
ABSTRACT
Methionine metabolism is dysregulated in multiple sclerosis (MS). The methyl donor betaine is depleted in the MS brain where it is linked to changes in levels of histone H3 trimethylated on lysine 4 (H3K4me3) and mitochondrial impairment. We investigated the effects of replacing this depleted betaine in the cuprizone mouse model of MS. Supplementation with betaine restored epigenetic control and alleviated neurological disability in cuprizone mice. Betaine increased the methylation potential (SAM/SAH ratio), levels of H3K4me3, enhanced neuronal respiration, and prevented axonal damage. We show that the methyl donor betaine and the betaine homocysteine methyltransferase (BHMT) enzyme can act in the nucleus to repair epigenetic control and activate neuroprotective transcriptional programmes. ChIP-seq data suggest that BHMT acts on chromatin to increase the SAM/SAH ratio and histone methyltransferase activity locally to increase H3K4me3 and activate gene expression that supports neuronal energetics. These data suggest that the methyl donor betaine may provide neuroprotection in MS where mitochondrial impairment damages axons and causes disability.
Introduction
It is clear that changes in epigenetic control are common to neurodegenerative disease. A re-emergence of silenced genes has been reported in Alzheimer’s disease (AD) indicating that methylation dependent transcriptional silencing is disrupted [Citation1]. In multiple sclerosis (MS), a dysregulation of methionine metabolism has been linked to epigenetic changes and mitochondrial defects [Citation2,Citation3]. Decreased concentrations of the methyl donor betaine have been shown to be correlated with reduced levels of H3K4me3 in neurons as well as mitochondrial deficits in MS cortical tissue. Additional studies have linked changes in methionine metabolism to MS [Citation4–Citation6] as well as other neurodegenerative diseases that exhibit mitochondrial impairment including AD and Parkinson’s disease [Citation7–Citation9].
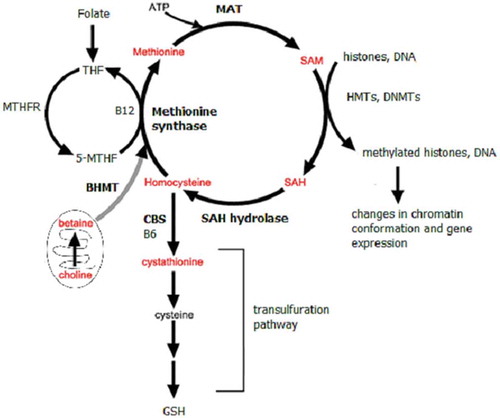
A dysregulation of methionine metabolism can alter chromatin by changing levels of methionine metabolites including the methyl donor S-adenosylmethionine (SAM). Chromatin modifying enzymes require small molecule metabolites including SAM, acetyl groups, and NAD+ to supply substrates for the enzymes that modify DNA and histones to alter chromatin structure in response to appropriate signals. It has previously been shown that metabolic enzymes including methionine adenosyltransferase (MAT), fumarase, pyruvate dehydrogenase complex, and nicotinamide mononucleotide adenylyl-transferase-1 (NMAT-1) are expressed in the nucleus where they synthesize the metabolites required by histone modifying enzymes [Citation10]. The methyl donor SAM is synthesized in the methionine cycle. SAM is the methyl donor for most methylation reactions in cells including histone and DNA methylation. Historically, it has been thought that methionine cycle enzymes and reactions were restricted to the cytoplasm and that SAM diffuses in to the nucleus where it supplies methyl groups for histone methyltransferases (HMTs) to methylate histones and for DNA methyltransferases (DNMTs) to methylate DNA. However, it has become increasingly clear that nuclear localization of biosynthetic enzymes is necessary to synthesize nuclear pools of metabolites required for epigenetic regulation of chromatin [Citation10]. Methionine metabolism enzymes including MAT and SAH hydrolase (AHCY) have been found in the nucleus [Citation11–Citation15], suggesting the existence of a nuclear methionine cycle.
The methionine metabolism enzyme betaine homocysteine methyltransferase (BHMT) is expressed in the liver and kidney as well as in both rodent and human brain in the cortex, hippocampus, and cerebellum [Citation16]. BHMT also been found to be localized to the cytoplasm and nucleus [Citation15]. Betaine, also known as trimethylglycine, provides the methyl group that is transferred to homocysteine in the BHMT catalysed reaction. BHMT catalysed remethylation of homocysteine to methionine contributes to the synthesis of SAM. The role of the BHMT-betaine methylation pathway has been studied extensively in the liver [Citation17], but very little is known about the role of BHMT in the brain. Previous studies, however, have shown that BHMT is important for neuronal cell survival and metabolism as Bhmt−/- mice exhibit neurological changes including decreased brain volume and defects in learning and memory [Citation16].
In MS, inflammation and mitochondrial impairment contribute to disease [Citation18,Citation19]. The cuprizone mouse model of MS mimics the demyelination, activation of microglia, and mitochondrial pathology observed in MS [Citation20,Citation21]. In the cuprizone mouse model, evidence suggests that there is a direct link between cuprizone treatment and changes in axonal mitochondria that results in cell death and demyelination [Citation22,Citation23]. In the present study we have investigated the effects of enhancing the BHMT-betaine methylation pathway by administering betaine in drinking water to assess potential neuroprotective effects in the cuprizone mouse model of MS.
Results
The nuclear BHMT-betaine methylation pathway
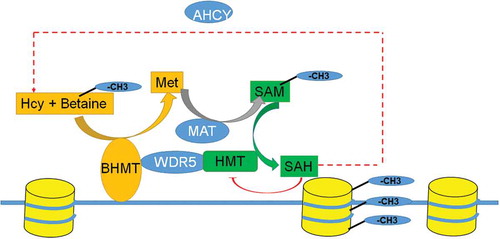
We investigated the role of the BHMT-betaine pathway in histone methylation and neuroprotection. The BHMT-betaine methylation pathway is important for neurodegenerative disease because it can maintain methionine metabolism and the methylation potential (SAM/SAH ratio) under oxidative conditions (). Our data show that betaine can regulate histone methylation in the nucleus and also supports mitochondrial health and viability. In the present study we have confirmed this in different experimental paradigms including the cuprizone mouse model of MS, human SH-SY5Y neuroblastoma cells, and in rat primary neurons (Figure S1).
