
ABSTRACT
While changes in DNA methylation are known to occur early in breast carcinogenesis and the landscape of breast tumour DNA methylation is profoundly altered compared with normal tissue, there have been limited efforts to identify DNA methylation field cancerization effects in histologically normal breast tissue adjacent to tumour. Matched tumour, histologically normal tissue of the ipsilateral breast (ipsilateral-normal), and histologically normal tissue of the contralateral breast (contralateral-normal) were obtained from nine women undergoing bilateral mastectomy. Laser capture microdissection was used to select epithelial cells from normal tissue, and neoplastic cells from tumour for genome-scale measures of DNA methylation with the Illumina HumanMethylationEPIC array. We identified substantially more CpG loci that were differentially methylated between contralateral-normal and tumour (63,271 CpG loci q < 0.01), than between ipsilateral-normal and tumour (38,346 CpG loci q < 0.01). We identified differential methylation in ipsilateral-normal relative to contralateral-normal tissue (9,562 CpG loci p < 0.01). In this comparison, hypomethylated loci were significantly enriched for breast cancer-relevant transcription factor binding sites including those for ESR1, FoxA1, and GATA3 and hypermethylated loci were significantly enriched for CpG island shore regions. In addition, progression of shore hypermethylation was observed in tumours compared to matched ipsilateral normal tissue, and these alterations tracked to several well-established tumour suppressor genes. Our results indicate an epigenetic field effect in surrounding histologically normal tissue. This work offers an opportunity to focus investigations of early DNA methylation alterations in breast carcinogenesis and potentially develop epigenetic biomarkers of disease risk.
Abbreviations
DCIS: ductal carcinoma in situ; GO: gene ontology; OR: odds ratio; CI: confidence interval; TFBS: transcription factor binding site; LOLA: Locus Overlap Analysis
Introduction
Breast cancer is the most diagnosed non-keratinocyte cancer and is responsible for the second highest number of cancer-related deaths among women in the United States, with over 40,000 women estimated to die from breast cancer in 2019 in the United States alone [Citation1]. It has been shown that the breast methylome varies greatly between histologically normal tissue and tumour tissue [Citation2], and such variation is related in part to age, parity, and alcohol consumption, among other factors [Citation3,Citation4]. Work assessing the methylome of ductal carcinoma in situ (DCIS) has demonstrated that alterations to DNA methylation occur early in breast carcinogenesis [Citation5,Citation6]. Due to the invasive nature of sample collection, molecular studies are conducted as a comparison between tumour and histologically normal tissue adjacent to the tumour (subsequently referred to as adjacent normal) tissue. However, given that alterations to DNA methylation have been identified at the early stages of breast carcinogenesis, it is currently unknown whether adjacent normal tissue is reflective of normal breast epithelium at the molecular level. This limitation may lead to an incomplete understanding of tumour development or an attenuation of the measured magnitude of epigenetic differences.
If a field effect of the tumour on adjacent histologically normal tissue occurs in breast cancer, the use of adjacent normal tissue as a referent sample in molecular studies would inhibit our ability to identify the earliest molecular alterations that occur in carcinogenesis. Therefore, assessment of possible field cancerization effects in breast cancer is critical to informing our understanding of early carcinogenesis.
A recent study demonstrated evidence of alterations to DNA methylation related to tumorigenesis in histologically normal tissue adjacent to breast tumours, suggestive of a field cancerization effect in breast cancer [Citation7]. However, this study was conducted comparing adjacent normal tissue in women with breast cancer to normal breast tissue in women without breast cancer. While comparing cancer-free control tissue from one set of study participants with adjacent normal tissue from an independent set of participants provides initial evidence of field cancerization effects, it may obscure the extent to which DNA methylation profiles reflect individual differences/exposures and thereby modify the field effect profile. To our knowledge, no study to-date has shown evidence of field effects in breast cancer using subject-matched tumour tissue, histologically normal tissue from the ipsilateral breast (ipsilateral-normal), and histologically normal tissue from the contralateral breast (contralateral-normal). Furthermore, most prior research assessing alterations to DNA methylation in breast tumours has leveraged bulk tissue samples which may confound results due to underlying differences in the cellular composition of collected tissue. In the present study, we present evidence of field effects in breast cancer, using subject-matched, laser capture microdissected tumour, ipsilateral-normal, and contralateral-normal samples from nine women undergoing bilateral mastectomy.
