
ABSTRACT
Hybrid male sterility (HMS) is a postzygotic reproductive isolation mechanism that enforces speciation. A bovine example of HMS is the yattle (also called dzo), an interspecies hybrid of taurine cattle (Bos taurus) and yak (Bos grunniens). The molecular mechanisms underlying HMS of yattle are not well understood. Epigenetic modifications of DNA methylation and P-element induced wimpy testis (PIWI)-interacting RNA (piRNAs) are important regulators in spermatogenesis. In this study, we investigated DNA methylation patterns and piRNA expression in adult testes in hybrid infertile yattle bulls and fertile cattle and yak bulls using whole genome bisulphite-seq and small RNA-seq. Promoter hypermethylation in yattle were associated with DNA methylation involved in gamete generation, piRNA metabolic processes, spermatogenesis, and spermatid development (P < 2.6 × 10−5). Male infertility in yattle was associated with the promoter hypermethylation-associated silencing of PIWI/piRNA pathway genes including PIWIL1, DDX4, PLD6, MAEL, FKBP6, TDRD1 and TDRD5. The downstream effects of silencing these genes were diminished production of 29- to 31- nucleotide pachytene piRNAs in yattle testes. Hypermethylation events at transposable element loci (LINEs, SINEs, and LTRs) were found in yattle. LINE-derived prepachytene piRNAs increased and SINE-derived prepachytene piRNAs were reduced in yattle testes. Our data suggests that DNA methylation affects the PIWI/piRNA pathway and is involved in gene expression and pachytene piRNA production during spermatogenesis in bovine HMS. DNA hypermethylation and disruption of piRNA production contributed to unsuccessful germ cell development that may drive bovine HMS.
Introduction
Hybrid male sterility (HMS) is a postzygotic reproductive isolation mechanism that enforces speciation by restricting gene flow between species or subspecies. It is thought to result from the acquisition of genetic incompatibilities [Citation1,Citation2]. HMS is a universal phenomenon in many eukaryotic interspecies hybrids [Citation3]. Domestic yaks (Bos grunniens) and domestic taurine cattle (Bos taurus) are closely related species that diverged 4.9 million years ago. The synteny of their genomes is as high as 94% [Citation4]. An example of HMS in bovines is the yattle (also called dzo), the interspecies hybrid of taurine cattle and yak. F1 hybrid yattle and F2 backcross males are sterile while females are fertile. Seminiferous tubules of yattle contain few primary spermatocytes at the leptotene to pachytene stage and no spermatocytes beyond this stage [Citation5–Citation7]. Thus, the male infertility of yattle may be due to arrest of meiosis at about the pachytene stage [Citation7]. Testes transcriptomics and proteomics show that downregulated genes primarily associated with meiosis and spermiogenesis are poorly expressed in sterile yattle [Citation8,Citation9]. However, the regulatory network that controls germline transcription in yattle is not understood.
Interspecific hybridization causes genomic stress that can lead to genetic instability in offspring. Accumulating evidence indicates that hybrids show extensive modifications of the genome and transcriptome, including novel gene expression pattern changes, chromosomal rearrangements, transposable element mobilization, miRNA and P-element induced wimpy testis (PIWI)-interacting RNA (piRNAs) deficiency, and DNA methylation changes (reviewed in [Citation10]). Epigenetic modifications of DNA methylation, histone modification, and piRNAs are important regulators in spermatogenesis [Citation11–Citation13]. DNA methylation involves the transfer of a methyl group from S-adenosyl-l-methionine to a cytosine residue and generally occurs in CpG dinucleotide sites [Citation14]. DNA methylation in promoters is usually restricted to genes in long-term, stable repressed states [Citation15] and promoter DNA hypermethylation generally suppresses transcription [Citation16]. The methylation state of promoters of genes involved in spermatogenesis has been studied. Hypermethylation of the promoter regions of methylenetetrahydrofolate reductase (MTHFR) and voltage dependent anion channel 2 (VDAC2) down-regulate MTHFR and VDAC2 expression, leading to nonobstructive azoospermia and potentially idiopathic infertility in humans [Citation17,Citation18]. Studies show that some meiosis genes have significantly reduced expression in testes of yattle and have hypermethylation in their promoters and CpG islands (CGI), including DAZL (deleted in azoospermia like), SYCP3 (synaptonemal complex protein 3), PIWIL1 (piwi-like RNA-mediated gene silencing 1), DDX4 (DEAD-box helicase 4), MEI1 (meiotic double-stranded break formation protein 1), and FKBP6 (FK506 binding protein 6) using bisulphite sequencing and RT-qPCR [Citation19–Citation23]. Genome-wide DNA methylation in testes of yattle and cattle has not been investigated.
Two studies demonstrated that the promoter hypermethylation-associated silencing of PIWI/piRNA pathway genes PIWIL1, PIWIL2, and TDRD1 is associated with male infertility [Citation24] and human testicular tumorigenesis [Citation25]. Promoter hypermethylation of PIWI/piRNA pathway genes diminished piRNA production and transposon loci such as LINE-1 are hypomethylated in human testicular tumorigenesis and spermatogenic disorders [Citation24,Citation25]. PIWI proteins are a subfamily of the Argonaute protein family that is expressed mainly in germline cells and forms specific RNA-induced silencing complexes (RISCs) with piRNAs that are termed piRISCs. PiRNAs are a class of small RNAs of 25–32 nucleotides (nt) that are highly abundant in testes [Citation26–Citation28] and predominantly contribute to spermatogenesis in a stage-specific manner [Citation29,Citation30]. During mouse spermatogenesis, two distinct populations of piRNAs associate with the PIWI proteins PIWIL1/MIWI, PIWIL2/MILI, and PIWIL4/MIWI2 at two different developmental stages: prepachytene piRNAs and pachytene piRNAs [Citation31]. Prepachytene piRNAs are mainly derived from transposable elements. They are associated with MILI and MIWI2 in the gonocyte stage and found in leptotene spermatocyte after birth. They have 26- to 27-nt MILI-bound modal lengths [Citation26] and a 28-nt MIWI2-bound modal lengths [Citation32]. Pachytene piRNAs originate from piRNA clusters in various regions of the genome and bind to both MILI and MIWI in pachytene spermatocytes to the round spermatid stage. They have 30-nt MIWI-bound modal lengths and are the majority of piRNAs in adult mouse testes [Citation26,Citation27]. In mammalian testes, piRISCs in germ granules regulate gene silencing and maintain genome stability by directing CpG DNA methylation at transposon loci [Citation13,Citation32–Citation34]. Prepachytene piRNAs direct DNA and histone methylation at transposon sequences during embryogenesis [Citation32,Citation35,Citation36]. Pachytene piRNAs post-transcriptionally repress transposons late in spermatogenesis [Citation33,Citation37] and ensure completion of meiosis and successful spermiogenesis [Citation34,Citation38,Citation39]. These results suggest that DNA methylation linked to the PIWI/piRNA pathway is crucial for regulating germline transcription and may contribute to male infertility disorders.
Whether DNA methylation and piRNA production are associated with bovine HMS is currently unclear. To address this matter, we investigated DNA methylation patterns and piRNA expression in hybrid infertile yattle bulls and fertile cattle and yak bulls using whole genome bisulphite-seq (WGBS) and small RNA-seq (sRNA-seq). Promoter hypermethylation of PIWI/piRNA pathway genes was associated with transcriptional silencing and diminished pachytene piRNA expression in yattle testes. Our data indicated that epigenetic disruption of the PIWI/piRNA pathway may drive bovine HMS.
