
ABSTRACT
The Presenilin1 (PSEN1) gene encodes the catalytic peptide of the γ-secretase complex, a key enzyme that cleaves the amyloid-β protein precursor (AβPP), to generate the amyloid-β (Aβ) peptides, involved in Alzheimer’s Disease (AD). Other substrates of the γ-secretase, such as E-cadherin and Notch1, are involved in neurodevelopment and haematopoiesis. Gene-specific DNA methylation influences PSEN1 expression in AD animal models. Here we evaluated canonical and non-canonical cytosine methylation patterns of the PSEN1 5ʹ-flanking during brain development and AD progression, in DNA extracted from the frontal cortex of AD transgenic mice (TgCRND8) and post-mortem human brain. Mapping CpG and non-CpG methylation revealed different methylation profiles in mice and humans. PSEN1 expression only correlated with DNA methylation in adult female mice. However, in post-mortem human brain, lower methylation, both at CpG and non-CpG sites, correlated closely with higher PSEN1 expression during brain development and in disease progression. PSEN1 methylation in blood DNA was significantly lower in AD patients than in controls. The present study is the first to demonstrate a temporal correlation between dynamic changes in PSEN1 CpG and non-CpG methylation patterns and mRNA expression during neurodevelopment and AD neurodegeneration. These observations were made possible by the use of an improved bisulphite methylation assay employing primers that are not biased towards non-CpG methylation. Our findings deepen the understanding of γ-secretase regulation and support the hypothesis that epigenetic changes can promote the pathophysiology of AD. Moreover, they suggest that PSEN1 DNA methylation in peripheral blood may provide a biomarker for AD.
Introduction
γ-secretase is an aspartyl protease that cleaves its substrates within their membrane-spanning domain. It is a tetrameric complex that includes PSEN1, Aph1 (Anterior pharynx-defective 1), NEP (Neprilysin) and Pen2 (Presenilin enhancer 2). PSEN1 is the active site of the enzyme, which catalyzes an unusual protein-cleavage event known as Regulated Intramembrane Proteolysis (RIP) [Citation1]. Although γ-secretase activity was initially described in the context of the AD, where it promotes the pathogenic cleavage of the Aβ peptide from its precursor AβPP [Citation2,Citation3], it was subsequently shown to cleave other substrates such as Notch1 and E-cadherin [Citation4] during normal embryonic development and neurodevelopment [Citation5–Citation7].
The fact that in addition to PSEN1, several other polymorphic genes that are critical for normal neural development, migration, and axonal extension (e.g., ApoE, Reelin, LRP, p35, CDK5) also convey risk for AD [Citation8], suggests that dysregulation of such genes in later life may cause or contribute to the pathogenesis of AD.
AD manifests as a familial disease with monogenic inheritance and early onset (Early Onset AD: EOAD), or as a sporadic, multifactorial, late-onset disease (Late Onset AD: LOAD). The latter accounts for more than 90% of all cases and its causes are unknown. It is increasingly accepted that epigenetic changes may promote LOAD [Citation9,Citation10], perhaps in response to environmental triggers [Citation11–Citation13]. Genomic and sequence-specific epigenetic changes have been extensively studied in animal models of EOAD, and to a lesser extent in human subjects with LOAD [Citation14]. The most commonly studied epigenetic modification in AD is DNA methylation. DNA methylation consists primarily of the enzymatic addition of methyl (-CH3) groups to the cytosine moieties, converting them to 5-methylcytosine (5mCs). DNA methylation of regulatory regions is generally responsible for the down-regulation or silencing of the associated genes [Citation15].
Our previous studies have helped elucidate how DNA methylation can regulate PSEN1 expression in animal and cellular models of AD [Citation16–Citation20]. For example, we showed that environmental conditions that impair one-carbon and methylation metabolism (namely, dietary deficiency of the vitamins B12, B6, and Folate) can result in hypomethylation of the PSEN1 promoter in TgCRND8 transgenic AD-model mice, with consequent worsening of its AD-like features [Citation17]. Conversely, supplementing the mice with the physiological methyl-donor S-adenosylmethionine (SAM) prevented PSEN1 hypomethylation and overexpression, thereby limiting Aβ accumulation in the brain and improving cognitive outcomes [Citation19]. Other studies confirmed the sensitivity of PSEN1 methylation to altered one-carbon metabolism in different AD models, underscoring the cross-talk between AβPP metabolism and DNA methylation [Citation21,Citation22]. In contrast to the animal studies, the few human studies of PSEN1 methylation in AD patients have been inconsistent. Whereas some evidence indicates that PSEN1 loci are hypomethylated in post-mortem brain of AD patients [Citation23] as with our findings in mice, other human studies have failed to confirm this in both post-mortem brain [Citation24] and in peripheral tissue [Citation25,Citation26].
An important limitation of earlier human studies is that they only assessed CpG methylation; however, methylation also occurs at cytosine moieties that are not followed by a guanosine (non-CpG methylation). We recently demonstrated that non-CpG methylation also correlates with gene expression [Citation27] including in the 5ʹ- region flanking the human PSEN1 gene [Citation28]. We interpret these contrasting results as reflecting a common technical bias resulting from the use of ‘methprimers’ or ‘methylation specific primers’ for the PCR amplification of bisulphite-treated DNA. Because these conventional primers are insensitive to non-CpG methylation, they cannot reveal epigenetic regulation by non-CpG sequences [Citation28,Citation29]. Consistent with this interpretation, several recent studies have shown that non-CpG methylation can regulate mRNA expression at gene-specific level [Citation30–Citation32] including in brain [Citation33,Citation34]. Moreover, we reported differential CpG and non-CpG methylation in mitochondrial DNA in human brain tissue from middle-aged (MA) healthy controls and AD patients [Citation35].
In light of the above, the present study was motivated by the need to characterize the patterns of PSEN1 DNA methylation and mRNA expression in murine and human brain at different stages of neurodevelopment and neurodegeneration. To this end, we analysed and compared CpG and non-CpG DNA methylation patterns in frontal brain cortex from transgenic TgCRND8 AD-model and control mice at different ages and stages of development, and post-mortem frontal cortex from foetal, adolescent, and middle-aged humans and from old AD patients and cognitively normal controls. A different cohort of AD patients and matched healthy controls was also studied to profile PSEN1 methylation in peripheral blood. This analysis aimed primarily to describe the methylation pattern of a large portion of the PSEN1 5ʹ-flanking region, in order to provide evidence of possible differentially methylated cytosines in the course of neurodevelopment and neurodegeneration of AD type. In order to achieve this result, we took advantage of the bisulphite assay performed using the previously described unbiased PCR step followed by cloning and Sanger sequencing [Citation28]. Disclosing the methylation status of specific cytosines in the 5ʹ-flanking of the gene, should permit more quantitative measurement of methylation in these specific cytosines in the future. In turn, this will allow the use of PSEN1 methylation as a possible biomarker in AD.
Results
PSEN1 methylation analyses
To evaluate the methylation pattern of the PSEN1 5ʹ-flanking region at a single cytosine level, we applied the bisulphite modification followed by cloning of the PCR products and Sanger sequencing. Taking advantage of PCR primers that are unbiased versus non-CpG methylation makes it possible to detect both CpG and non-CpG methylation status [Citation28]. The technique is described in detail in the Materials and Methods section. PSEN1 methylation pattern has been assessed on DNA isolated from prefrontal cortex of mice (AD TgCRND8 and WT 129Sv) and humans, during the course of neurodevelopment and neurodegeneration. The experimental layout is depicted in .
