–172.
- Ngoka LCM. Sample prep for proteomics of breast cancer: proteomics and gene ontology reveal dramatic differences in protein solubilization preferences of radioimmunoprecipitation assay and urea lysis buffers. Proteome Sci. 2008;6:30.
- Pasquali L, Gaulton KJ, Rodriguez-Segui SA, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46(2):136–143.
- Forn M, Muñoz M, Tauriello DV, et al. Long range epigenetic silencing is a trans-species mechanism that results in cancer specific deregulation by overriding the chromatin domains of normal cells. Mol Oncol. 2013;7(6):1129–1141.
- Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–570.