Results
Global view of WGBS data
Yattle were interspecies hybrid of taurine cattle (B. Taurus, ♂) and yaks (B. grunniens, ♀) used in this study, and males were sterile. To investigate DNA methylation patterns in hybrid infertile bulls and fertile parent bulls, testicular tissues from one infertile yattle and one fertile cattle were used for WGBS. A total of 109.39 G raw bases were generated for the yattle sample and 120.91 G for the cattle sample. After data filtering, 358,346,324 clean reads were generated for yattle and 395,585,807 for cattle. The bisulphite sequencing conversion rates for the groups were as high as 99.9%. Mapping reads covered 77.62% (278,148,416 reads) of the cattle genome (Bos_taurus_UMD_3.1.1) for yattle and 81.65% (322,995,811) for cattle. Sequencing depths were 25.40× for yattle and 29.43× for cattle. Approximately 3% of all genomic cytosine sites were methylated in yattle and cattle. Methylation in yattle and cattle occurred in three sequence contexts: CpG, CHG (where H is A, C, or T), and CHH. Only CpG sites were used for further analysis.
Identification of DMRs between sterility and fertility testes
To highlight aberrant methylated CpG sites in yattle testes and allow downstream processing and analyses, differential methylation regions (DMRs) were determined. We identified 47,252 hypermethylated and 108,231 hypomethylated DMRs for CpG in yattle testes compared to cattle. We used the top 10,000 significant DMRs of CpG for further analysis.
The length distribution of the top 10,000 DMRs was major from 250 bp to 900 bp ()). The 10,000 DMRs were annotated in 24,857 genomic regions including CGI, CGI shores, promoters, transcription start sites (TSSs), 5ʹ untranslated regions (utr5), exons, introns, utr3, transcription termination sites (TESs), and repeats ()). We mapped 70.22% of hypermethylated and 55.83% of hypomethylated DMRs to genomic regions with no gene annotation (intergenic regions). Low percentages of DMRs, specifically 1.96% (446) of hypermethylated and 3.64% (78) of hypomethylated, DMRs mapped to promoter regions of known genes ()). Among the top 10,000 DMRs, 9142 were hypermethylated and 858 were hypomethylated in yattle. The methylation level of DMRs was higher in yattle than in cattle as seen with heatmaps and boxplots (). The chromosomal distribution of hypermethylated and hypomethylated DMRs is shown using Circos in ). DMRs were not evenly distributed in the genome. The ends of chromosomes had preferentially hypermethylated or hypomethylated DMRs in yattle. Chromosomes 1, 7, 8, 9, and X exhibited fewer hypomethylated DMRs. The top 10,000 DMRs of CpG were annotated in 9255 repeats. Mainly short interspersed elements (SINEs), long interspersed elements (LINEs), long terminal repeats (LTRs) retrotransposons were more hypermethylated in yattle than cattle testes ()). LINE1, LINE (RET) and SINE (Core) mRNA were extensively expressed in all tested tissues (), left). RT-qPCR showed that LINE1, LINE (RET) and SINE (Core) mRNA expression were reduced in yattle testes (), right).
Figure 1. Characteristics of differential methylation regions (DMRs) between adult yattle and cattle testes.
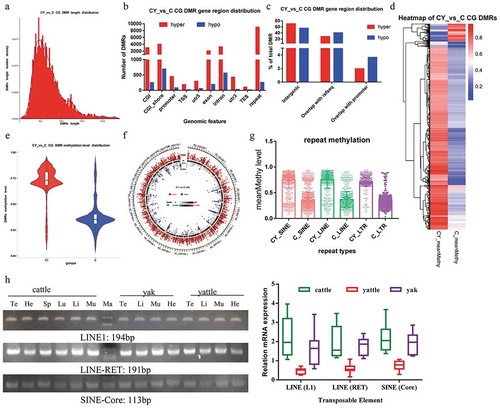
Overlap between methylated promoters and CpG islands
Although the effect of DNA methylation in intergenic regions is less clear, aberrant methylation in promoter regions is linked to altered chromosomal state and thus to gene transcription silencing [Citation16]. In this study, 3206 DMRs (2956 hypermethylated and 250 hypomethylated) were at CGIs and 524 DMRs (446 hypermethylated and 78 hypomethylated) were at promoters. Among them, 264 hypermethylated and 28 hypomethylated gene promoters overlapped with CGIs, and were 59.2% (264/446) of the hypermethylated and 35.9% (28/78) of the hypomethylated promoters. These results indicated that more genes exhibited differential methylation in promoters that were not associated with CGIs. Therefore, aberrant promoter methylation was not restricted to CGIs.
Promoter hypermethylation of piRNA metabolism and spermatogenesis was associated with male sterility in yattle
When regions of a DMR and a specific gene functional element overlapped, the corresponding gene was selected as a DMR-related, differentially methylated gene (DMG). A total of 7710 CpG DMRs were annotated to 2848 DMGs, and more DMGs were observed in gene bodies than in promoters ()). Among the DMGs, 2362 were more prone to hypermethylation in yattle than cattle ()). The 419 DMGs displayed hypermethylation in their promoters ()).
Figure 2. Promoter hypermethylation in yattle involved in PIWI/piRNA pathway and spermatogenesis.
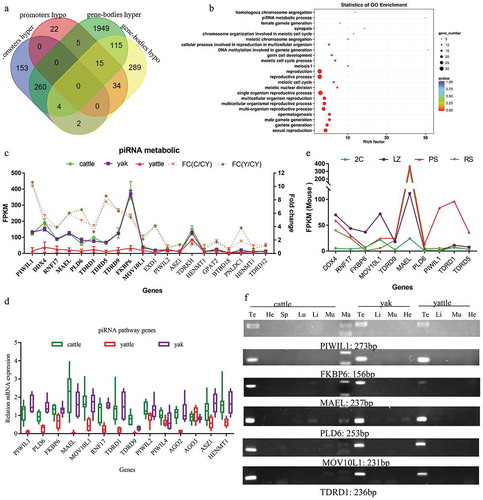
To examine the potential function of 419 promoter-hypermethylated genes, we performed functional analysis. Significantly gene ontology (GO)-enriched terms were in spermatogenesis, meiotic nuclear division, meiosis I, meiotic chromosome segregation, DNA methylation involved in gamete generation, piRNA metabolic process, spermatid development, synapsis, and homologous chromosome segregation (, P < 2.3 × 10−5, FDR P < 0.04). Although only one yattle and one cattle sample were used for WGBS, and not from yak. Our data identified hypermethylated promoters and CGIs for DAZL, SYCP3, PIWIL1, DDX4 and FKBP6 in yattle testes compared to cattle. Hypermethylated promoters and CGIs for these genes were identified in yattle compared to yak and cattle using bisulphite sequencing in previous studies [Citation19–Citation23]. This result gave us confidence in our WGBS data. These findings suggested that hypermethylation of genes at promoters in testes of yattle was involved in spermatogenesis, meiosis I, gamete generation and piRNA metabolic process.