Figure 1. Experimental layout.
All the following results related to PSEN1 promoter methylation in mouse and human brain were neatly discriminated by Principal Component Analysis (PCA, see ‘Materials and Methods’ section) with no superposition between different groups. Three relevant factors (components) were extracted by PCA pointing to three main independent variation sources: overall methylation; genotype in mice or age/AD in humans; sex. The cytosines with a score > |2| for discriminant components are considered relevant for the interpretation as they correspond to scores outside the 95% confidence interval.
PSEN1 methylation in embryonic mouse brain
To assess PSEN1 methylation pattern at neurodevelopment age in mice, we measured the methylation percent of CpG and non-CpG (CpA, CpT, CpC) cytosine moieties in the 5ʹ-flanking region (from cytosine 1212–1869) of the PSEN1 gene in cortical brain tissue from both wild-type 129Sv (Wt) and TgCRND8 (Tg) embryos (,), respectively). The methylation profile was largely coherent between the two conditions giving rise to a first principal component PC1 explaining 93% of total variability. This factor is a ‘size component’ (all positive near to unity correlation with all sites) and thus can be interpreted as an ‘overall methylation’ score. Figure S1 (supplementary material) reports the first two component loadings (correlation coefficients between components and methylation profiles) and demonstrates the distinct separation of the classes.
Figure 2. PSEN1 promoter CpG and non-CpG methylation in mice brain.
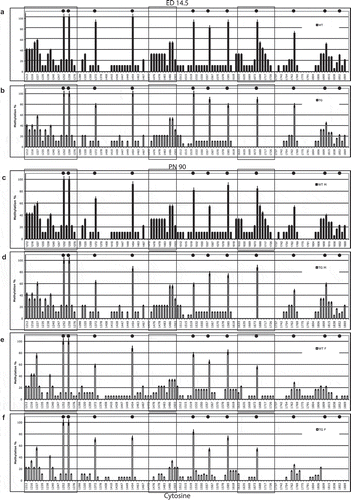
The eleven CpG sites present in this region are markedly methylated (>60% of methylation) except for the two proximal sites (30–50% of methylation). Focusing selectively on the CpG methylation profile, this is consistent with the profile previously observed in adult mice that were subjected to a dietary intervention that impaired methylation metabolism [Citation16,Citation18]. Non-CpG methylation ranges from 10% to 60% at several sites, particularly in the distal part of the analysed region. Two areas of methylated non-CpG cytosines are located at bases 1212–1276 and 1474–1503. A third area (1620–1726) is methylated in Wt but hypomethylated in Tg mice. Specifically, PC2 (genotype) is responsible for significant (p < 0.05) hypomethylation at cytosines 1620,1633,1634,1688,1605,1717.
Sex-specific PSEN1 methylation in adult mouse brain
We analysed PSEN1 5ʹ-flanking DNA methylation separately in males and females 129Sv and TgCRND8 mice at PN90, in order to discover possible differences between sexes. At PN90 TgCRND8 mice already display amyloid plaque spreading and cognitive decline [Citation16,Citation19], which represents the AD-like neurodegeneration that is absent in the WT mice.
As in embryos, PC1 (overall methylation) explains 88.6% of variability. Because genotype appeared to have a different effect on males and females, we applied a four component PCA which revealed a significant genotype*sex effect in females. No overall significant differences were observed between Wt and Tg males (,) respectively) or between Wt and Tg females (,), respectively). Figure S2 reports the third and fourth component loadings that allow separation into classes based on genotype (Fig. S2A) and sex (Fig. S2B).
Interestingly, significant hypomethylation of one of the non-CpG areas in the proximal region (cytosines 1618–1742, specifically at cytosines 1618, 1620, 1633, and 1634, p < 0.05) was observed in Tg mice and in Wt and Tg females. Two distal cytosines (1217, 1227) also show hypomethylation. Moreover, hypomethylation (20% vs. 0%) was evident for the 4 proximal CpG sites (,), cytosines: 1674, 1764, 1813, 1852, and for the non-CpG sites 1688, 1705, and 1717) (p < 0.05), as previously observed in conditions of altered methylation potential [Citation19,Citation20], and for overall non-CpG sites. We found no significant difference in DNA methylation at PN30 by either sex or genotype (data not shown). The differentially methylated region studied here putative binding sites for several transcription factors according to an analysis using the CONSITE database (http://consite.genereg.net/cgi-bin/consite). Interestingly, the location of three putative Snail1 binding sites in this proximal region is reminiscent of a similar finding of Snail1 sites that are situated within differentially methylated non-CpG cytosine clusters in the mouse myogenin gene [Citation27].
Differential methylation patterns predict PSEN1 expression in mice
After observing differentially methylated CpG and non-CpG sites in the PSEN1 5ʹ-flanking in mice cortex, we then analysed the gene expression by Real-Time PCR to assess whether it was correlated to the methylation status. As predicted, PSEN1 mRNA expression was comparable in Wt and TgCRND8 embryonic brain ()), given the consistency of CpG and non-CpG methylation in the 5ʹ-flanking region for both genotypes.
Figure 3. PSEN1 mRNA Expression in mice brain.
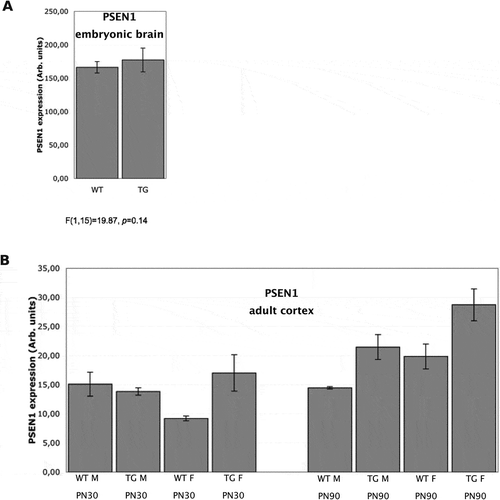
In adult mice, PSEN1 expression was evaluated in the cortex at 30 and 90 days ()). At PN30, PSEN1 expression was significantly higher in TgCRND8 females than in Wt females (p < 0.01). At PN90, PSEN1 expression was higher in TgCRND8 mice than in any mice at PN30 regardless of sex (p < 0.01), whereas PSEN1 expression in Wt mice at PN90 did not differ from PN30. Interestingly, a sex X age effect was observed at PN90, as female mice showed higher expression than males of the same genotype (p < 0.01). A significant inverse correlation was observed between the extent of DNA methylation and gene expression (r = −0.7953, p < 0.05) using Spearman’s rank correlation analysis. This inverse correlation is even more significant (r = −0.8841, p < 0.01) when analysing only the ‘proximal region’ at cytosines 1618–1726.
PSEN1 immunohistochemistry in mice
Finally, we assessed the presence and distribution of PSEN1 protein in mice brain by immunohistochemistry. As expected, PSEN1 brain immunostaining for protein expression was similar in Wt (129Sv) and TgCRND8 embryos (Fig. S3). However, contrary to our expectation, qualitative immunohistochemistry revealed only limited qualitative differences in the overall pattern of PSEN1 protein expression in adult mice (PN30, PN90) (Fig. S4A). Nevertheless, subtle sub-regional differences (Table S1) were evident in the six cortical layers (Fig. S4B) and in various cortical sub-fields (Fig. S4C). For instance, PSEN1 expression was noticeably lower throughout the six layers of the somatosensory cortex in PN30 TgCRND8 male mice as compared to age-matched Wt controls, whereas both Wt and TgCRND8 PN30 female mice show similar reductions of PSEN1 staining in layer IV and VI (Fig. S4B). In PN90 mice, a general reduction in neuronal PSEN1 expression, was particularly pronounced in female TgCRND8 mice (Fig. S4B). However, qualitative staining for PSEN1 expression in the entorhinal cortex showed little variability by age, sex, and genotype (Fig. S4C), and did not decline with age, as can be seen by the large number of PSEN1-positive neurons detected in PN90 TgCRND8 female mice (Fig. S4C).