Figure 1. Schematic depicts methionine metabolism and the BHMT-betaine methylation pathway.
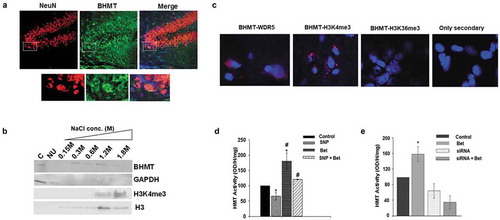
BHMT is expressed in the brain, in the cortex, hippocampus, and cerebellum where it is localized to both the cytoplasm and the nucleus in neurons ()). To better understand the role of BHMT in the nucleus, we performed chromatin fractionation of nuclei from human SH-SY-5Y neuroblastoma cells followed by Western blotting for BHMT and other markers (histone H3, H3K4me3, GAPDH) ()). In these experiments, nuclear extracts were fractionated with increasingly higher NaCl concentrations (0.15–1.8 M NaCl). Proteins bound more tightly to chromatin elute in the higher NaCl fractions (1.2 M, 1.8 M NaCl). We found that BHMT is present in several chromatin fractions including tight chromatin fractions (1.2–1.8 M NaCl fractions) which overlaps with the presence of H3K4me3 ()). As expected, histone H3 is also present in chromatin fractions and GAPDH was present in the cytoplasmic fraction and in the nuclear unbound fraction (NU) that isn’t bound to chromatin. We then performed in situ fluorescence studies and found that Bhmt interacts with the WD repeat domain 5 (Wdr5) subunit [Citation24] of the Set/MLL HMT that methylates H3K4me3 [Citation25] in mouse brain sections ()). To determine if the BHMT-betaine pathway could regulate HMT activity we treated primary neuronal cultures with betaine and measured HMT activity. We found that treating primary neurons with 1 mM betaine overnight as previously described [Citation2] increases HMT activity over control cells by 1.8 fold ((d)). We then treated cells with the nitric oxide (NO) donor sodium nitroprusside (SNP) to increase reactive nitrogen species (RNS) in order to mimic the inflammatory environment that exists in the MS brain [Citation2]. In MS, microglia, which are the resident innate immune cells of the CNS, are activated. These cells synthesize NO which increases RNS in the MS brain. We found that HMT activity decreased by 40% under conditions of excess RNS after treatment with SNP and betaine restored HMT activity back to control levels in SNP treated cells ()). We then knocked down Bhmt expression in rat primary neurons with an siRNA. These data show that betaine mediated regulation of HMT activity is dependent on Bhmt ()).
Figure 2. BHMT is expressed in neurons where it interacts with chromatin and regulates HMT activity.
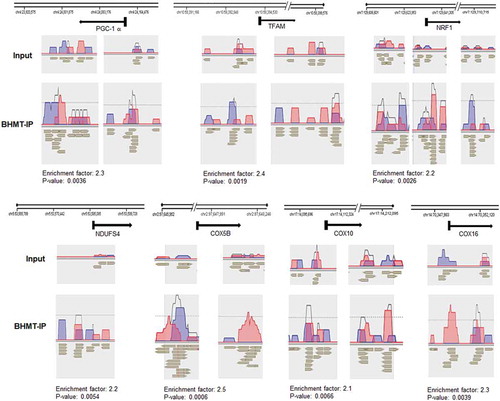
To determine where BHMT was bound to chromatin, we then performed ChIP-seq with chromatin isolated from human SH-SY5Y neuroblastoma cells and an antibody to BHMT. We found that BHMT was enriched predominately within 4 kb the transcription start sites (TSS) of genes (Figure S2) and bound to the sequence motif GCTGGGA (Figure S3). Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway analysis identified categories of genes enriched for BHMT (). The top categories identified were (1) metabolic genes, (2) oxidative phosphorylation genes, (3) Alzheimer’s disease genes, (4) Parkinson’s disease genes, (5) Huntington’s disease genes. All of these groups contain nuclear encoded mitochondrial genes. Several transcription factors that regulate mitochondrial genes including the mitochondrial biogenesis factor peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α also called PPARGC1a) [Citation26] and over 70 mitochondrial genes were enriched for BHMT over 1.5 fold (p < 0.01) (Table S1). These data are consistent with our previous study showing that mitochondrial genes were enriched for H3K4me3 with betaine treatment [Citation2]. Visualization with NGS Strand software version 3.3 shows that BHMT is enriched near TSS and in genic and intronic regions of transcription factors that regulate mitochondrial gene transcription (PGC-1α, mitochondrial transcription factor A [TFAM], and nuclear respiratory factor 1 [NRF-1]) and in nuclear encoded mitochondrial electron transport chain subunit genes (NADH: ubiquinone oxidoreductase subunit S4 [NDUFS4], cytochrome c oxidase subunit 5B [COX5B], cytochrome c oxidase assembly factor haem A:farnesyltransferase [COX10], and cytochrome c oxidase assembly factor [COX16]) ().
Table 1. BHMT is enriched at regulatory regions of mitochondrial genes.
Figure 3. ChIP-seq shows that BHMT is enriched at transcriptional regulators of mitochondrial genes and at mitochondrial genes.
Betaine restores methionine metabolism and epigenetic control in an MS mouse model
We have previously shown that betaine concentrations are decreased in the MS cortex and are linked to mitochondrial impairment [Citation2]. To determine whether similar alterations in methionine metabolism are modelled in the cuprizone mouse model of MS, we analysed methionine metabolite concentrations in the brains of control mice and cuprizone fed mice by LC-MS/MS. Cuprizone induces oligodendrocyte cell death, demyelination, activation of microglia, and neuronal mitochondrial defects, all of which occur in MS. To establish the efficacy of the cuprizone treatment, we performed immunohistochemistry with antibodies to the microglial marker Iba1 and to nitrotyrosine. Microglial cell activation can be detected by changes in morphology. Iba1+ microglia in cuprizone brains were more ameboid, confirming that microglia were in an activated state (). An increase in nitrotyrosine was also observed in cuprizone brains indicating increased RNS and protein nitration (). Black and gold staining was performed to determine the extent of demyelination in our cuprizone mice. These mice exhibited marked demyelination of corpus calossum and cortex as shown in . We found that the activity of the methionine synthase enzyme is reduced in the brains of cuprizone mice () indicating that the increased RNS in the cuprizone mouse model is sufficient to inhibit methionine synthase [Citation27–Citation29].