Promoter hypermethylation of PIWI/piRNA pathway genes in yattle were associated with transcriptional silencing
Basing on GOTERM_DIRECT analysis, promoter-hypermethylated genes were significantly enriched for DNA methylation involved in gamete generation (GO: 0043046), piRNA metabolic process (GO: 0034587), chromatoid body (GO: 0033391), and pi-body (GO: 0071546). Fold-enrichment factors were higher than 30 (Table S1). PIWIL1, PIWIL2, PIWIL4, DDX4, MAEL, PLD6, TDRD1, TDRD5, TDRD9, TDRD12, RNF17, FKBP6, MOV10L1, ASZ1, TDRKH, HENMT1, EXD1, GPAT2, PNLDC1, BTBD18, and HSP90 were involved the piRNA pathway (reviewed in [Citation13,Citation40]). Among these genes, well-known piRNA metabolic genes DDX4, MAEL, PLD6, PIWIL2, TDRD1, TDRD5, RNF17, FKBP6, and MOV10L1 were hypermethylated in promoters and CGIs (Table S1). PIWIL1 and TDRD9, were hypermethylated in exons or near CGIs. The mRNA levels of PIWIL1, DDX4, MAEL, PLD6, TDRD1, TDRD5, TDRD9, RNF17, FKBP6 and MOV10L1 genes were significantly lower in yattle than cattle and yaks (), fold change > 2). The mRNA levels were from unpublished RNA-Seq data (), n = 3 for cattle, yaks, and yattle, including same samples as the WGBS) and RT-qPCR data (), n = 8 for cattle, yaks, and yattle, including same samples as the WGBS and sRNA-seq).
In mice, DDX4, RNF17, FKBP6, MOV10L1, TDRD9, MAEL genes were turned on in leptotene and zygotene spermatocytes (LZ; meiotic prophase I early stages). Peak expression of PIWIL1, PLD6, TDRD1, TDRD5 was in pachytene spermatocytes (PS; medium meiotic prophase) (), data from [Citation41]). We were unable to obtain highly purified spermatogenic cell populations to investigate stage-specific expression of PIWI/piRNA genes in bovine. RT-PCR agarose-gel results showed that PIWIL1, PIWIL4, DDX4, RNF17, FKBP6, MOV10L1, MAEL, PLD6, TDRD1, TDRD5 and TDRD9 were uniquely expressed in adult testes of cattle, yak and yattle ()). Overall, promoter hypermethylation of PIWI/piRNA pathway genes was associated with transcriptional silencing in yattle.
Small RNA sequencing data
The disruption of genes associated with the piRNA processing machinery is directly related to spermatogenesis failure due to maturation arrest, resulting in male sterility in mouse models (reviewed in [Citation13,Citation40]). To determine promoter hypermethylation-associated transcriptional silencing of PIWI/piRNA pathway genes and effects on piRNA production, we analysed small RNA-seq (sRNA-seq) libraries from two testicular tissues samples from yattle, cattle, and yaks by deep sequencing. After removing low-quality reads and adaptors, we obtained an average of 19.67 million clean reads from cattle libraries, 17.68 million from yattle libraries, and 20.36 million from yak libraries (Table S2). For sRNA analysis, we used an average 19.32 million reads for cattle, 17.29 million for yattle, and 19.99 million for yaks, ranging from 18 to 35 nt (Table S2). Clean reads from 18 to 35 nt were mapped to the cattle genome (Bos_taurus_UMD_3.1.1). Mapping read coverage was above 91% for cattle and yattle and about 88% for yaks (Table S2).
For each sRNA-seq library, more than 94% reads were 21 to 32 nt. The frequency of percentages of approximately 22- and 30-nt modal lengths were different for yattle, cattle and yaks ()). In cattle and yak fertile bulls, the average frequencies of percentages were 11% of total sRNA reads for 21–24 nt, 19% for 25–28, and 65% for 29–32 modal lengths. However, for hybrid infertile yattle, the average frequency percentage for 21- to 24-nt modal length was up to 51% and the 29- to 32-nt modal length decreased to 23% of total sRNA reads ()).
Figure 3. Reduced piRNA levels in adult yattle testes.
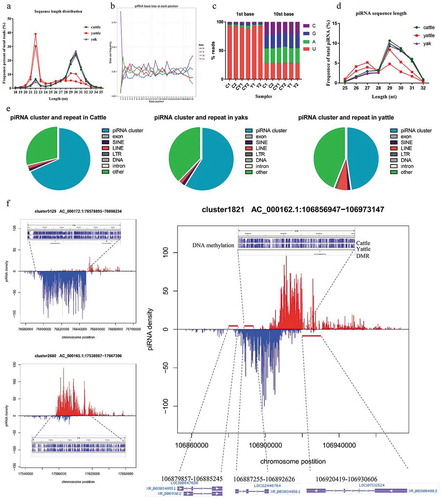
Pachytene piRNA production was reduced in adult yattle testes
In this study, piRNA sequences were identified using a k-mer scheme [Citation42]. We obtained an average of 0.65 million distinct piRNA species for cattle, 0.56 million for yattle, and 0.68 million for yaks. An average of 5.29 million piRNA sequence reads were obtained for cattle, 1.80 million for yattle, and 5.36 million for yaks (Table S2). Primary piRNAs show an antisense and 1 U bias, secondary piRNAs loaded onto AGO3 show 10-nt complementarity at their 5ʹ ends with PIWI-bound piRNAs and possess a 10A bias that are referred to as the ping-pong signature [Citation27,Citation43]. PiRNA has a strong bias for U at the first position in this study ()). More than 93.0% of piRNA sequences began with U at the 5ʹ-end in cattle and yak testes ()). The percent of 1 U bases decreased about 2.25% and 4.93% from two biological replicates in yattle. Meanwhile, the percent of 10A bases ()) and mRNA expression of AGO3 ()) were no significantly different between three groups. These results indicated that piRNA 5′ formation and the ping-pong cycle were normal in cattle, yaks and yattle.
In mice, modal lengths are 26–27 nt for MILI (PIWIL2)-bound piRNAs (prepachytene piRNAs) [Citation26,Citation32,Citation44], 28 nt for MIWI2 (PIWIL4)-bound piRNAs [Citation32], and 30 nt for MIWI (PIWIL1)-bound piRNAs (pachytene piRNAs) [Citation31,Citation45]. Because no datasets were available for small RNAs from different stages of bovine spermatogenesis, we used length of reads to computationally separate prepachytene from pachytene piRNAs. Pachytene piRNAs are the vast majority of piRNAs in adult mouse testes and accumulate rapidly at the pachytene stage of meiosis [Citation31]. More than 73% of piRNAs with 29- to 31-nt modal lengths were present in adult cattle and yak testes. The relative percentages of 29- to 31-nt pachytene piRNAs were diminished in yattle, while 26- to 27-nt modal length piRNAs were increased in yattle testes ()). Together, these data established that pachytene piRNAs production was diminished in adult yattle testes.
Characteristics of piRNA clusters in bovine adult testes
Previous work showed that piRNAs are primarily generated from hundreds of unique genomic loci (piRNA clusters) through a primary processing pathway [Citation27,Citation31,Citation46]. PiRNA clusters contain a ten to thousands of piRNAs, varying in size from 1 to 100 kb [Citation47,Citation48]. Using a coverage cut-off of two and a minimum piRNA cluster length cut-off of 200, we identified 9222 cluster regions from 200 bp to 181.5 kb. Putative cluster-piRNAs mapped uniquely in more than 68% of total piRNAs in cattle and 59% in yaks ()). Also, 3% piRNAs mapped to repeats of SINEs, LINEs, LTRs retrotransposons. Only 48% of putative piRNAs were located in piRNA clusters in yattle. Up to 5.48% of piRNAs were in LINEs in yattle, with 1.22% in cattle and 1.27% in yaks ()).