PSEN1 methylation and mRNA expression in blood in adult mice
In order to investigate possible parallel regulation of PSEN1 in brain and blood, we measured PSEN1 5ʹ-flanking methylation and gene expression in whole blood taken from the same mice that were used for brain isolation. However, CpG and non-CpG DNA methylation and mRNA expression of PSEN1 did not differ in the blood of adult (PN90) mice, regardless of sex and genotype (data not shown).
PSEN1 methylation in human brain
To study PSEN1 methylation and expression in the human brain, we follow a similar experimental approach to that which we used in mice, examining postmortem brain tissue from embryos, young individuals, and old individuals, with and without AD. DNA methylation analysis of the 5ʹ-flanking region of human PSEN1 in cortex from post-mortem foetal brain (–) showed high CpG and low non-CpG methylation, that did not differ significantly at the three time points studied (p > 0.05): GW 14 ()), GW22 ()), GW 40 ()). The three ‘GW’ samples show higher overall methylation, with PC1 accounting for 77% of variance, with respect to the older groups. PC2 also separates the child vs. young samples (accounting for 12% of the variance) and PC3 embryos vs. young (accounting for 10% of the variance).
Figure 4. PSEN1 promoter CpG and non-CpG methylation in Human Brain during neurodevelopment.
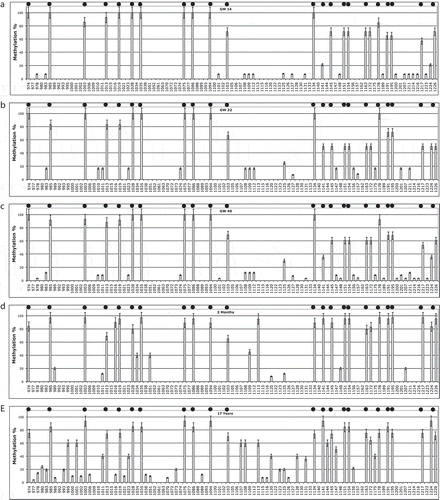
At 2 months and 17 years of age (,), respectively) CpG methylation remains high and non-CpG methylation remains low, although with the 2 month subjects more non-CpG sites are methylated and show intermediate values. Significantly higher methylation (cytosines 995, 1001, 1107, 1113, 1141, 1144, 1224) is evident (PC2, p < 0.05) at 17 years of age. We can, therefore, observe a specific ageing-associated drift leading to hypermethylation in the PSEN1 5ʹ-flanking proximal region, from foetal age to adolescence.
The embryonic and young brains represent different stages of neurodevelopment, in contrast with the brains from old adults with and without AD. Figure S5 reports the first three component loadings (correlation coefficients between components and methylation profiles). These demonstrates the clear separation of the classes along PC2 (Fig. S5A), while PC1 has very elevated loading shared by all the groups; PC3 separates the very young (2 months) from the 17 years old (Fig. S5B and C).
PSEN1 DNA methylation profile clearly changes in post-mortem cortex from old subjects, as shown in . In post-mortem cortex of MA controls, both CpG and non-CpG methylation are diffusely present at 30% of average value ()), a finding comparable to the pattern observed at 17 years. Looking at the DNA methylation profile of PSEN1 5ʹ-flanking in AD subjects, it is possible to observe very significant general hypomethylation both at CpG and non-CpG sites (PC1, p < 0.001) both in patients at Braak stages I–II/A and V–VI/C vs. controls (,), respectively). The PCA did not show sex*health status differences, arguing against differential methylation between males and females in this study. Figure S6A reports the first two component loadings (correlation coefficients between components and methylation profiles). It shows the clear separation of the classes along PC2, in contrast to PC1 that has very elevated loading shared by all the profiles. AD I–II and V–II show similar profiles due to the very low methylation of many cytosines. Specific cytosine moieties demethylated in AD are the 974, 977, 995, 1001, 1011, 1107, 1108, 1113, 1120, 1144 (p < 0.001)
Figure 5. PSEN1 promoter CpG and non-CpG methylation in Human Brain during ageing and AD.
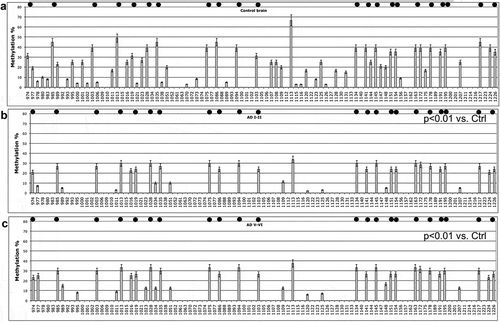
DNA digestion with methylation-sensitive endonucleases was performed as a control on random samples as previously described [Citation27,Citation28] and confirmed the observed epigenetic drift (not shown).
CpG and non-CpG methylation predict PSEN1 mRNA expression in human brain
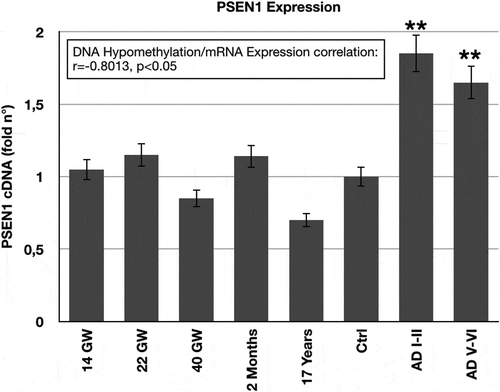
As in mice, we analysed the correlation of the methylation status to gene expression in human brain tissue. PSEN1mRNA expression, assessed by Real-Time PCR, corresponds to the DNA methylation profile of the 5ʹ-flanking region. shows that the only significant variation is evident as PSEN1 up-regulation (p < 0.01) in the post-mortem cortex of LOAD patients (both at Braak stages I–II/A and V–VI/C). A significant inverse correlation was observed between the extent of DNA methylation and gene expression (r = −0.8013, p < 0.05) by Spearman’s rank correlation analysis.
Figure 6. PSEN1 mRNA Expression in Human Brain.
PSEN1 methylation in human blood
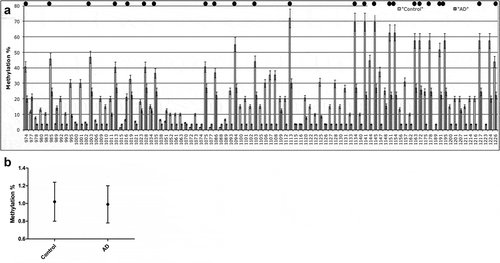
Since we could not obtain blood samples from the same subjects used for brain samples, CpG and non-CpG methylation of PSEN1 5ʹ-flanking was measured in whole blood DNA in a separate cohort of LOAD patients and healthy-matched controls. PCA of the methylation profile after bisulphite analysis using MIPs ()) showed that PSEN1 is significantly and generally hypomethylated at both CpG and non-CpG moieties in LOAD patients versus controls, although the difference is not as large as in brain samples. Figure S6B shows the first two component loadings. In brain, the distinct neat separation of the classes along PC2 is clearly seen.