Figure 4. The cuprizone mouse model mimics the activation of microglia, increased RNS, and methionine metabolism changes exhibited in MS.
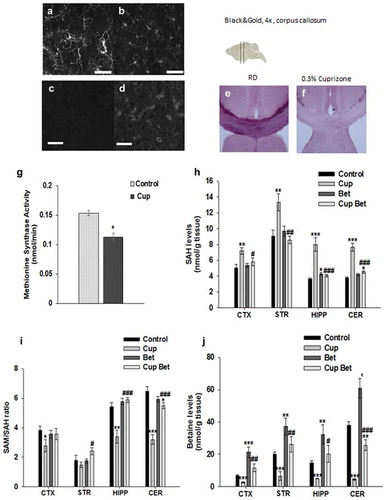
We then measured the concentration of methionine cycle metabolites including methionine, SAM, S-adenosylhomocysteine (SAH), cystathionine, choline, and betaine by LC-MS/MS in the brains of control and cuprizone fed mice. Methionine metabolites were measured in cortex, striatum, hippocampus, and cerebellum in mice fed a regular diet and in mice on the 0.3% cuprizone diet with and without betaine (1% in drinking water). Similar to what we previously reported in MS [Citation2], we found a 20–40% increase in levels of SAH in brain regions of cuprizone mice (). The concentration of cystathionine was also significantly increased in cuprizone treated mice consistent with changes reported in MS previously [Citation2] (Figure S4). Increased cystathionine also points to a build-up of SAH and homocysteine since these metabolites can be diverted to cystathionine. While methionine and SAM levels were maintained (Figure S4) the increase in SAH was substantial and resulted in a 25–50% decrease in the SAM/SAH ratio, also known as the methylation potential, in brain regions (cortex, hippocampus, cerebellum) of cuprizone fed mice compared to controls ()). When methionine synthase is inhibited, SAH builds up and inhibits histone and DNA methylating enzymes. Inhibition of methionine synthase can be overcome by activation of the BHMT-betaine pathway to remethylate homocysteine to methionine in cells that express BHMT. However, similar to what we previously reported in post-mortem MS brain tissue, levels of betaine were also decreased in cuprizone fed mice ()). This could reflect a depletion of betaine in an attempt to maintain the methylation potential under oxidative conditions (). Levels of choline were also decreased in brains of cuprizone fed mice (Figure S4) consistent with reductions in betaine, since betaine can be obtained by oxidation of choline. To determine whether supplementation with betaine can overcome the block of methionine synthase and reduction in the SAM/SAH ratio in the presence of increased RNS, we administered betaine in drinking water and measured methionine metabolite concentrations in control and cuprizone fed mice. We found that betaine was able to bring levels of methionine metabolites including betaine, SAH, and the SAM/SAH ratio back to control levels in cuprizone fed mice ().
Betaine promotes transcriptional programmes that support mitochondria under oxidative conditions
Betaine administration has been shown to support neuronal respiration under conditions of increased RNS in cell culture by increasing levels of H3K4me3 and expression of mitochondrial genes [Citation2,Citation30]. To determine whether betaine could support levels of H3K4me3 in vivo we performed immunohistochemistry with antibodies to NeuN to mark neurons, and H3K4me3. We quantitated levels of H3K4me3 in neurons in brains of mice on regular diet, cuprizone, betaine supplemented, and in cuprizone + betaine supplemented mice and found that H3K4me3 is reduced in cuprizone mice. Levels of H3K4me3 were brought back near control levels in cuprizone + betaine treated mice ()).
Figure 5. Betaine treatment rescues epigenetic control and supports mitochondria.
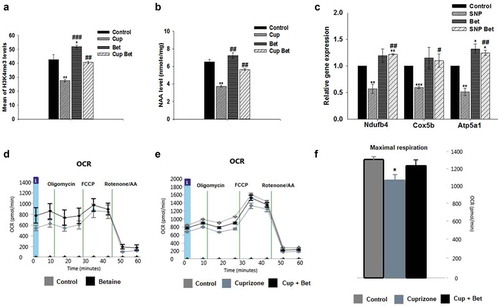
Levels of the neuronal mitochondrial metabolite N-acetylaspartate (NAA) are linked to mitochondrial health and viability in neurons/axons [Citation31,Citation32]. To determine whether betaine can support neuronal mitochondria and protect axons in vivo, we also measured the effects of betaine on NAA levels in cuprizone fed mice. We found that similar to what has been previously reported in MS [Citation2,Citation31], NAA levels were decreased on average by 40% in the brains of cuprizone mice ()). Betaine administration rescued levels of NAA in cuprizone mice back to control levels ()). To confirm that betaine can restore appropriate transcriptional control to support mitochondria under oxidative conditions in neurons, we modelled the oxidative environment that exists in the cuprizone mouse model by treating rat primary neurons with SNP to increase NO and RNS [Citation2]. We then supplemented the cell culture media with betaine (1 mM) and measured the expression of mitochondrial genes by qRT-PCR. We found that betaine treatment increased the expression of mitochondrial complex I, IV, and V genes under oxidative conditions ()).
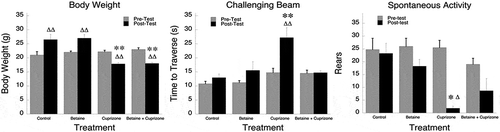
Seahorse respirometry revealed that while betaine didn’t significantly change mitochondrial respiration in control mice ()), basal and maximal respiratory capacity were increased in cuprizone mice who were given betaine in drinking water for the last 4 weeks of cuprizone feeding (). Betaine administration also improved sensorimotor performance in cuprizone mice (). Cuprizone fed mice took significantly longer to traverse the challenging beam, but betaine alleviated this disability in these mice. The effects of betaine didn’t change the weight loss exhibited by cuprizone mice indicating that the protective effects of betaine weren’t linked to any change in weight.
Figure 6. Betaine alleviates sensorimotor disability in cuprizone mice.