To increase confidence in piRNA clusters, the cut-off for piRNA clusters was set to transcripts per kilobase of exon model per million mapped reads (TPM) ≥50 for two biological replicates of cattle and yak samples. We identified 288 high-confidence piRNA clusters. The piRNA clusters showed a nonuniform distribution in chromosomes and strong strand asymmetry. Most clusters encoding piRNAs were mainly on only one strand; there were named monodirectional clusters. In some clusters, one strand changed abruptly to another strand; there were named bidirectional clusters [Citation27,Citation30]. For example, cluster 2680 (chr8:17538987–17667396) and cluster 5129 (chr15:76578895–76698234) were found mainly on one strand, while cluster 1821 (chr5:106856947–106973147) was a bidirectional cluster. We found no overlap between DMRs and piRNA cluster regions ()).
Top pachytene piRNA clusters were diminished in adult yattle testes
We used read lengths to computationally separate prepachytene from pachytene piRNA clusters. Clusters were considered prepachytene if the fraction of 26-nt reads was larger than the fraction of 30-nt reads, and vice versa for pachytene clusters [Citation49]. Using only this parameter, we found among 288 clusters, 32 with the length of presumed prepachytene piRNA clusters (26–28 nt), and 256 (88.9%) with the length of presumed pachytene piRNA clusters (29–31 nt) mapped to intergenic regions. As a pachytene piRNA cluster example, the top 50 high-expression piRNA from cluster 5129 were diminished in adult yattle testes. A few piRNAs of cluster 5129 were mapped to repeat sequences ()). Cluster 5129 was in an intergenic region and repeat sequences showed scattered distribution in this genomic region ()). These results were consistent with the finding that pachytene piRNAs are poor in transposon sequences [Citation26,Citation27]. In contrast, the top 50 high-expression piRNAs from prepachytene piRNA cluster 3647 were increased in adult yattle testes ()). The genomic region of cluster 3647 was highly enriched in LINE sequences ()). By read numbers, pachytene clusters had about 10 times more piRNA reads than prepachytene reads.
Figure 4. Cluster-derived piRNAs were diminished in yattle testes.
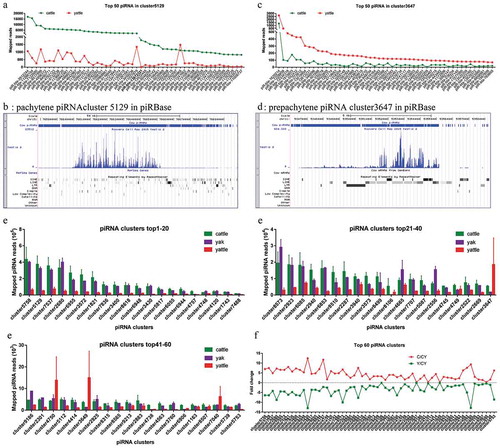
An average of 66.6% piRNAs reads were derived from the 288 high-confidence piRNA clusters in adult cattle and an average of 57.5% from yak testes. Yattle testes had a specific reduced percentage of 40.6% of these cluster-piRNAs. In cattle and yak testes, the top 60 high-confidence piRNA clusters were the majority (>97%) of the 288 cluster-derived piRNAs and were diminished in yattle (), Table S2). In yattle testes, piRNAs from these top 25 piRNA-producing clusters were uniformly reduced by an average of six-fold ()).
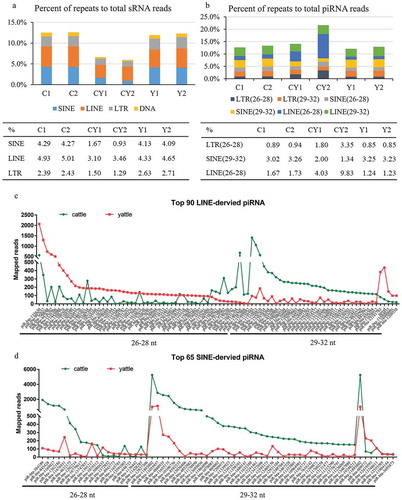
LINE-derived prepachytene piRNAs were increased in yattle testes
PIWI/piRNA complexes recruit methylation factors to silence transposons in male germline stem cells. Thus, the piRNA-related machinery is directly involved in regulation of repetitive elements [Citation32,Citation35,Citation36]. We annotated transposable elements (TEs) in the cattle reference genome. From sRNA libraries, an average 2.32 million reads (12.92% of total mapped reads) from cattle, 1.02 million (6.55%) from yattle, and 2.15 million (12.24%) from yaks were aligned to repeat reads (satellite, SINE, LTR, LINE, and DNA). Mapped SINE, LTR, and LINE reads to total sRNA reads were significantly lower in yattle than in cattle and yaks (), Table S2). The percent of piRNA originating from LINEs was increased in yattle ()). We classified repeat sequences of noncluster regions by length into 26–28 nt and 29–32 nt. The percent of 26-to 28-nt LINE-derived piRNA reads to total piRNA reads was increased in yattle ()). Among the top 90 high expression LINE-derived piRNAs, expression of 26- to 28-nt piRNAs was higher in yattle than cattle ()). Expression of 29- to 32-nt LINE-derived and SINE-derived piRNAs was significantly reduced in yattle ()). We found no overlap between DMRs and LINE-derived and SINE-derived piRNA regions (data not shown).
Figure 5. Transposable element-driving piRNAs in adult cattle, yak and yattle testes.
Discussion
Understanding the process of speciation is a central problem in biology and animal genetics. Interspecies HMS is studied for speciation because sterility is a postzygotic isolation barrier to free gene flow between organisms; thus, effectively isolating distinct species [Citation1,Citation2]. A related question in HMS is why genes that cause fertility in a pure-species genetic background fail to produce fertility in a hybrid genetic background [Citation1]. To address this matter, gene expression arrays have been used to establish associations between misregulation of gene expression and sterility in hybrids between closely related species. In Drosophila, sterile hybrids are more variable in gene expression than fertile males and most misregulated genes are underexpressed [Citation50,Citation51]. Interspecies sterile Drosophila hybrids have mostly postmeiotic problems during spermatogenesis. This connection between sterility phenotype and tissue-specific misregulation of gene expression suggests that downregulation of postmeiotic spermatogenesis genes in sterile hybrids is a causative factor of HMS [Citation1,Citation51].