Figure 7. PSEN1 promoter CpG and non-CpG methylation in Human peripheral Blood during ageing and AD.
In contrast, MS-HRM analysis performed in the same DNA samples using primers designed with the ‘methprimer’ software [Citation25] showed very low average PSEN1 methylation levels (approximately 1% in average) for both LOAD and control samples, and no difference between groups ()). The large difference in methylation detected by the two techniques (~30% methylation vs. ~1%) is largely dependent on the primers used as previously discussed [Citation28] and on the non-CpG methylation.
Discussion
Environment-induced epigenetic alterations can influence brain function at both gene-specific and genomic levels. Because it is known to be regulated by methylation, it is involved in neurodevelopment and neurodegeneration and it is central to the pathophysiology of AD, PSEN1 provides an important example of how gene-specific epigenetic regulation can influence neurodegeneration. Moreover, the PSEN1 promoter includes several methylated cytosine moieties that are not organized in canonical CpG islands. This architecture could make PSEN1 potentially more prone to dynamic, environmentally induced changes in its methylation pattern, as compared to promoters that are characterized by CpG island-associated cytosines that usually undergo stable methylation-dependent silencing [Citation13].
Despite evidence from animal models and cell studies that epigenetic regulation of this gene is associated with AD-like pathology [Citation16–Citation20,Citation36], only a handful of studies have investigated human PSEN1 methylation in AD [Citation23,Citation24,Citation35]. Given the role of the γ-secretase complex (in which PSEN1 protein represents the catalytic site) in both neurodevelopment and AD type neurodegeneration [Citation6,Citation8], we aimed to describe the dynamics of CpG and non-CpG DNA methylation of the PSEN1 promoter in AD transgenic mice and in humans. A key finding of our work is that cortical PSEN1 expression appears stable during human embryonic development (at least during the periods that we could analyse) but it is downregulated by 17 years of age and then upregulated in older adults and in AD patients. The methylation profile of the 5ʹ-flanking region of the PSEN1 gene corresponds to its expression; we observed increased methylation at 17 years and progressive hypomethylation in elders and in AD patients at Braak stages I–II/A and V–VI/C.
A recent study demonstrated that cortical PSEN1 expression in mice is tightly regulated by histone modification at different stages of neurodevelopment [Citation37]. Because we wanted to chart the implications of PSEN1 methylation in humans, we limited the comparative analysis in embryonic mouse cortex to one time point (ED14.5). Since it has not been previously studied in mice [Citation37], we cannot compare it to data on histone modification. It would be of interest, in the future, to compare histone modifications and DNA methylation at the same developmental stages in mice. Our data show that PSEN1 mRNA expression and methylation patterns are similar in Tg and Wt mice at ED14.5. PSEN1 expression increases in adult TgCRND8 mice (PN90) when the deposition of AD-like amyloid pathology is already apparent in this strain, in contrast to embryos and young mice (PN30) and to age-matched Wt littermates that do not yet display the pathology. Despite these differences, we did not find an effect of either age or genotype in the statistical analyses correlating DNA methylation status with gene expression in mice. The significant hypomethylation found at cytosines 1618–1742 in a region proximal to the transcription start site suggests that this non-CpG region becomes hypomethylated earlier than the proximal CpG sites, but that this event is not sufficient to induce gene expression. This result indicates that PSEN1 upregulation in TgCRND8 adult mice (unlike in Wt mice) is induced directly by the overexpression of the mutated AβPP transgene rather than by epigenetic control. Thus, the lack of correlation between DNA methylation and gene expression in this mouse model is not surprising. On the other hand, when looking for possible sex differences, we found female Tg and Wt mice express PSEN1 more highly than males at PN90, and this overexpression was associated with DNA cytosine hypomethylation in the proximal region. Such sex-specific epigenetic regulation would be extremely interesting and could help accounting for the higher prevalence of AD among women than among men [Citation38]. The fact that the statistical analysis (PCA) did not result in any statistically significant sex-specific differences in human samples, either in DNA methylation or in gene expression, is most likely due to the relatively small sample size and is worthy of further investigation.
Immunohistochemical analyses detected only slight differences in overall PSEN1 expression in female versus male mice, although sub-regional differences (i.e., different expressions within the cortex layers and in sub-fields) suggest the existence of specific cell-population differences in the expression of neuronal PSEN1, an issue that deserves further investigation.
A second key finding is that non-CpG methylation is prominent both in mouse and human brain and that not only CpG but non-CpG methylation is associated with gene expression. We previously demonstrated that dynamic non-CpG methylation is temporally correlated with the expression of the myogenin gene during muscle differentiation in cultured mouse myoblasts and in muscle in vivo [Citation27]. In two very recent studies, we demonstrated the involvement of non-CpG methylation in regulating of IL1β gene in the brain of patients with Tuberous Sclerosis Complex (TSC) [Citation39], and of IL1β and IL6 using tissue from the same MA and AD subjects analysed in the present work [Citation40]. We believe that, together with the data presented here, these are the first demonstrations linking DNA methylation at non-CpG promoter loci to the expression of their corresponding genes, in human brain.
These observations were made possible by our seminal demonstration of a bias in the PCR step following the bisulphite modification in conventional DNA methylation studies and our development of unbiased PCR primers (non-CpG Methylation Insensitive Primers, MIPs) that reveal true non-CpG methylation [Citation28]. As we described in detail in those studies, MIPs are designed to account for the possibility that non-CpG cytosines might be methylated and therefore protected from bisulphite conversion. In other words, they are designed to anneal with regions with no cytosines. If this is not possible, to account for the presence of few (1 to 3) non-CpG cytosines, the MIPs are designed as ‘degenerated’ primers containing a mixture of C/A or G/T at the non-CpG sites. In contrast, conventional primers used in bisulphite-modified DNA amplification are designed with the idea that non-CpG cytosines are unmethylated and therefore will necessarily be converted by bisulphite reaction. Thus, such primers will fail to anneal if some cytosines in the target sequence are methylated, which introduces a bias against the amplification and detection of sequences containing methylated non-CpG. The discrepant methods can explain the inconsistency between the present study and previous findings by a member of our group where PSEN1 methylation was analysed in the brain of patients affected by AD and other neurodegenerative dementias using biased PCR primers [Citation24].
The presence and role of non-CpG methylation, which was previously considered to be limited to stem cells or embryonic cells, is supported by a growing body of evidence demonstrating this epigenetic modification at both gene-specific and genomic levels [Citation30–Citation33,Citation39–Citation41]. The fact that non-CpG methylation is mainly generated by the de novo methylase activity of DNMT3, which does not require cell replication, and which is expressed in brain, is compatible with changeable methylation in the relatively quiescent neural tissue. Although unlike neurons, glial cells are actively proliferating, our previous work indicated that these cells are less involved in the methylation-dependent regulation of PSEN1 gene expression [Citation42].