Discussion
Previous studies have shown that treatment with the methyl donor betaine can activate neuroprotective transcriptional programmes [Citation2,Citation29,Citation33]. We have extended these findings and now show that betaine mediated neuroprotection involves a novel mechanism that involves BHMT interactions with chromatin. The regulation of chromatin structure involves modifications to DNA and histones. These modifications require metabolites such as SAM, acetyl groups, and NAD+ to supply substrates for the enzymes that modify DNA and histones to alter chromatin structure. It has previously been shown that metabolic enzymes including MAT, fumarase, pyruvate dehydrogenase complex, and NMAT are expressed in the nucleus where they synthesize metabolites required by histone modifying enzymes [Citation10]. Our data add to the list of metabolic enzymes previously believed to be restricted to the cytoplasm that have now been found to be present in the nucleus as well. We have found that BHMT is expressed in the nucleus and interacts with Wdr5 [Citation24], a component of the Set/MLL HMT that methylates H3K4 [Citation25]. These data suggest the existence of a nuclear methionine cycle that regulates the activity of HMTs at specific genomic regions to regulate gene expression.
Nuclear BHMT may be important for removing the toxic build-up of methionine metabolites homocysteine and SAH (). Increased levels of these metabolites inhibit the activity of methyltransferase enzymes [Citation34,Citation35]. Nuclear BHMT can remove excess homocysteine and SAH locally to prevent Set/MLL inhibition and N-homocysteinylation of histone H3 which blocks H3K4 methylation [Citation36]. Our data suggest that chromatin bound BHMT may facilitate the removal of excess nuclear homocysteine and SAH at specific genomic regulatory regions thereby increasing the SAM/SAH ratio and HMT activity. While cytoplasmic BHMT could also act to remove excess homocysteine and SAH, having BHMT bound to specific regulatory regions on chromatin would allow for local control of the Set/MLL methyltransferase activity, levels of H3K4me3, and gene expression. BHMT activity may be important in cells like neurons that have limited metabolism through the transulfuration pathway, which limits the toxic build-up of SAH and homocysteine by first converting homocysteine to cystathionine.
Figure 7. Model for BHMT-betaine mediated regulation of mitochondrial gene expression.
In addition to removing toxic levels of homocysteine and SAH in the nucleus, BHMT bound to chromatin would also contribute to producing a nuclear pool of SAM to methylate histones at specific sites by HMTs. This would require MAT, the enzyme that converts methionine to SAM, to also be expressed in the nucleus. In fact, nuclear MAT has been found and its expression is correlated with levels of histone methylation [Citation13,Citation14]. The MAT enzyme converts methionine to SAM for methylation reactions and like BHMT it has been shown to be localized to both the cytoplasm and nucleus [Citation13,Citation14]. The interaction of MAT alone on chromatin can synthesize SAM locally, but wouldn’t allow cells to eliminate SAH and homocysteine build-up in the nucleus.
Cuprizone is known to be a copper chelator and was believed to cause MS like pathology in mice by damaging oligodendrocytes by impairing activity of copper dependent enzymes including cytochrome oxidase. However, recently it has been shown that cuprizone alters brain metabolism independently of its ability to chelate copper [Citation37]. Consistent with our data, Taraboletti et al. [Citation37] demonstrated that cuprizone treatment also resulted in a dysregulation of one carbon and methionine metabolism and in levels of metabolites necessary for energy in the corpus callosum and in the hippocampus. Our data confirm this finding and show that cuprizone treatment models the methionine metabolism changes previously reported in MS [Citation2,Citation3,Citation38]. These findings suggest that a dysregulation of methionine metabolism and epigenetic regulation of chromatin may be central to MS pathology and that the cuprizone model mimics these pathological changes. Similar to MS, where the availability of methyl donors is reduced, in the brains of cuprizone mice the methyl donor betaine is depleted considerably and the SAM/SAH ratio, a measure of methylation potential, is reduced. Changes in methionine metabolism and the SAM/SAH ratio can lead to changes in methylation of downstream substrates including histones and result in aberrant gene expression [Citation39,Citation40]. We found that downstream methylation reactions and mitochondrial metabolism were also altered in the brains of cuprizone treated mice. Dietary supplementation with betaine in drinking water restored methionine metabolism and brought the SAM/SAH ratio, levels of H3K4me3, and mitochondrial gene expression back to what was observed in control mice on the regular diet.
Deficiencies in or supplementation with dietary factors including the methyl donor betaine and other methionine metabolites can alter epigenetic signatures and gene expression programmes [Citation41], however it is unclear how the specificity of changes in methylation of DNA or histones due to these metabolites is conferred. Supplementation with metabolites that feed in to the methionine cycle do not appear to increase methylation globally throughout the genome but, instead increase methylation at specific genomic regions [Citation38,Citation42,Citation43]. Very little is known concerning how dietary supplementation and changes in the SAM/SAH ratio can regulate specific subsets of genes. Our data indicate that BHMT interactions with chromatin confer specificity for methylation of histone H3 at regulatory regions of genes involved in mitochondrial respiration. Betaine administration can activate the methylation of H3K4me3 and gene expression through chromatin bound BHMT activity. These data have important implications for the development of new therapeutic strategies for neurodegenerative diseases such as MS.
Materials and Methods
Immunohistochemistry
Mice (C57Bl/6) at 13 weeks of age were euthanized by cervical dislocation and isolated brains were fixed in 4% PFA and cryoprotected with 10%, 20% and 30% sucrose-PBS solution. 30 µm of coronal sections were cut with a cryostat. Sections were collected in PBS and used for immunohistochemistry with antibodies to Bhmt (sc-69708) (Santa Cruz Biotechnology, Dallas, TX), H3K4me3 (ab8580), and NeuN (ab177487) (Abcam, Cambridge, MA). We also performed immunostaining with anti-nitrotyrosine (06–284) (Millipore, Burlington, MA) and Iba1 (019-19741) (Wako, Richmond, VA) to examine increased RNS and the degree of microglia activation, respectively. Sections were blocked in blocking buffer (5% normal donkey serum in PBS containing 0.1% Triton-X-100 for 1 hour at room temperature and incubated overnight at 4°C with primary antibody to Bhmt (1:250), H3K4me3 (1:250), NeuN (1:500), anti-nitrotyrosine (1:500), or Iba1 (1:100) made in blocking buffer. After washing, sections were incubated in secondary antibody donkey anti-rabbit Alexa-488 (1:400) ([ab150073]Abcam, Cambridge, MA) or donkey anti-chicken Alexa-555 (1:400) ([ab150170]Abcam, Cambridge, MA) for 3 hours at 4°C. Sections were washed three times with PBS and mounted with Vectashield mounting medium containing DAPI to label nuclei. Images were acquired with an Olympus Fv1000 confocal microscope equipped with five laser lines (HeCd 442 nm, Ar 488 and 514 nm, HeNe 543 nm and HeNe 633 nm). Image stacks were z-projected with ImageJ (National Institutes of Health, Bethesda, MD) and channels merged to show colocalized signals.