We hypothesized that gene repression by hypermethylation of male meiosis specific genes, particularly the PIWI/piRNA pathway genes for pachytene piRNA production PIWIL1, DDX4, PLD6, MAEL, FKBP6, RNF17, MOV10L1, TDRD1, TDRD5, and TDRD9, probably drive bovine HMS. WGBS and GOTERM_DIRECT biological process analysis showed promoter hypermethylated genes in sterile yattle were significantly enriched in ‘DNA methylation involved in gamete generation (GO: 0043046)’ and ‘piRNA metabolic process (GO: 0034587)’ (), Table S1). Therefore, abnormal DNA methylation impacted spermatogenesis failure in yattle. Gene-promoter DNA hypermethylation generally suppresses transcription [Citation16]. In several mutant mouse models, PIWIL1 [Citation27], DDX4 [Citation52,Citation53], MAEL [Citation54], PLD6 [Citation55], TDRD1 [Citation56,Citation57], TDRD5 [Citation58], TDRD9 [Citation52], RNF17 [Citation59], FKBP6 [Citation60], MOV10L1 [Citation61,Citation62], BTBD18 [Citation63] and TDRKH [Citation64] are important in pachytene piRNA production and spermatogenesis. Pachytene piRNAs accumulate rapidly at the pachytene stage of meiosis [Citation31]. Pachytene piRNAs may promote spermatogenesis by regulating mRNAs and long noncoding RNAs in mouse testes and ensure completion of meiosis and successful spermiogenesis [Citation34,Citation38,Citation39]. Indeed, promoter and CGI hypermethylation associated with silencing PIWI/piRNA pathway genes PIWIL1, DDX4, PLD6, MAEL, FKBP6, RNF17, MOV10L1, TDRD1, TDRD5, and TDRD9 (), resulted in diminished production of pachytene piRNAs in yattle testes () and ). This molecular connection with histologic observations of seminiferous tubules indicated that male infertility of yattle may be due to meiosis arrest at about the pachytene stage [Citation7]. In contrast, PIWIL2, PIWIL4, EXD1, TDRD12 are involved in prepachytene piRNA production [Citation65–Citation68]. PNLDC1 is essential for piRNA 3'-end trimming and transposon silencing during spermatogenesis in mice [Citation69]. HENMT1 is required for piRNA 3'-methylation and stability [Citation70]. Corresponding to the functions of these genes, mRNA levels of BTBD18, TDRKH, PIWIL2, PIWIL4, EXD1, TDRD12, PNLDC1, HENMT1 and piRNA length were not significantly different between yattle, cattle and yaks () and )). These results indicated that promoter hypermethylation of specific pachytene piRNA-biogenesis genes had a direct impact on pachytene piRNA production. In addition, all loss-of-function mutations of PIWI/piRNA pathway genes reported so far in mice lead to male sterility, while females are fertile. These results indicate the importance of this pathway in male germline development in mammals (reviewed in [Citation13]). PIWIL1, FKBP6, DDX4, MAEL, TDRD5 and TDRD9 were uniquely expressed in adult testes of cattle, yak and yattle ()). This result may explain why males were sterile while females were fertile in yattle. Finally, PIWI/piRNA pathway genes predominantly contribute to spermatogenesis in a stage-specific manner [Citation29,Citation30]. Transcriptome analysis of highly purified mouse spermatogenic cell populations showed that DDX4, RNF17, FKBP6, MOV10L1, TDRD9, MAEL genes were turned on in leptotene and zygotene spermatocytes (meiotic prophase I early stages), while the expression peak of PIWIL1, PLD6, TDRD1, TDRD5 was in pachytene spermatocytes (medium meiotic prophase) [Citation41] ()). Although no bovine datasets of gene expression from different stages of spermatogenesis were available, higher expression is seen for PIWIL1 in leptotene and zygotene spermatocytes and PIWIL2 in spermatogonial stem cells and leptotene spermatocytes during human spermatogenesis [Citation71]. Thus, PIWI/piRNA pathway genes may conservatively contribute to bovine spermatogenesis in a stage-specific manner. Overall, promoter hypermethylation of male-specific and pachytene piRNA biogenesis-specific genes have a direct impact on pachytene piRNA production, which may have contributed to pachytene arrest in yattle. Studies report that promoter hypermethylation-associated silencing of PIWI/piRNA pathway genes PIWIL1, PIWIL2, and TDRD1 and piRNA production are associated with male infertility [Citation24] and human testicular tumorigenesis [Citation25]. Our study showed that DNA hypermethylation of PIWI/piRNA pathway genes was associated with diminished pachytene piRNA production in bovine HMS.
The Dobzhansky-Muller incompatibility model explains HMS as the result of incompatible epistatic interactions between divergent genomes. This model of genetic incompatibilities has long provided a useful theoretical framework for speciation genetics. However, only one mammalian gene, PRDM9, is responsible for sterility phenotypes in male hybrids of certain mouse subspecies [Citation72,Citation73]. In addition, interspecific hybridization is a genomic stress condition that leads to activation of TEs in animals. TE insertions can disrupt genes or regulatory sequences and can cause chromosomal rearrangements, threatening host genome integrity [Citation74]. To mitigate these deleterious effects, mechanisms of TE control are especially important in the germline. In mice, CpG DNA methylation and PIWI/piRNA complexes safeguard the male germline genome from TEs [Citation75]. From gonocytes to leptotene spermatocytes, MILI and MIWI2 are associated with transposon sequence-rich TE piRNAs, and guide specific de novo DNA methylation to suppress TEs and maintains germline genome integrity [Citation32,Citation36,Citation48]. In contrast, MIWI and MILI are highly expressed in pachytene spermatocytes and associated with pachytene piRNA species [Citation76]; both are essential for adult spermatogenesis in mice [Citation77,Citation78]. MIWI and MILI carry out LINE1 transposon silencing functions in pachytene spermatocytes, likely through slicer activity in a piRNA-independent manner [Citation33,Citation37]. In our study, we found that 26- to 27-nt modal length piRNAs ()) and 26–28 nt LINE-driver piRNAs expression () and )) were increased in yattle. Meanwhile, the LINEs and SINEs were hypermethylated ()) and the LINE and SINE mRNA expression ()) were reduced in yattle. We found no overlap between top 10,000 DMRs and 288 high-confidence piRNA cluster regions ()). PIWIL1 expression and pachytene piRNAs production were diminished in yattle testes. These results suggested higher TE piRNAs expression may affect TE DNA methylation and lead to suppress TEs expression in yattle. However, the regulation mechanisms between TE piRNAs and DNA methylation could not be confirmed in the whole testis data.
Conclusions
In this work, we used WGBS and sRNA-seq to investigate DNA methylation and piRNA production differences in the testes of yattle, cattle and yaks. Promoter hypermethylation genes were significantly enriched in DNA methylation involved in gamete generation, piRNA metabolic process, chromatoid body, and pi-body. We confirmed that promoter hypermethylation-associated silencing of PIWI/piRNA pathway genes had the potential to reduce pachytene piRNA production in yattle. Our data indicated that epigenetic disruption of the entire PIWI/piRNA pathway was involved with bovine HMS. Gene repression by hypermethylation of male meiosis-specific genes and PIWI/piRNA pathway genes is probably a driver of bovine HMS.
Materials and methods
Sample collection
Testes tissue samples from taurine cattle (B. taurus, n = 8), yak (B. grunniens, n = 8), and yattle (B. taurus ♂ × B. grunniens ♀, n = 8) were used to obtain genomic DNA and total RNA. Detail information is in our previous study [Citation7]. Yaks and yattle bulls were from the Hongyuan Bovine Breeding Farm in Sichuan, China. Hybrid taurine cattle were from a beef farm in Chongqing, China. The genetic relationship between individuals was unclear. Animals were slaughtered after sexual maturity (4.5 years for yaks and yattle and 2.5 years for taurine cattle). Fat and fascia surrounding testes were resected and two cross-sections of testicular tissue from the middle of the testes were obtained by fine-scale dissection. Crosscut slice was snap frozen in liquid nitrogen for transport and stored at −80°C. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Southwest University (Chongqing, China).