Although we validated the use of the MIPs in a technical paper [Citation28] before applying them in several different studies [Citation27,Citation39,Citation40], a caveat remains regarding the possibility that incompletely converted DNA could be amplified by degenerated primers, resulting in a false positive signal for methylated cytosines. We therefore confirmed that our bisulphite assay is effective in efficiently converting DNA by comparing it with other bisulphite based assays (see Materials and Methods and reference [Citation28]). Moreover, we digested genomic DNA with non-CpG methylation-sensitive restriction endonucleases to confirm non-CpG methylation by an independent technique. Finally, one must consider, on theoretical grounds, that if degenerated primers were to amplify non-bisulphite modified DNA, then one should obtain the same ‘flat’ results both on hypermethylated and hypomethylated regions inside the same sequence and on both hypermethylated and hypomethylated samples, resulting in the lack of methylation differences. In contrast, the presence of methylated and unmethylated contiguous cytosines, as we observed, lends confidence that the samples are fully converted.
A limitation of the comparing mice and humans is that mouse stages do not correspond precisely to those of human ageing. All the AD transgenic mouse models, including the TgCRND8, used here, model the familial, genetic, early-onset AD, where the overexpression of amyloid does not require environmental triggers. In contrast, the human AD samples are from subjects with sporadic late-onset AD. This is the most prevalent form of the disease, accounting for high socio-economic impact, and it is likely to be sensitive to environmental, epigenetic triggers [Citation38]. However, the genetic and environmental causes of AD may not be exclusive and they can interact. We previously demonstrated that when a modifiable risk factor for AD such as dietary methylation deficiency and hyperhomocysteinemia, is experimentally superimposed on the genetic model, the resulting DNA hypomethylation can induce PSEN1 overexpression [Citation20]. Thus, despite their limitations, the mouse results reported here are valuable as the first temporal description of epigenetic PSEN1 regulation during neurodevelopment, normal ageing and progressive EOAD-like neurodegeneration.
The observed PSEN1 promoter hypomethylation in the blood samples from healthy and AD subjects of the second cohort opens the door to developing novel epigenetic biomarkers for AD. It is likely that members of our group previously failed to detect significant differences in blood PSEN1 methylation between AD cases and controls with MS-HRM method [Citation25,Citation26], because those studies used what we have now identified as biased PCR primers that are insensitive to non-CpG methylation. Similarly, other studies have failed to detect differences between brain and lymphocyte DNA methylation when the analysis was limited to CpG sites [Citation43]. To show that primers that are insensitive to non-CpG methylation can bias the detection of PSEN1 DNA methylation, we also amplified all the blood DNA samples with ‘methprimers’ using the MS-HRM technique, following the same protocol that members of our group had used in the past [Citation25]. This protocol failed to detect differences in PSEN1 methylation between AD patients and controls, indicating that failing to account for non-CpG methylation biases the results. However, the unbiased MIPs in the present study allowed us to detect differential methylation of both CpG and non-CpG in the studied PSEN1 promoter region [Citation28]. While blood and brain were obtained from different subjects, in this study our preliminary findings of consistent patterns of differential methylation between cases and controls in both brain and blood DNA argue for further study in a larger cohort using DNA from blood and post-mortem brain tissue obtained from the same individuals to validate this potential biomarker.
The observation that several regions of the analysed PSEN1 promoter display differential methylation is consistent with recent findings that variably methylated probes (VMPs) associated with AD did not overlap with differentially methylated probes, and highlights the relevance of methylation variability associated with AD pathology [Citation44]. Although other papers have demonstrated differential methylation of PSEN1 in old adults with and without dementia [Citation23], some recent genome-wide analysis failed to evidence linking differentially methylated loci to PSEN1 [Citation45,Citation46]. On the one hand, such discrepancies underscore the need for additional unbiased studies, since current genome-wide approaches typically analyse only a few CpG sites per gene. The methylation profiles we obtained through the unbiased analysis of both CpG and non-CpG sites, show clear evidence that not all the cytosines are methylated and not all are modulated in AD patients versus controls.
In agreement with the DNA methylation patterns, our data also demonstrate that PSEN1 mRNA upregulation in the cortex of AD patients is statistically correlated to promoter hypomethylation, although whether PSEN1 is always upregulated in LOAD remains to be clarified. These data suggest that PSEN1 might be physiologically partially methylated and repressed after development, and should remain in this state during adult life and ageing, whereas, the hypomethylation that is associated with elevated gene expression observed in AD reflects a pathological process. Our novel finding in this small study population cannot rule out the possibility that PSEN1 upregulation is limited to some sub-group of AD patients and requires confirmation in larger cohorts of well-characterized patients and controls. It would be interesting if future methylation and expression analyses on PSEN1 gene in human subjects could also examine the relation of CpG and non-CpG DNA methylation to biomarkers and mediators of methylation metabolism such as SAM, S-adenosylhomocysteine, homocysteine, methionine, vitamin B12, folate and choline. In this regard, a previous investigation of a cohort of 200 individuals revealed that PSEN1 CpG methylation in blood DNA correlates with circulating folate and homocysteine levels [Citation47]. It would be interesting to determine the diagnostic value of combining circulating concentrations of the methylation metabolites, with blood PSEN1 methylation and PSEN1 expression in a multidimensional index of AD and its progression. It is worth noting that developing PSEN1 methylation as a possible biomarker might benefit from more quantitative techniques (i.e., pyrosequencing) [Citation48]. Unfortunately, the current state-of-the-art of pyrosequencing typically requires the use of sequencing primers internal to the investigated DNA sequence, which are designed with proprietary pyrosequencing software. This hinders the design of unbiased sequencing primers with respect to the possible modification of the non-CpG sites, and restricts efforts to assess non-CpG methylation. Until such time that new unbiased pyrosequencing assays are validated and become widely available, cloning, and Sanger sequencing will remain the preferred method for assessing sequence-specific non-CpG methylation. We attempted, in the present study, to check whether the MIPs we used to amplify bisulphite-modified DNA might also work in pyrosequencing reactions, in order to confirm the methylation values observed through the cloning and Sanger sequencing. As described in the Materials and Methods section, these primers, unsurprisingly, were very inefficient, in pyrosequencing, probably due to their length and because their target sequence is situated at the extremity of the PCR fragment, whereas the primers normally used for pyrosequencing are internal. Nevertheless, we succeeded in sequencing a short 20bp-stretch at the 3ʹ of the region, which confirmed the reliability of the methylation data obtained by cloning and Sanger sequencing (Fig. S7). Therefore, an important contribution of the present study is that it accurately describes the site-specific methylation pattern (i.e., at the level of single cytosines) of a large (450 bp) portion of the proximal PSEN1 5ʹ-flanking region. This mapping allowed us to identify specific cytosines that show differential methylation in AD patients vs. controls. The logical continuation of this work would be to reproduce and expand our findings in larger cohorts, using more precisely quantitative assays (i.e., bisulphite modification + pyrosequencing) to develop diagnostically and prognostically useful biomarkers on the basis of differential methylation in PSEN1 and other genes.
The findings in human foetal brain must be interpreted with caution because of the small number of cases analysed. However, due to the very scarce availability of this material, we consider our data as a good starting point for possible further analyses.
Methylation analysis on specific neuronal and non-neuronal cell populations showed that methylation profiles and the related changes can diverge in different cell types as neuronal and glial cells [Citation34,Citation41]. The different cell-type ratios in mice and human samples could have affected the results in the two species since it is known by cell-type databases (http://neuroexpresso.org, http://www.brainrnaseq.org, http://dropviz.org) that microglia can be responsible for sex-specific PSEN1 expression. However, we previously showed that PSEN1 expression in glial cancer cells, unlike in neuronal cells, seems not to depend on DNA methylation [Citation42]. These data indirectly indicate that whole-tissue homogenates have limitations for assessing methylation changes sensitively and specifically. Although we are aware that cell-specific methylation patterns are of the highest interest to disclose how PSEN1 is modulated in the brain and could help to understand the molecular mechanisms of AD, in the present work we aimed to first demonstrate possible methylation differences between patients and controls. Although the admixture of different cell types represented in the tissue homogenates could dilute differences that might have been observed at cell type-specific level [Citation42], this would tend to underestimate the full extent of case–control differences in the methylation status of PS1 expressing cells.