Relative H3K4me3 was compared between control, cuprizone, betaine, and betaine-cuprizone treated mice. We measured the mean intensity of H3K4me3 from at least 50 cells from the cortical area right above the corpus callosum. Image stacks were captured sequentially for each channel to prevent bleed through and spanned the sections. An image mask was created using the NeuN channel as a guide to include the entire nucleus. The thresholded image mask was then used to clip the H3K4me3 and the pixels within the unclipped region were summed. This technique measures the amount of H3K4me3 fluorescence from within individual NeuN+ nuclei.
Cell culture
Primary neurons were isolated from the brain cortices of Sprague Dawley rat E18 embryos as per guidelines of IACUC. Enriched neuronal cell culture was maintained using neurobasal medium ([21103049] Gibco, Waltham, MA) supplemented with B-27, glutamine ([35050-061] Gibco, Waltham, MA), 50 µg/ml penicillin, 50 µg/ml streptomycin ([30-002-C1] Corning, Tewksbury, MA) at 37° C in a humidified 5% CO2 incubator. Cells were harvested between days 9–10 of culture. Human SH-SY5Y neuroblastoma cells were cultured in a 1:1 mixture of DMEM and F-12 medium ([D8437] Sigma-Aldrich, St. Louis, MO) with 10% FBS ([89510-188] VWR, Radnor, PA), 50 µg/ml penicillin, 50 µg/ml streptomycin (Corning, Tewksbury, MA) at 37° C in a humidified 5% CO2 incubator. Cells were grown in 10 cm petri dishes in confluence up to 90%.
In situ fluorescence
To show the interaction between Wdr5, Bhmt, and H3K4me3, we performed in situ fluorescence by using the Duolink In Situ Fluorescence kit ([DUO92101] Sigma-Aldrich, St. Louis, MO). In brief, mouse brain sections were incubated with primary antibodies to Wdr5 and Bhmt ([sc-393080; sc-69708] Santa Cruz Biotechnology, Dallas, TX) or H3K4me3 ([ab8580] Abcam, Cambridge, MA) diluted at 1:250. Sections were then incubated with secondary antibodies conjugated with oligonucleotides (PLA probe MINUS and PLUS) ([DUO92002; DUO92004] Sigma-Aldrich, St. Louis, MO). The oligonucleotide will hybridize to the PLA probes and make a closed circle if proteins are in closed proximity (40 nm). Amplification solution was then added which contains polymerase, nucleotides, and fluorescent nucleotides. The oligonucleotide arm of one of the PLA probes acts as a primer for a rolling circle amplification reaction using the ligated circle as a template, generating a concatemeric product, which was detected by fluorescence microscopy.
Histone methyltransferase activity
Histone methyltransferase activity was measured specifically for histone H3K4 methylation in sodium nitroprusside (SNP) and betaine treated primary neurons. The HMT assays were performed by using EpiQuick histone methyltransferase assay kit ([P-3002-2] Epigentek, Farmingdale, NY). The effects of betaine on restoring Set/MLL HMT activity under conditions of increased RNS was measured. To increase RNS, cells were treated with the NO donor SNP as previously described [Citation2]. In brief, nuclear extracts were prepared from control cells or cells treated with 400 μM SNP, 1 mM betaine, or SNP + betaine overnight and 4 ug of protein was added to substrate and assay buffer and incubated for 1 and a half hours at 37C in a 96 well plate. After washing, capture antibody was added followed by the detection antibody and developing solution. The absorbance was measured at 450 nm within 10 min and HMT activity was measured in O.D./h/mg protein. An siRNA oligo to Bhmt or a non-targeting control siRNA was transfected in to rat primary neurons to show that betaine related increases in HMT activity are Bhmt dependent (siRNA targeting Bhmt sequence UUAGAACGCUUAAAUGCUG) (Dharmacon, Lafayette, CO). The non-targeting control siRNA sequence was AUGCGACUAAACACAUCAA. The siRNA knockdown efficiency was approximately 60%.
Salt gradient extraction and tight chromatin fractionation
Neuronal cultures were homogenized with cytosolic buffer (10 mM HEPES,10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 0.2% NP-40, 1 mM PMSF, phosphatase and protease inhibitors) and centrifuged at 5000 g for 10 min at 4 ° C. After centrifugation, the pellet containing the nuclei was suspended in soluble nuclear buffer (3 mM EDTA, 0.2 mM EGTA, 1 mM PMSF, phosphatase and protease inhibitors) [Citation44] and centrifuged again at 5000 g. The pellet was taken for sequential salt gradient extraction with NaCl (0.3, 0.45, 0.6, 1.2 and 1.8 M) in 1.5 M Tris-HCl. Protein fractions were analysed by Western blotting with antibodies to BHMT ([sc-69708] Santa Cruz Biotechnology, Dallas, TX), histone H3 or H3K4me3 ([ab1791; ab8580] Abcam, Cambridge, MA), and GAPDH ([mab374] Millipore, Burlington, MA).
ChIP-seq
To identify genes regulated by BHMT, ChIP-seq was performed with chromatin isolated from human SH-SY5Y neuroblastoma cells and an antibody to BHMT ([sc-69708] Santa Cruz Biotechnology, Dallas, TX). ChIP-seq was performed in duplicate and BHMT immunoprecipitated sequencing reads were compared to input DNA. For ChIP, cells were fixed with 37% formaldehyde solution for 10 min. After washing with PBS, fixation was stopped by adding 1x glycine stop-fix solution for 5 min. Neuroblastoma cells were lysed to release the nuclei and centrifuged at 2400 x g at 4°C for 10 min to pellet the nuclei. Chromatin was resuspended in RIPA buffer containing protease inhibitors ([R0278] Sigma-Aldrich, St. Louis, MO) and was sheared by sonication with a focused ultrasonicator (Covaris) for 8 min to obtain chromatin fragments 300–400 bp in length. Fragment size was confirmed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and 10 µl of sheared sample (input DNA) was stored at 20°C to be used as a control.