Whole-genome bisulphite sequencing
We analysed one cattle (fertile) and one yattle (infertile) testicular DNA sample for genome-wide DNA methylation by WGBS. DNA extraction, library preparation and sequencing was performed at Novogene (Beijing, China). DNA was purified with tissue DNA extraction kits (Tiangen, cat: DP304). DNA concentrations were determined by a Qubit 2.0 fluorometer (Life Technologies, USA). Libraries were prepared from 5.2 mg genomic DNA spiked with 26 ng lambda DNA fragmented by sonication to 200–300bp with a Covaris S220. Libraries were prepared using Illumina TruSeq DNA methylation kits (Illumina, USA). DNA was terminal-repaired and A-ligated, followed by ligation of methylated adapters according to the Illumina TruSeq protocol. Libraries were bisulphite converted twice using EZ DNA methylation gold kits (Zymo Research, cat: D5005). Library concentration was quantified by a Qubit 2.0 flurometer (Life Technologies) and quantitative PCR. Insert sizes were determined on an Agilent Bioanalyzer 2100 system. Libraries were analysed in separate lanes on a HiSeq 4000 (Illumina, USA) in paired-end mode using 150-bp sequence length. Raw sequencing data were deposited to NCBI in SRA.
Processing WGBS data
Sequence data processing was according to procedures of Novogene (Beijing). Following sequencing, we used FastQC (fastqc_v0.11.5, Babraham Institute, UK) to perform basic statistics on raw read quality. We obtained high-quality reads after trimming low-quality bases from the 3′ end and adapter sequences from the 5′ end using Trimmomatic 0.36 software and parameters SLIDINGWINDOW: 4:15; LEADING:3, TRAILING:3; ILLUMINACLIP: adapter.fa: 2:30:10; and MINLEN:36. Resulting clean reads were aligned to the reference B. taurus cattle genome (UMD_3.1.1,) using Bismark alignment software (version 0.16.3) [Citation79], using the stringent option -X 70 with slightly relaxed parameters for mapping (score-min L, 0, −0.2). Initial data analysis was by Bowtie2 [Citation80]. Methylation level (ML) for each C site was defined as: ML = mC/(mC+umC). This study focused on CpG sites.
DMRs were identified based on a P-value cut-off of 0.05 using DDS software [Citation81]. A sliding-window approach (selecting every 200 bp as a window with 100-bp as step lengths) was used to identify DMRs in the genome. Regions with length greater than 50 bp, corrected P-values less than 1e-05, and read coverage greater than 3 were regarded as DMRs. According to DMR distribution through the genome, DMRs were annotated as promoter, exon, intron, CGI, CGI shore, repeat, TSS, TES. We defined genes related to DMRs as having gene body regions (from TSS to TES) or promoter regions (2kb upstream from the TSS) that overlapped DMRs. When a region where a DMR and a specific gene function element overlapped, the corresponding gene was selected as the DMR-related DMG.
GO enrichment and KEGG pathway analyses were conducted using the DAVID 6.8 (https://david.ncifcrf.gov/) for DMGs to investigate biological processes and functions [Citation82]. GO terms and KEGG were corrected using the Benjamini method. Corrected P-values less than 0.05 were considered significantly enriched for DMGs. Graphs for mRNA expression from mRNA-seq were produced with GraphPad Prism 7.0.
Small RNA sequencing
We analysed testicular RNA sample from fertile cattle (n = 2) and yaks (n = 2), and infertile yattle (n = 2) for miRNA and piRNA using small RNA-seq. Total RNA extraction, library preparation and sequencing were performed at Novogene (Beijing, China). Total RNA from testes were extracted using TRIzol reagent (Invitrogen, cat: 15596026). The quantity and purity of total RNA were analysed by Nanodrop one (ThermoFisher Scientific, USA) and agarose gel electrophoresis. RNA concentration was measured using Qubit RNA Assay Kits in a Qubit 2.0 fluorometer (Life Technologies). RNA integrity was assessed using RNA Nano 6000 Assay Kits for the Agilent Bioanalyzer 2100 system (Agilent Technologies, USA) with RNA integrity number >8.0. Total RNA at 3 μg per sample was used as input for small RNA libraries. Sequencing libraries were generated using NEBNext Multiplex Small RNA Library Prep Sets for Illumina (NEB, cat: E7300 S). Libraries were purified on 8% polyacrylamide gels (100 V, 80 min) and quantified using an Agilent Bioanalyzer 2100 system using DNA High Sensitivity Chips. Library preparations were sequenced on an Illumina Hiseq 2500 platform, and 125-bp single-end reads generated. Raw data were deposited to NCBI in SRA.
Processing of sRNA sequencing data
Sequence data processing was according to procedures at Novogene (Beijing). Clean reads were obtained by removing reads containing poly-N; with 5′-adapter contaminants; without 3′-adapters or insert tags; containing poly A, T, G or C; or having low quality in raw data. Simultaneously, Q20, Q30, and GC-content of raw data were calculated. A length range of 18–35 nt for clean reads was selected for all downstream analyses.
Small RNA tags were mapped to the reference sequence B. taurus cattle genome (UMD_3.1.1) using Bowtie (v0.12.9, -v 0 v 0 -k) [Citation83] without mismatches to analyse expression and distribution. Small RNAs were mapped to single annotations by performing annotations in the order: miRNAs > rRNAs > tRNAs > snRNAs > snoRNAs > repeats > genes > novel miRNAs. Mapped small RNA tags were used to search for known miRNAs using miRBase21.0 as a reference.
Processing of piRNA and piRNA clusters
PiRNA and piRNA clusters were analysed based on ‘repeat’ and ‘other’ sRNA data of 25–32 nt. PiRNA sequences were identified using a k-mer scheme [Citation42]. To obtain the first base preference map of piRNAs of different lengths, AUCG statistics were performed on identified piRNA sequences. Identified piRNA sequences were aligned with the reference genome to obtain the distribution of piRNAs on the chromosome using Bowtie (-v 0 -k 1). The coverage of piRNA on the reference sequence was obtained using samtools depth. PiRNA-generating genes were obtained from the sam file based on alignments of piRNAs mapped to exons and with coverage ≥ 5. We annotated TEs in the cattle reference genome using RepeatMasker (version open-4.0.1).
Parameters for identifying piRNA clusters were coverage ≥ 2 piRNAs, minimum piRNA cluster length 200 nt, and threshold distance of interval length 10,000 nt. Neighbouring genes within piRNA clusters were detected based on the range of each piRNA cluster, considering sequences 2000 bp upstream and downstream. Differential expression of piRNA clusters among yattle, cattle and yaks testes were measured with DESeq2 [Citation84], setting padj < 0.05 as the cut-off. The piRNA name was identified in piRBase using the ‘Seq To Name’ tool [Citation85]. Graphs for first-base bias and piRNA cluster expression differences were produced with GraphPad Prism 7.0.
mRNA expression arrays
mRNA expression profiles of target genes in different tissues was performed from testes, muscle, liver, spleen, heart, and lung tissues using reverse transcription PCR (RT-PCR) as described in previous studies [Citation7,Citation86]. The mRNA expression of the target genes was determined by reverse transcription quantitative PCR (RT-qPCR) in cattle, yaks, and yattle testes as described in previous studies [Citation7,Citation86]. Primers for all these genes were designed using Primer3 software and synthetized by Genewiz (Suzhou, China, Table S3). Relative mRNA expression was analysed with the system’s relative quantification software based on the 2delta-delta-Ct method (Bio-Rad). Graphs for mRNA expression differences were produced with GraphPad Prism 7.0.