Finally, it will certainly be valuable to study other epigenetic marks in these samples. For example, DNA hydroxymethylation would be of particular interest. Because it is an intermediate in dynamic DNA demethylation, profiling it could provide crucial insight into reversible, regulatory epigenetic control of gene expression. Indeed, just as we prepared the present research for submission, a very interesting paper reported the parallel comparison of DNA methylation and hydroxymethylation in AD [Citation49] and pointed out that CpG hypermethylation may be overestimated due to the presence of 5-hydroxymethylcytosines that are not differentially detected during the standard bisulphite modification. Such uncertainty warrants further clarification. In this context, our findings that implicate non-CpG methylation as an important epigenetic event in neurodevelopment and neurodegerentation, should spur efforts to elucidate the putative regulatory function of non-canonical DNA methylation.
Conclusions
Approaching the analysis of PSEN1 promoter methylation by an unbiased assay capable of revealing both CpG and non-CpG methylation, allowed us to document, for the first time, that hypomethylation of the gene is a common feature of ageing and AD. We also showed that PSEN1 expression is associated with its methylation status, in this cohort. These findings have important implications: First, they increase our knowledge of epigenetic influences on Aβ processing and accumulation and support the hypothesis that epigenetic changes are involved in the pathophysiology of AD. Second, the finding of differential PSEN1 methylation in peripheral blood opens the door to developing this assay as a potential biomarker for the disease. Third, if causal, our findings would provide a starting point for developing epigenetic therapies for AD. We hope that these results will foster further analysis in larger cohorts, including a comparison of parallel brain and blood samples and correlating the methylation data with environmental determinants of biological methylation as folates, B vitamins, homocysteine, and S-adenosylhomocysteine.
Materials and methods
Mice
All procedures were carried out according to the European Communities Council Directives (86/609/EEC and 2010/63/EU) and Italian national legislation on animal experimentation (D.Lgs 116/92) and formally approved by the Italian Ministry of Health.
TgCRND8 transgenic mice (kindly provided by D. Westaway, University of Toronto) overexpress a double mutant form of human amyloid precursor protein 695 (APP695, carrying the ‘Swedish’ KM670/671NL and the ‘Indiana’ V717F mutations). The 5-fold higher expression of the transgene, compared to the endogenous AβPP, results in the production and accumulation of ‘human’ Aβ40 and Aβ42 as the mouse ages, the toxic Aβ42 being the more prevalent species [Citation50]. Heterozygous transgenic mice (TgCRND8±) and wild-type (129Sv, Wt) littermate, used as controls, were obtained from the same breeding. An equal number of female and male mice were used in order to explore previously reported sex differences [Citation51,Citation52]. Experimental groups were formed and processed as follows: (a) Embryonic brains were collected from TgCRND8 and 129Sv control littermate at Embryonic Day (ED) 14.5, N = 12 per each genotype for DNA and RNA isolation (cortex) and N = 10 for histological analyses; (b) adult brains (cortex) were collected at postnatal (PN) day 30 and PN90, N = 24 (12 males and 12 females) per each genotype for DNA and RNA isolation (cortex) and N = 12 (6 males and 6 females) for histological analyses.
Human subjects
Human brain tissue was obtained and used in accordance with the Declaration of Helsinki, relevant Spanish law and the AMC Research Code provided by the Medical Ethics Committees. The local ethics committees of all participating centres approved the study.
MA, LOAD stages I–II/A and LOAD stages V–VI/C human brain tissue (frontal cortex, N = 15 per group) was obtained from the Institute of Neuropathology Brain Bank (HUB-ICO-IDIBELL Biobank, Barcelona, Spain) and Clinic Hospital-IDIBAPS Biobank (Barcelona, Spain). Details of the tissue collection protocol and the characterization of control and LOAD cases are described elsewhere [Citation40,Citation53] and summarized in Table S1.
Prenatal and postnatal human brain tissue (frontal cortex) was obtained from the archives of the Departments of Neuropathology of the Academic Medical Centre (AMC, University of Amsterdam): 14, 22, and 40 gestational weeks (GW), N = 2 per each group; 2 months, N = 2; 17 years, N = 2.
Archived blood samples for 20 LOAD patients (9 males and 11 females, mean age 75.5 ± 5.4 years) and 20 healthy-matched controls (8 males and 12 females, mean age 75.6 ± 6.2 years) were randomly selected from a previously described cohort [Citation25]. RNA samples were not available from this cohort. Each subject gave informed written consent for PSEN1 methylation analysis. The Ethics Committee of the Pisa University Hospital approved the study (Protocol number 3618/2012) that was performed in accordance with the Declaration of Helsinki. Details of subject diagnosis and tissue collection have been published elsewhere [Citation25].
CpG and non-CpG DNA methylation study
Assessment of CpG and non-CpG DNA methylation was performed by bisulphite DNA modification and genomic sequencing using non-CpG Methylation-Insensitive Primers (MIPs), previously described (HSPS1BisF1 and HSPS1BisR1 for the human DNA, MMPS1BisF1, and MMPS1BisR1 for the mouse DNA) [Citation27–Citation29]. These primers recognize DNA sequences with unpredicted non-CpG methylation and their binding is not negatively selected in case of non-CpG methylation. Briefly, DNA was extracted from brain tissues and whole blood using the DNeasy Blood and Tissue Kit (Qiagen, cat. 69504) and the Qiacube instrument. The EpiTect Bisulphite kit was used for bisulphite treatment and PCR products obtained were cloned using the PCR Plus Cloning Kit (Qiagen, cat. 59104 and 231224, respectively). At least 10 clones were analysed per experimental condition using M13 primers for sequencing. Sequencing reactions of purified plasmid DNA were performed by the cycle sequencing method using the ABI PRISM 3130xl genetic analyser (Applied Biosystems). Methylation status of any single cytosine in each sequenced clone was annotated. For each experimental sample, methylation % of every single cytosine was calculated as the number of methylated cytosines divided by the number of sequenced clones x 100 [Citation28].
In order to clarify if the detection of PSEN1 methylation status could be biased by using primers that do not allow for the possibility of non-CpG methylation, PSEN1 methylation in human blood DNA samples was also assessed with the methylation-sensitive high-resolution melting (MS-HRM) technique, using ‘methprimers’ and PCR conditions detailed elsewhere [Citation25]. Briefly, MS-HRM allows to detect the average methylation status of a limited number of CpGs sites since the melting temperature (analysed by Real-Time PCR) of the PCR product changes depending on the presence of methylated cytosines after bisulphite modification.
GenBank accession numbers, primer names, sequences, and position and expected products size of the MIPs used for bisulphite analysis were previously described [Citation17]. The primers used allowed us to assess the methylation status of the plus (5ʹ->3ʹ) DNA strand. The graphical structures of the mouse and human PSEN1 promoters, reporting regulatory regions and primers binding sites, were also previously described [Citation17].
CpG moieties are indicated in figures by a black dot over the related columns.