ChIP was performed with the ChIP-IT Express kit provided by Active Motif. ChIP reactions were set up by sequential addition of 25 µl protein G magnetic beads, 20 µl of ChIP buffer, 50 µg of sheared chromatin, 2 µl of protease inhibitor mixture, distilled water to makeup the final volume 200 µl, and 4 µg of BHMT antibody followed by incubation on an end-to-end rotor at 4°C overnight. Beads were separated on a magnetic stand, washed, and then resuspended in 50 µl of elution buffer. Chromatin was cleaned up by treatment with proteinase K followed by phenol-chloroform extraction and EtOH precipitation. Library preparation and sequencing was performed at Novogene Genome Sequencing Company. Libraries were constructed with the NEBNext® Ultra™ II DNA kit. Qualified libraries were sequenced on an Illumina HiSeq Platform using a single-end 50 run (1 × 50 bases) and 40 million reads/sample were generated. Data trimming removed reads with low quality (low quality bases > 50%), reads with unsure bases (N ratio more than 15%), and discarded sequences less than 18 nucleotides after trimming adaptor sequences. Sequences were mapped to the human reference genome build HG 38 with BWA and mapping quality was calculated with MAPQ [Citation45]. Peak calling was done with MACS2 software and data were visualized with IGV 2.5.2 software and Strand NGS version 3.3 software (Agilent Technologies, Santa Clara, CA). Single gene analysis provided gene information, including distance to TSS, coding region, location in the genome, and overlap type (intron or exon). The enrichment factor for each gene was determined through MACS analysis. If a gene had multiple sites of enrichment, an average fold enrichment was calculated. Statistical significance of BHMT enrichment for genes assigned to KEGG pathways [Citation46] was performed with a hypergeometric Fisher’s exact test. False discovery rate (FDR) correction was done with the Benjamini and Hochberg test.
Cuprizone treatment
We obtained male C57Bl/6 mice (6 weeks of age) from The Jackson Laboratory. Cuprizone (bis-cyclohexanone-oxaldihydrazone) 0.3% (w/w) was introduced into pellet food (Teklad, Madison, WI). After the first week of acclimatization, control mice were fed a regular diet and another set of mice were fed a diet containing 0.3% cuprizone for 6 weeks. The food was changed daily, and mice were weighed weekly to monitor for weight loss. Demyelination was assessed by histochemistry with black and gold staining as described [Citation47]. Brain sections were washed with saline followed by incubation in 0.3% black-gold solution for 10–12 minutes at 60 ° C. After washing with saline, sections were fixed in 1% sodium thiosulphate for 3 minutes. Sections were washed with saline three times, and mounted on glass slide using glycerol. Images were taken using bright field microscopy. Methionine synthase activity was assessed in the brains of control and cuprizone mice with a cobalamin-dependent method [Citation48]. For betaine treated mice, betaine was administered in drinking water (1% betaine) for the last 4 weeks during the 6 week cuprizone feeding.
Analysis of methionine metabolites by LC-MS/MS
Stable-isotope dilution liquid chromatography-electrospray ionization (ESI) tandem mass spectrometry (LC-ESI-MS/MS) was performed to determine the concentrations of SAM, SAH, methionine, cystathionine, choline and betaine in brain regions including cortex, striatum, hippocampus, and cerebellum from eight mice from each group including control mice on regular diet, regular diet + betaine treated mice, cuprizone fed, and cuprizone + betaine treated mice as previously described [Citation49]. Calibrators and internal standards (2H3-SAM, 2H4-SAH, 2H3-methionine, 2H4-cystathionine, 2H3-choline, 2H3-betaine, were included in each analytical run for calibration. Samples were prepared by the addition of 140 µL mobile phase A containing 5–25 µmol/L labelled-isotope internal standards to 20 µl of plasma. Samples were filtered through microcentrifugal filter units (Microcon YM-10, 10 kDa NMWL (Millipore, Burlington, MA) by centrifugation for 20 min at 14,800 x g at 4ºC. Chromatographic separation was achieved on a EZ-faast 250 × 2.0 mm 4µ AAA-MS analytical column (Phenomenex, Torrance, CA) maintained at 40°C at a flow of 200 μL/min with a binary gradient with a total run time of 10 minutes. Solvents for HPLC were: (A) 4 mM ammonium acetate, 0.1% formic acid, 0.1% heptafluorobutyric acid (pH 2.5); (B) 100% methanol and 0.1% formic acid. The initial gradient condition was 75% A: 25% B and was increased in a linear fashion to 100%B in 7 min. The compounds were detected by multiple reaction monitoring (MRM) using positive-ESI. Sample separation and injection was performed by a Nexera LC System (Shimadzu, Columbia, MD) interfaced with a 5500QTRAP® LC-MS/MS (Sciex, Framing ham, MA). All data was collected using Analyst software version 1.6.2. Statistical significance of changes in average methionine metabolite concentrations between groups was determined with a two-tailed T-test with p ≤ 0.05 considered significant.
Measuring NAA concentration by HPLC
The neuronal mitochondrial metabolite NAA was quantitated in brains of mice by high performance liquid chromatography (HPLC) as previously described [Citation32]. Cortices were dissected from brains of three mice from each group including control, betaine treated, cuprizone treated, and cuprizone + betaine treated mice. Brain tissue was homogenized in ice-cold 90% methanol using pellet pestle, and centrifuged twice at 14,000 rpm for 10 min at 4°C. The supernatant was dried by speed-vac and the powder was then dissolved in 0.5 ml deionized H2O. The solution was then added to an AG50 W x 8 poly-pre column ([7316214] Bio-Rad, Hercules, CA). The column was washed with 1 ml of deionized H2O, and all the eluate was collected, lyophilized, and stored at 4°C. For HPLC analysis, each sample was resuspended in 300 μl deionized H2O. A Whatman partisil 10 SAX anion-exchange column (4.6 mm x 250 mm) was used in an Agilent 1100 Series HPLC Value System (Agilent Technologies, Santa Clara, CA). The mobile phase was 0.1 M KH2PO4 and 0.025 M KCl at pH 4.5. After washing the column with 50% acetonitrile and 50% deionized H2O, the column was conditioned with at least 20–30 column volumes of new mobile phase. Retention data were collected at a flow rate of 1.5 ml/min. The flow was monitored with an Agilent 1100 series UV detector at 214 nm. Retention time was 5.10 minutes and was determined with an NAA standard ([00920-SG] Sigma-Aldrich, St. Louis, MO). Peak areas were acquired with Agilent Chemstation software. NAA concentrations were determined in triplicate and statistical significance (p < 0.05) was determined with a Student’s T-test.