Author summary
Domestic yaks (Bos grunniens) and domestic taurine cattle (Bos taurus) are closely related species. The interspecies F1 hybrid yattle and F2 backcross males are sterile. Previous studies showed that the male infertility of yattle may be due to arrest of meiosis at about the pachytene stage and meiosis gene expression is dysregulated. Epigenetic modifications of DNA methylation, histone modification, and piRNAs are important regulators in spermatogenesis. In this study, we investigated DNA methylation patterns and piRNA expression in adult testes in hybrid infertile yattle bulls and fertile cattle and yak bulls using whole genome bisulphite-seq and small RNA-seq. We confirmed that promoter DNA hypermethylation-associated silencing of PIWI/piRNA pathway genes had the potential to reduce pachytene piRNA production during spermatogenesis in yattle. Our data indicated that epigenetic disruption of the entire PIWI/piRNA pathway was involved with yattle male sterility. DNA hypermethylation of male meiosis-specific genes and disruption of piRNA production contributed to unsuccessful germ cell development that may drive bovine hybrid male sterility.
Authors’ contributions
GZ and FZ conceived and supervised the project. GZ, LW, HC, JG, YW, and JZ performed most experiments. JG, JZ, ZL, and WH provided support or technical assistance. GZ, LW, YW, and ZL contributed to bioinformatics data. GZ wrote the manuscript with major inputs from LW, YW, JZ, and FZ. All authors approved the final manuscript.
Ethics declaration
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Southwest University (Chongqing, China).
Supplemental Material
Download MS Word (21 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Civetta A. Misregulation of gene expression and sterility in interspecies hybrids: causal links and alternative hypotheses. J Mol Evol. 2016 May;82(4–5):176–182.
- Mallet J. Hybrid speciation. Nature. 2007 Mar 15;446(7133):279–283.
- Maheshwari S, Barbash DA. The genetics of hybrid incompatibilities. Annu Rev Genet. 2011;45:331–355.
- Qiu Q, Zhang G, Ma T, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012 Jul 1;44(8):946–949.
- Lou YN, Liu WJ, Wang CL, et al. Histological evaluation and PRDM9 expression level in the testis of sterile male cattle-yaks. Livestock Sci. 2014;160:208–213.
- Yan P, Xiang L, Guo X, et al. The low expression of Dmrt7 is associated with spermatogenic arrest in cattle-yak. Mol Biol Rep. 2014 Nov;41(11):7255–7263.
- Zhang GW, Wu Y, Luo Z, et al. Comparison of Y-chromosome-linked TSPY, TSPY2, and PRAMEY genes in Taurus cattle, yaks, and interspecific hybrid bulls. J Dairy Sci. 2019 May 15;102(7):6263–6275.
- Cai X, Yu S, Mipam T, et al. Comparative analysis of testis transcriptomes associated with male infertility in cattleyak. Theriogenology. 2017 Jan;15(88):28–42.
- Yu S, Cai X, Sun L, et al. Comparative iTRAQ proteomics revealed proteins associated with spermatogenic arrest of cattleyak. J Proteomics. 2016 Jun;16(142):102–113.
- Michalak P. Epigenetic, transposon and small RNA determinants of hybrid dysfunctions. Heredity (Edinb). 2009 Jan;102(1):45–50.
- Stewart KR, Veselovska L, Kelsey G. Establishment and functions of DNA methylation in the germline. Epigenomics. 2016 Oct;8(10):1399–1413.
- Rajender S, Avery K, Agarwal A. Epigenetics, spermatogenesis and male infertility. Mutat Res. 2011 May-Jun;727(3):62–71.
- Iwasaki YW, Siomi MC, Siomi H. PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem. 2015;84:405–433.
- Chen T, Li E. Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol. 2004;60:55–89.
- Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001 Aug 10;293(5532):1068–1070.
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012 May 29;13(7):484–492.
- Khazamipour N, Noruzinia M, Fatehmanesh P, et al. MTHFR promoter hypermethylation in testicular biopsies of patients with non-obstructive azoospermia: the role of epigenetics in male infertility. Hum Reprod. 2009 Sep;24(9):2361–2364.
- Xu A, Hua Y, Zhang J, et al. Abnormal hypermethylation of the VDAC2 promoter is a potential cause of idiopathic asthenospermia in men. Sci Rep. 2016 Nov 28;6:37836.
- Liu Z, Li Q, Pan Z, et al. Comparative analysis on mRNA expression level and methylation status of DAZL gene between cattle-yaks and their parents. Anim Reprod Sci. 2011 Jul;126(3–4):258–264.
- Wang S, Pan Z, Zhang Q, et al. Differential mRNA expression and promoter methylation status of SYCP3 gene in testes of yaks and cattle-yaks. Reprod Domest Anim. 2012 Jun;47(3):455–462.
- Gu Y, Li Q, Pan Z, et al. Molecular cloning, gene expression and methylation status analysis of PIWIL1 in cattle-yaks and the parental generation. Anim Reprod Sci. 2013 Aug;140(3–4):131–137.
- Luo H, Zhou Y, Li Y, et al. Splice variants and promoter methylation status of the Bovine Vasa Homology (Bvh) gene may be involved in bull spermatogenesis. BMC Genet. 2013 Jul;1(14):58.
- Li B, Luo H, Weng Q, et al. Differential DNA methylation of the meiosis-specific gene FKBP6 in testes of yak and cattle-yak hybrids. Reprod Domest Anim. 2016 Dec;51(6):1030–1038.
- Heyn H, Ferreira HJ, Bassas L, et al. Epigenetic disruption of the PIWI pathway in human spermatogenic disorders. PloS One. 2012;7(10):e47892.
- Ferreira HJ, Heyn H, Garcia Del Muro X, et al. Epigenetic loss of the PIWI/piRNA machinery in human testicular tumorigenesis. Epigenetics. 2014 Jan;9(1):113–118.
- Aravin A, Gaidatzis D, Pfeffer S, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006 Jul 13;442(7099):203–207.
- Girard A, Sachidanandam R, Hannon GJ, et al. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006 Jul 13;442(7099):199–202.
- Lau NC, Seto AG, Kim J, et al. Characterization of the piRNA complex from rat testes. Science. 2006 Jul 21;313(5785):363–367.
- de Mateo S, Sassone-Corsi P. Regulation of spermatogenesis by small non-coding RNAs: role of the germ granule. Semin Cell Dev Biol. 2014 May;29:84–92.
- Kim VN. Small RNAs just got bigger: piwi-interacting RNAs (piRNAs) in mammalian testes. Genes Dev. 2006 Aug 1;20(15):1993–1997.
- Li XZ, Roy CK, Dong X, et al. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell. 2013 Apr 11;50(1):67–81.
- Aravin AA, Sachidanandam R, Bourc’his D, et al. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 2008 Sep 26;31(6):785–799.
- Reuter M, Berninger P, Chuma S, et al. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature. 2011 Nov 27;480(7376):264–267.
- Zhang P, Kang JY, Gou LT, et al. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res. 2015 Feb;25(2):193–207.
- Pezic D, Manakov SA, Sachidanandam R, et al. piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev. 2014 Jul 1;28(13):1410–1428.
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 2008 Apr 1;22(7):908–917.
- Di Giacomo M, Comazzetto S, Saini H, et al. Multiple epigenetic mechanisms and the piRNA pathway enforce LINE1 silencing during adult spermatogenesis. Mol Cell. 2013 May 23;50(4):601–608.
- Goh WS, Falciatori I, Tam OH, et al. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev. 2015 May 15;29(10):1032–1044.
- Gou LT, Dai P, Yang JH, et al. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 2014 Jun;24(6):680–700.
- Ozata DM, Gainetdinov I, Zoch A, et al. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet. 2019 Feb;20(2):89–108.