We validated the bisulphite modification by analysing different bisulphite kits and assays [Citation28,Citation29] using genomic DNA obtained from brain tissue that showed, at the preliminary analysis, low and high (both CpG and non-CpG) methylation, to ensure that cytosine conversion was complete so that we could exclude false-positive results. In particular, an alternative commercial Kit (EZ DNA Methylation Kit, Zymo-Research, cat. D5001), a standard bisulphite procedure and a modified method with ammonium bisulphite [Citation29] were used. In all these cases, the methylation patterns we found were similar; a representative comparison between the four methods is shown for in Fig. S8 (Human brain, elder healthy controls). For negative control of bisulphite modification, we used unmethylated purified PCR products of the PSEN1 gene promoter, obtained by amplifying genomic template DNA with the same MIPs primers used for bisulphite PCR. The same purified PCR products were methylated in vitro with SssI methylase (New England Biolabs) that methylates only cytosines in CpG dinucleotides to be used as positive controls. We adopted all the possible cautions and controls to be sure that no methodological troubles could bias our analysis. DNA digestion with methylation-sensitive endonucleases was performed as a control on random samples as previously described [Citation27,Citation28] and confirmed the bisulphite results (not shown). Representative agarose gel used for the resolution of the PCR products to be cloned are showed in Fig. S9 to demonstrate that PCR signals were not on saturation, since saturation could imply the amplification of non-fully-converted DNA.
Finally, in order to verify whether the Cloning + Sanger sequencing approach resulted in reliable methylation values, we tried to pyrosequence some differentially methylated samples (i.e., bisulphite-modified DNA obtained from human brain by 4 MA health controls and 4 AD I–II subjects) using the biotinylated MIPs. Unfortunately, as expected, these primers are suboptimal for the pyrosequencing application and we could only obtain short (about 20 bp) sequences from only 2 Controls and 2 AD samples, with the reverse primers. Nevertheless, these partial sequences confirmed the methylation values previously obtained with Cloning + Sanger sequencing (Ctrl: R = 0.95; AD I–II: R = 0.90; Fig. S7). Pyrosequencing methylation analysis was performed on the Pyro-Mark Q24 (Qiagen). The level of methylation was manually analysed by the operator since the dedicated Software only calculates methylation for CpG sites.
Gene expression
Total RNA was extracted using the RNeasy Lipid Tissue mini kit (Qiagen, cat. 74804) and the Qiacube instrument; 1 μg of total RNA was used for cDNA synthesis and Real-Time PCR reactions were performed as previously described [Citation16]. cDNA amounts were normalized to β-actin and expressed as fold-increase over control samples. Expression levels were also normalized to Gapdh and 18S obtaining similar results (not shown). Oligonucleotides used as primers in PCR reactions were previously described [Citation27,Citation42,Citation54].
Immunohistochemistry
Coronal sections (thickness, 8 μm) obtained at −2,00 mm from bregma were processed as previously described [Citation51,Citation52,Citation55]. Briefly, antigen retrieval was achieved by pre-treating sections with 10 mM sodium citrate pH 6.0 in a microwave oven for 5 min; then, sections were incubated overnight at 4°C with a mouse monoclonal primary antibody against PSEN1 (#MAB5232, Millipore; 1:500 final dilution in PBS), washed 5 times with PBS, and stained using the mouse Vectastain Elite ABC Kit (Vector Laboratories Inc., cat. PK-6100) according to manufacturer’s instructions. Images were acquired using a Zeiss Axioplan microscope equipped with Sony nex-3N mirrorless camera (Sony Europe Limited) and processed using ImageJ NIH software version 1.48v.
Monoclonal mouse antibodies and published procedures were used to evaluate tau pathology and Aβ-deposition were previously published [Citation52].
Statistical analysis
Results obtained by bisulphite assay were evaluated by Principal Component Analysis (PCA) using SAS statistical software version 10.1.
PCA is the most widespread multivariate analysis technique. The aim of PCA is to project an initial multivariate space spanned by N variables into a p-dimensional component space (with p ≪N) preserving the major part of initial information. Here we extracted the components from the data matrix having sample methylation profiles as variables and cytosines as statistical units. Each variable (sample) has component loadings that range between −1 (maximal negative correlation) to 1 (maximal positive correlation), component scores have by construction zero mean and unit standard deviation. In all the analysed cases, we identified a by far major ‘size’ component (Factor1) explaining around 80–90% of total information. This result points to a strict invariance of the ‘profile’ (relative proportion of methylation along the sequence) typical of a given profile. When we observe a significant between-classes discrimination on Factor1, this corresponds to an effect on the total amount of methylation. On the contrary, when the discrimination happens on a shape component, it implies a change in profile altering the relative balance among different positions with no relation with total methylation.
All the examined cases were neatly discriminated by PCA with no superposition between different groups; the probes with a scores > |2| for discriminant components are considered as relevant for the interpretation, given they correspond to the most influenced by the discriminant component (being component values corresponding to a Z-score a value > |2| can be interpreted as a p < 0.05).
To assess the differences in gene expression, one-way ANOVA for mouse embryos and human samples and three-way ANOVA for adult mice were used, with Tukey’s post-hoc test to evaluate the statistical significance of reported differences. The general model for mice included 2 genotypes (Wt or Tg) x 2 sexes (female or male) x 2 ages (1 or 3 months). The correlation between DNA methylation and PSEN1 gene expression was calculated by Spearman’s rank correlation analysis. P values less than 0.05 were considered statistically significant.
Author’s contribution
AF, NM, VN, and RAC performed the experiments with animals, the mRNA expression, and the DNA methylation studies. ML, AS and FC contributed to the DNA methylation analysis and to the mRNA expression analysis in blood. MTF and GGK performed the immunohistochemical analyses. IF, FC and EA contributed to the collection of tissue samples. The overall experimental design was conceived and supervised by AF, AMT, and SS. AMT and AF prepared the manuscript for submission. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Work on human subjects:
Adult post-mortem brains used in the present study were obtained from the Institute of Neuropathology and Brain Bank (HUB-ICO-IDIBELL Biobank) following the guidelines of the Declaration of Helsinki, and according to the Spanish and Catalonian Autonomous regulations on this matter, and the approval of the local Ethics Committee of the Bellvitge University Hospital.
Embryonic and young post-mortem brains used in this study were obtained from the AMC, University of Amsterdam, and used in accordance with the Declaration of Helsinki and the AMC Research Code provided by the Medical Ethics Committee. The local ethical committees of all participating centres gave permission to undertake the study.
Blood samples were obtained from the University of Pisa (details can be found in reference) [Citation25]. The Ethics Committee of the Pisa University Hospital approved the study (Protocol number 3618/2012) that was performed in accordance with the Declaration of Helsinki.
Work on animals:
Wild-type and transgenic animals used in the present study were bred at the Sapienza University of Rome. All procedures were carried in accordance with the European Communities Council Directive (86/609/EEC 2010/63/EU) and approved by the Italian Ministry of Health and by the local ethical committee.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental Material
Download Zip (5 MB)Acknowledgments
The authors are grateful to Viviana Ciraci, Ph.D. and to Giampiero Palladino, Ph.D., for the help with the experimental procedures. The authors would also like to thank Dr Alessandro Giuliani at Istituto Suoeriore di Sanità, Rome, Italy, for the great help with the execution of the statistical analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Hass MR, Sato C, Kopan R, et al. Presenilin: RIP and beyond. Semin Cell Dev Biol. 2009;20:201–210.
- Edbauer D, Winkler E, Regula JT, et al. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488.
- Iwatsubo T. The gamma-secretase complex: machinery for intramembrane proteolysis. Curr Opin Neurobiol. 2004;14:379–383.