Respirometry
A Seahorse Bioscience XF-24 Respirometer (Agilent Technologies, Santa Clara, CA) was used to measure mitochondrial respiration. Mitochondria were isolated from the brains of mice by homogenizing cortices in 70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, and 0.5% BSA with a glass homogenizer at 4°C. The cell suspension was then centrifuged at 1000 X g for 10 min at 4°C. The supernatant was centrifuged again at 8000 X g for 10 min at 4°C, the pellet was resuspended and total protein concentration was determined using Bradford Assay reagent ([500-0113-0115] Bio-Rad, Hercules, CA). For the assay, mitochondria were diluted 10 times in cold 70 mM sucrose, 220 mM mannitol, 2 mM HEPES, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, and 0.2% BSA and were then plated on the XF assay plate in equal concentration (5 ug). 50ul of mitochondrial suspension was delivered to each well (except for background correction wells). The XF cell culture microplate was centrifuged at 2000 X g for 20 min at 4°C to adhere the mitochondria. The plate was then transferred to the XF analyser, and the experiment was initiated. Basal oxygen consumption rates (OCR) were monitored initially. To measure maximal respiration, oligomycin (2.5 μg/ml) was injected to inhibit ATP synthase, followed by the uncoupler carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (4 μM). This eliminates the proton gradient across the inner mitochondrial membrane and respiratory control exerted by the proton gradient so that electron transport is stimulated and oxygen is consumed at the maximal rate. OCR was monitored over time and was calculated in pmol/min. Average changes in basal and maximal respiration were determined after subtracting nonmitochondrial oxygen consumption, measured after addition of rotenone (4 μM) and antimycin A (2 μM) to inhibit Complex I and III respectively. Measurements were from four mice from each group. Statistical significance was determined by a two-tailed Student’s t test, with p < 0.05 considered significant.
Behavioural tests
Mice were fed regular diet or cuprizone diet for 4 weeks. Some mice were given betaine (1%) in drinking water that was changed every other day. There were 4–6 mice per group. Mice were supplemented with betaine after 2 weeks of the feeding regimen and continued on betaine for the duration (2 more weeks). After 4 weeks, the challenging beam traversal was performed to test motor performance and coordination. The beam is constructed out of Plexiglas and consists of four sections (25 cm each, 1 m total length). The beam begins at a width of 3.5 cm and is gradually narrowed to 0.5 cm by 1 cm increments. Animals were trained to traverse the length of the beam starting at the widest section and ending at the narrowest, most difficult, section. The narrow end of the beam leads directly into the animal’s home cage. Mice were then videotaped while traversing the grid-surfaced beam for a total of five trials [Citation50,Citation51]. Videotapes were viewed and rated in slow motion for number of steps made by each animal, and time to traverse across five trials by an investigator blinded to the mouse genotype. Time to traverse and number of steps were calculated for wildtype and mutant mice across all five trials and averaged. Spontaneous activity was measured by placing animals in a small transparent cylinder (height, 15.5 cm, diameter, 12.7 cm) and videotaping activity for three minutes [Citation50,Citation51]. The number of rears, forelimb and hindlimb steps, and time spent grooming were measured by the investigator blinded to genotype. Statistical significance was determined with a 4 × 2 mixed design ANOVA, Tukey’s HSD post hoc (Body Weight and Challenging Beam) and Mann-Whitney U and Wilcoxin Sign Rank (Spontaneous activity).
Supplemental Material
Download MS Word (2.2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Frost B, Hemberg M, Lewis J, et al. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17(3):357–366.
- Singhal NK, Li S, Arning E, et al. Changes in methionine metabolism and histone H3 trimethylation are linked to mitochondrial defects in multiple sclerosis. J Neurosci. 2015;35(45):15170–15186.
- Singhal NK, Freeman E, Arning E, et al. Dysregulation of methionine metabolism in multiple sclerosis. Neurochem Int. 2018;112:1–4.
- Reynolds EH, Bottiglieri T, Laundy M, et al. Vitamin B12 metabolism in multiple sclerosis. Arch Neurol. 1992;49(6):649–652.
- Miller A, Korem M, Almog R, et al. Vitamin B12, demyelination, remyelination and repair in multiple sclerosis. J Neurol Sci. 2005;233(1–2):93–97.
- Kocer B, Engur S, Ak F, et al. Serum vitamin B12, folate, and homocysteine levels and their association with clinical and electrophysiological parameters in multiple sclerosis. J Clin Neurosci. 2009;16(3):399–403.
- Irizarry MC, Gurol ME, Raju S, et al. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology. 2005;65(9):1402–1408.
- Licking N, Murchison C, Cholerton B, et al. Homocysteine and cognitive function in Parkinson’s disease. Parkinsonism Relat Disord. 2017;44:1–5.
- Fahmy EM, Elfayoumy NM, Abdelalim AM, et al. Relation of serum levels of homocysteine, vitamin B12 and folate to cognitive functions in multiple sclerosis patients. Int J Neurosci. 2018;128(9):835–841.
- Gibson BA, Kraus WL. Small molecules, big effects: a role for chromatin-localized metabolite biosynthesis in gene regulation. Mol Cell. 2011;41(5):497–499.
- Radomski N, Kaufmann C, Dreyer C. Nuclear accumulation of S-adenosylhomocysteine hydrolase in transcriptionally active cells during development of Xenopus laevis. Mol Biol Cell. 1999;10(12):4283–4298.
- Radomski N, Barreto G, Kaufmann C, et al. Interaction of S-adenosylhomocysteine hydrolase of Xenopus laevis with mRNA (guanine-7-)methyltransferase: implication on its nuclear compartmentalisation and on cap methylation of hnRNA. Biochim Biophys Acta. 2002;1590(1–3):93–102.
- Reytor E, Pérez-Miguelsanz J, Alvarez L, et al. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J. 2009;23(10):3347–3360.