- da Cruz I, Rodriguez-Casuriaga R, Santinaque FF, et al. Transcriptome analysis of highly purified mouse spermatogenic cell populations: gene expression signatures switch from meiotic-to postmeiotic-related processes at pachytene stage. BMC Genomics. 2016 Apr;19(17):294.
- Zhang Y, Wang X, Kang L. A k-mer scheme to predict piRNAs and characterize locust piRNAs. Bioinformatics. 2011 Mar 15;27(6):771–776.
- Brennecke J, Aravin AA, Stark A, et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007 Mar 23;128(6):1089–1103.
- Robine N, Lau NC, Balla S, et al. A broadly conserved pathway generates 3ʹUTR-directed primary piRNAs. Curr Biol. 2009 Dec 29;19(24):2066–2076.
- Li XZ, Roy CK, Moore MJ, et al. Defining piRNA primary transcripts. Cell Cycle. 2013 Jun 1;12(11):1657–1658.
- Gan H, Lin X, Zhang Z, et al. piRNA profiling during specific stages of mouse spermatogenesis. Rna. 2011 Jul;17(7):1191–1203.
- O’Donnell KA, Boeke JD. Mighty Piwis defend the germline against genome intruders. Cell. 2007 Apr 6;129(1):37–44.
- Aravin AA, Sachidanandam R, Girard A, et al. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007 May 4;316(5825):744–747.
- Gainetdinov I, Skvortsova Y, Kondratieva S, et al. Two modes of targeting transposable elements by piRNA pathway in human testis. Rna. 2017 Nov;23(11):1614–1625.
- Michalak P, Noor MA. Genome-wide patterns of expression in Drosophila pure species and hybrid males. Mol Biol Evol. 2003 Jul;20(7):1070–1076.
- Michalak P, Noor MA. Association of misexpression with sterility in hybrids of Drosophila simulansand D. mauritiana. J Mol Evol. 2004 Aug;59(2):277–282.
- Wenda JM, Homolka D, Yang Z, et al. Distinct roles of RNA helicases MVH and TDRD9 in PIWI slicing-triggered mammalian piRNA biogenesis and function. Dev Cell. 2017 Jun 19;41(6):623–637 e9.
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, et al. MVH in piRNA processing and gene silencing of retrotransposons. Genes Dev. 2010 May;24(9):887–892.
- Castaneda J, Genzor P, van der Heijden GW, et al. Reduced pachytene piRNAs and translation underlie spermiogenic arrest in Maelstrom mutant mice. Embo J. 2014 Sep 17;33(18):1999–2019.
- Gao Q, Frohman MA. Roles for the lipid-signaling enzyme MitoPLD in mitochondrial dynamics, piRNA biogenesis, and spermatogenesis. BMB Rep. 2012 Jan;45(1):7–13.
- Wang J, Saxe JP, Tanaka T, et al. Mili interacts with tudor domain-containing protein 1 in regulating spermatogenesis. Curr Biol. 2009 Apr 28;19(8):640–644.
- Reuter M, Chuma S, Tanaka T, et al. Loss of the Mili-interacting Tudor domain-containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat Struct Mol Biol. 2009 Jun;16(6):639–646.
- Ding D, Liu J, Midic U, et al. TDRD5 binds piRNA precursors and selectively enhances pachytene piRNA processing in mice. Nat Commun. 2018 Jan 9;9(1):127.
- Wasik KA, Tam OH, Knott SR, et al. RNF17 blocks promiscuous activity of PIWI proteins in mouse testes. Genes Dev. 2015 Jul 1;29(13):1403–1415.
- Xiol J, Cora E, Koglgruber R, et al. A role for Fkbp6 and the chaperone machinery in piRNA amplification and transposon silencing. Mol Cell. 2012 Sep 28;47(6):970–979.
- Zheng K, Xiol J, Reuter M, et al. Mouse MOV10L1 associates with Piwi proteins and is an essential component of the Piwi-interacting RNA (piRNA) pathway. Proc Natl Acad Sci U S A. 2010 Jun 29;107(26):11841–11846.
- Vourekas A, Zheng K, Fu Q, et al. The RNA helicase MOV10L1 binds piRNA precursors to initiate piRNA processing. Genes Dev. 2015 Mar 15;29(6):617–629.
- Zhou L, Canagarajah B, Zhao Y, et al. BTBD18 regulates a subset of piRNA-generating loci through transcription elongation in mice. Dev Cell. 2017 Mar 13;40(5):453–466 e5.
- Ding D, Liu J, Dong K, et al. Mitochondrial membrane-based initial separation of MIWI and MILI functions during pachytene piRNA biogenesis. Nucleic Acids Res. 2019 Mar 18;47(5):2594–2608.
- Pandey RR, Homolka D, Olotu O, et al. Exonuclease domain-containing 1 enhances MIWI2 piRNA biogenesis via its interaction with TDRD12. Cell Rep. 2018 Sep 25;24(13):3423–3432 e4.
- Carmell MA, Girard A, van de Kant HJ, et al. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell. 2007 Apr;12(4):503–514.
- Pandey RR, Tokuzawa Y, Yang Z, et al. Tudor domain containing 12 (TDRD12) is essential for secondary PIWI interacting RNA biogenesis in mice. Proc Natl Acad Sci U S A. 2013 Oct 8;110(41):16492–16497.
- Manakov SA, Pezic D, Marinov GK, et al. MIWI2 and MILI have differential effects on piRNA biogenesis and DNA methylation. Cell Rep. 2015 Aug 25;12(8):1234–1243.
- Ding D, Liu J, Dong K, et al. PNLDC1 is essential for piRNA 3ʹ end trimming and transposon silencing during spermatogenesis in mice. Nat Commun. 2017 Oct 10;8(1):819.
- Lim SL, Qu ZP, Kortschak RD, et al. HENMT1 and piRNA stability are required for adult male germ cell transposon repression and to define the spermatogenic program in the mouse. PLoS Genet. 2015 Oct;11(10):e1005620.
- Wang M, Liu X, Chang G, et al. Single-cell RNA sequencing analysis reveals sequential cell fate transition during human spermatogenesis. Cell Stem Cell. 2018 Oct 4;23(4):599–614 e4.
- Mihola O, Trachtulec Z, Vlcek C, et al. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science. 2009 Jan 16;323(5912):373–375.
- Davies B, Hatton E, Altemose N, et al. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature. 2016 Feb 11;530(7589):171–176.
- Hedges DJ, Deininger PL. Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res. 2007 Mar 1;616(1–2):46–59.
- Ishizu H, Siomi H, Siomi MC. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012 Nov 1;26(21):2361–2373.
- Vourekas A, Zheng Q, Alexiou P, et al. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat Struct Mol Biol. 2012 Aug;19(8):773–781.
- Deng W, Lin H. miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev Cell. 2002 Jun;2(6):819–830.
- Kuramochi-Miyagawa S, Kimura T, Ijiri TW, et al. Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development. 2004 Feb;131(4):839–849.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011 Jun 1;27(11):1571–1572.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012 Mar 4;9(4):357–359.
- Feng H, Conneely KN, Wu H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014 Apr;42(8):e69.
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
- Wang J, Zhang P, Lu Y, et al. piRBase: a comprehensive database of piRNA sequences. Nucleic Acids Res. 2019 Jan 8;47(D1):D175–D180.
- Wu Y, Zhang WX, Zuo F, et al. Comparison of mRNA expression from Y-chromosome X-degenerate region genes in taurine cattle, yaks and interspecific hybrid bulls. Animal genetics. 2019 Dec;50(6):740-743.