- Lleó A. Activity of gamma-secretase on substrates other than APP. Curr Top Med Chem. 2008;8:9–16.
- Lathia JD, Mattson MP, Cheng A. Notch: from neural development to neurological disorders. J Neurochem. 2008;107:1471–1481.
- Jurisch-Yaksi N, Sannerud R, Annaert W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim Biophys Acta. 2013;1828:2815–2827.
- Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–597.
- Bothwell M, Giniger E. Alzheimer’s disease: neurodevelopment converges with neurodegeneration. Cell. 2000 Aug 4;102(3):271–273.
- Lardenoije R, Iatrou A, Kenis G, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64.
- Sanchez-Mut JV, Gräff J. Epigenetic alterations in Alzheimer’s disease. Front Behav Neurosci. 2015;9:347.
- Migliore L, Coppedè F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res. 2009;667:82–97.
- Fuso A, Scarpa S. One-carbon metabolism and Alzheimer’s disease: is it all a methylation matter? Neurobiol Aging. 2011;32:1192–1195.
- Nicolia V, Lucarelli M, Fuso A. Environment, epigenetics and neurodegeneration: focus on nutrition in Alzheimer’s disease. Exp Gerontol. 2015;68:8–12.
- Mastroeni D, Grover A, Delvaux E, et al. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging. 2011;32:1161–1180.
- Eden S, Cedar H. Role of DNA methylation in the regulation of transcription. Curr Opin Genet Dev. 1994;4:255–259.
- Fuso A, Seminara L, Cavallaro RA, et al. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204.
- Fuso A, Nicolia V, Pasqualato A, et al. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging. 2011;32:187–199.
- Fuso A, Nicolia V, Cavallaro RA, et al. DNA methylase and demethylase activities are modulated by one-carbon metabolism in Alzheimer’s disease models. J Nutr Biochem. 2011;22:242–251.
- Fuso A, Nicolia V, Ricceri L, et al. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol Aging. 2012;33:1482.e1–16.
- Fuso A, Cavallaro RA, Nicolia V, et al. PSEN1 promoter demethylation in hyperhomocysteinemic TgCRND8 mice is the culprit, not the consequence. Curr Alzheimer Res. 2012;9:527–535.
- Schrötter A, Pfeiffer K, El Magraoui F, et al. The amyloid precursor protein (APP) family members are key players in S-adenosylmethionine formation by MAT2A and modify BACE1 and PSEN1 gene expression-relevance for Alzheimer’s disease. Mol Cell Proteomics. 2012;11:1274–1288.
- Li W, Liu H, Yu M, et al. Folic acid administration inhibits amyloid β-peptide accumulation in APP/PS1 transgenic mice. J Nutr Biochem. 2015;26:883–891.
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One. 2008;3:e2698.
- Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J Neuropathol Exp Neurol. 2009;68:880–891.
- Tannorella P, Stoccoro A, Tognoni G, et al. Methylation analysis of multiple genes in blood DNA of Alzheimer’s disease and healthy individuals. Neurosci Lett. 2015;600:143–147.
- Carboni L, Lattanzio F, Candeletti S, et al. Peripheral leukocyte expression of the potential biomarker proteins Bdnf, Sirt1, and Psen1 is not regulated by promoter methylation in Alzheimer’s disease patients. Neurosci Lett. 2015;605:44–48.
- Fuso A, Ferraguti G, Grandoni F, et al. Early demethylation of non-CpG, CpC-rich, elements in the myogenin 5ʹ-flanking region: a priming effect on the spreading of active demethylation. Cell Cycle. 2010;9:3965–3976.
- Fuso A, Ferraguti G, Scarpa S, et al. Disclosing bias in bisulfite assay: methPrimers underestimate high DNA methylation. PLoS One. 2015;10:e0118318.
- Fuso A, Scarpa S, Grandoni F, et al. A reassessment of semiquantitative analytical procedures for DNA methylation: comparison of bisulfite- and HpaII polymerase-chain-reaction-based methods. Anal Biochem. 2006;350:24–31.
- Patil V, Ward RL, Hesson LB. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics. 2014;9:823–828.
- Ziller MJ, Müller F, Liao J, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389.
- Nishino K, Hattori N, Sato S, et al. Non-CpG methylation occurs in the regulatory region of the Sry gene. J Reprod Dev. 2011;57:586–593.
- Guo JU, Su Y, Shin JH, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215–222.
- Kozlenkov A, Roussos P, Timashpolsky A, et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014;42:109–127.
- Blanch M, Mosquera JL, Ansoleaga B, et al. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Pathol. 2016;186:385–397.
- Lin HC, Hsieh HM, Chen YH, et al. S-Adenosylhomocysteine increases beta-amyloid formation in BV-2 microglial cells by increased expressions of beta-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology. 2009;30:622–627.
- Kumar A, Thakur MK. Epigenetic regulation of presenilin 1 and 2 in the cerebral cortex of mice during development. Dev Neurobiol. 2015;75:1165–1173.
- Niu H, Álvarez-Álvarez I, Guillén-Grima F, et al. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia. 2016;32:pii:S0213-4853(16)30003–2.
- Fuso A, Iyer AM, van Scheppingen J, et al. Promoter-specific hypomethylation correlates with IL-1β overexpression in Tuberous Sclerosis Complex (TSC). J Mol Neurosci. 2016;59:464–470.
- Nicolia V, Cavallaro RA, López-González I, et al. DNA methylation profiles of selected pro-inflammatory cytokines in Alzheimer disease. J Neuropathol Exp Neurol. 2017;76:27–31.
- Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905.
- Fuso A, Cavallaro RA, Zampelli A, et al. gamma-Secretase is differentially modulated by alterations of homocysteine cycle in neuroblastoma and glioblastoma cells. J Alzheimers Dis. 2007;11:275–290.
- Yu L, Chibnik LB, Yang J, et al. Methylation profiles in peripheral blood CD4+ lymphocytes versus brain: the relation to Alzheimer’s disease pathology. Alzheimers Dement. 2016;12:942–951.
- Huo Z, Zhu Y, Yu L, et al. DNA methylation variability in Alzheimer’s disease. Neurobiol Aging. 2018;76:35–44.
- Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17:1164–1170.
- De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–1163.
- Grossi E, Stoccoro A, Tannorella P, et al. Artificial neural networks link one-carbon metabolism to gene-promoter methylation in Alzheimer’s disease. J Alzheimers Dis. 2016;53(4):1517–1522.
- BLUEPRINT consortium. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34:726–737.
- Smith AR, Smith RG, Pishva E, et al. Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer’s disease. Clin Epigenetics. 2019;11(1):52.
- Chishti MA, Yang DS, Janus C, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570.
- Canterini S, Bosco A, Carletti V, et al. Subcellular TSC22D4 localization in cerebellum granule neurons of the mouse depends on development and differentiation. Cerebellum. 2012;11:28–40.
- Palladino G, Nicolia V, Kovacs GG, et al. Sexually dimorphic expression of reelin in the brain of a mouse model of Alzheimer disease. J Mol Neurosci. 2017;61:359–367.
- López-González I, Schlüter A, Aso E, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. 2015;74:319–344.
- Fuso A, Nicolia V, Cavallaro RA, et al. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol Cell Neurosci. 2008;37:731–746.
- Piscopo P, Canterini S, Carletti V, et al. Sex effect on presenilins expression in post-natal rat brain. Adv Biosci Biotechnol. 2013;4:1086–1094.