- Katoh Y, Ikura T, Hoshikawa Y, et al. Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol Cell. 2011;41(5):554–566.
- Delgado M, Garrido F, Pérez-Miguelsanz J, et al. Acute liver injury induces nucleocytoplasmic redistribution of hepatic methionine metabolism enzymes. Antioxid Redox Signal. 2014;20(16):2541–2554. Epub 2014 Jan 3.
- Prieur EAK, Pjetri E, Zeisel SH, et al. Reduced brain volume and impaired memory in betaine homocysteine S-methyltransferase knockout mice. Appl Physiol Nutr Metab. 2017;4211:1228–1231. Epub 2017 Jul 17 .
- Kathirvel E, Morgan K, Nandgiri G, et al. Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: a potential mechanism for hepatoprotection by betaine. Am J Physiol Gastrointest Liver Physiol. 2010;299(5):G1068–77.
- Broadwater L, Pandit A, Azzam S, et al. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim Biophys Acta. 2011;1812:630–641.
- Witte ME, Mahad DJ, Lassmann H, et al. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20(3):179–187.
- Torkildsen Ø, Brunborg LA, Myhr KM, et al. The cuprizone model for demyelination. Acta Neurol Scand. 2008;Suppl,188:72–76.
- Hagemeyer N, Boretius S, Ott C, et al. Erythropoietin attenuates neurological and histological consequences of toxic demyelination in mice. Mol Med. 2012;18:628–635.
- Acs P, Selak MA, Komoly S, et al. Distribution of oligodendrocyte loss and mitochondrial toxicity in the cuprizone-induced experimental demyelination model. J Neuroimmunol. 2013;262(1–2):128–131.
- Xuan Y, Yan G, Wu R, et al. The cuprizone-induced changes in 1H-MRS metabolites and oxidative parameters in C57BL/6 mouse brain: effects of quetiapine. Neurochem Int. 2015;90:185e192.
- Grebien F, Vedadi M, Getlik M, et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat Chem Biol. 2015;11(8):571–578.
- Sun J, Zhao Y, McGreal R, et al. Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers. Epigenetics Chromatin. 2016;9(1):37.
- Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124.
- Nicolaou A, Ast T, Velasco Garcia C, et al. In vitro NO and N2O inhibition of the branch point enzyme vitamin B12 dependent methionine synthase from rat brain synaptosomes. Biochem Soc T. 1994;22:296S.
- Nicolaou A, Kenyon SH, Gibbons JM, et al. In vitro inactivation of mammalian methionine synthase by nitric oxide. Eur J Clin Invest. 1996;26:167.
- Mukherjee R, Brasch NE. Kinetic studies on the reaction between cob(I)alamin and peroxynitrite: rapid oxidation of cob(I)alamin to cob(II)alamin by peroxynitrous acid. Chemistry. 2011;17(42):11723–11727.
- Lee I. Betaine is a positive regulator of mitochondrial respiration. Biochem Biophys Res Commun. 2015 9;456(2):621–625.
- Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998;20(4–5):271–276.
- Li S, Clements R, Sulak M, et al. Decreased NAA in gray matter is correlated with decreased availability of acetate in white matter in postmortem multiple sclerosis cortex. Neurochem Res. 2013;38(11):2385–2396.
- Chai GS, Jiang X, Ni ZF, et al. Betaine attenuates Alzheimer-like pathological changes and memory deficits induced by homocysteine. J Neurochem. 2013;124(3):388–396.
- Hoffman DR, Cornatzer WE, Duerre JA. Relationship between tissue levels of S-adenosylmethionine, S-adenosylhomocysteine, and transmethylation reactions. Can J Biochem. 1979;57:56–65.
- Kennedy BP, Bottiglieri T, Arning E, et al. Elevated S-adenosylhomocysteine in Alzheimer brain: influence on methyltransferases and cognitive function. J Neural Transm (Vienna). 2004 4;111:547–567. Epub 2004 Feb 4.
- Xu L, Chen J, Gao J, et al. Crosstalk of homocysteinylation, methylation and acetylation on histone H3. Analyst. 2015 May 7;140(9):3057–3063. Epub 2015 Mar 25.
- Taraboletti A, Walker T, Avila R, et al. Cuprizone intoxication induces cell intrinsic alterations in oligodendrocyte metabolism independent of copper chelation. Biochemistry. 2017;56(10):1518–1528.
- Gardner LA, Desiderio DM, Groover CJ, et al. LC-MS/MS identification of the one-carbon cycle metabolites in human plasma. Electrophoresis. 2013;34(11):1710–1716.
- Chen NC, Yang F, Capecci LM, et al. Regulation of homocysteine metabolism and methylation in human and mouse tissues. Faseb J. 2010;24(8):2804–2817.
- Mentch SJ, Mehrmohamadi M, Huang L, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015;22(5):861–873.
- Zeisel S. Choline, other methyl-donors and epigenetics. Nutrients. 2017;9(5):pii: E445.
- Fuso A, Nicolia V, Pasqualato A, et al. Changes in presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging. 2011a;32(2):187–199.
- Fuso A, Nicolia V, Cavallaro RA, et al. DNA methylase and demethylase activities are modulated by one-carbon metabolism in Alzheimer’s disease models. J Nutr Biochem. 2011b;22(3):242–251.
- O’Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell. 2011;20(5):606–619.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760.
- Kanehisa M, Araki M, Goto S., et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
- Schmued L, Bowyer J, Cozart M, et al. Introducing black-gold II, a highly soluble gold phosphate complex with several unique advantages for the histochemical localization of myelin. Brain Res. 2008;1229:210–217.
- Drummond JT, Jarrett J, González JC, et al. Characterization of nonradioactive assays for cobalamin-dependent and cobalamin-independent methionine synthase enzymes. Anal Biochem. 1995;228(2):323–329.
- Inoue-Choi M, Nelson HH, Robien K, et al. Plasma S-adenosylmethionine, DNMT polymorphisms, and peripheral blood LINE-1 methylation among healthy Chinese adults in Singapore. BMC Cancer. 2013;13(1):389.
- Fleming SM, Salcedo J, Fernagut PO, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24(42):9434–9440.
- Fleming SM, Salcedo J, Hutson CB, et al. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-synuclein. Neuroscience. 2006;142(4):1245–1253.