Results
Global trends in DNA methylation suggest differences between ipsilateral-normal and contralateral-normal breast tissue
Unsupervised hierarchical clustering using mean methylation beta values of the top 10,000 most variable probes across the three tissue types showed clear separation between tumour and normal (ipsilateral or contralateral) tissue (). Among the cluster of normal tissue, some samples clustered by subject (subjects 4, 7, and 8; ) while some samples appeared to separate by the breast from which they were collected (ipsilateral or contralateral). When assessing the methylation status of Alu and long interspersed nucleotide element-1 (LINE-1) repetitive elements, there was a statistically significant decrease in median Alu element methylation in tumour relative to contralateral-normal tissue (p = 0.03) while no statistically significant differences were observed in ipsilateral-normal tissue relative to tumour or contralateral-normal tissue (Additional file 1: Figure S1). However, there was visual suggestion of a trend in decreased Alu element methylation across contralateral-normal, ipsilateral-normal, and tumour tissue respectively. No statistically significant differences were observed in the methylation status of LINE-1 elements across tissue types.
Figure 1. Heatmap with unsupervised hierarchical clustering based on Manhattan distance of tumour, ipsilateral-normal, and contralateral-normal methylation profiles.
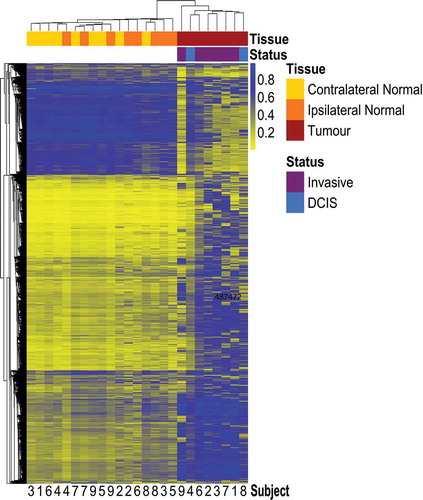
Table 1. Selected subject and tumour characteristics.
Contralateral-normal tissue provides a distinct reference epigenome compared to ipsilateral-normal tissue
At an FDR cut-off of q < 0.01, a total of 38,346 CpG loci were identified as differentially methylated in tumour tissue relative to ipsilateral-normal tissue ()). However, when comparing tumour tissue to contralateral-normal tissue, 63,271 CpG loci were identified as differentially methylated ()), a 65% increase. Furthermore, among those differentially methylated loci, 33,657 (88% of those identified as differentially methylated using ipsilateral-normal as the referent tissue and 53% of those identified as differentially methylated using contralateral-normal as there referent tissue) were identified in both comparisons ()) with consistent directionality of association in tumour relative to the respective referent tissue.
Figure 2. Epigenome-wide association analyses identifying CpG sites that are significantly differentially methylated in (a) tumour relative to contralateral-normal tissue and (b) tumour relative to ipsilateral-normal tissue. The beta coefficient reflects the difference in M-value associated with tissue type, adjusted for subject age and parity. Red dashed lines indicate a significance threshold of q < 0.01 and black dashed lines indicate a significance threshold of q < 0.05. CpGs are coloured (red) by sites identified as significant in the tumour versus contralateral-normal tissue (q < 0.01) analysis. (c) Overlap (green) between significantly differentially methylated CpG loci (q < 0.01) in tumour versus contralateral-normal and tumour versus ipsilateral-normal tissue. (d) Epigenome-wide association analysis identifying CpG sites that are significantly differentially methylated in ipsilateral-normal relative to contralateral-normal tissue. Red dashed lines indicate a significance threshold of p < 0.01 and black dashed lines indicate a significance threshold of p < 0.05.
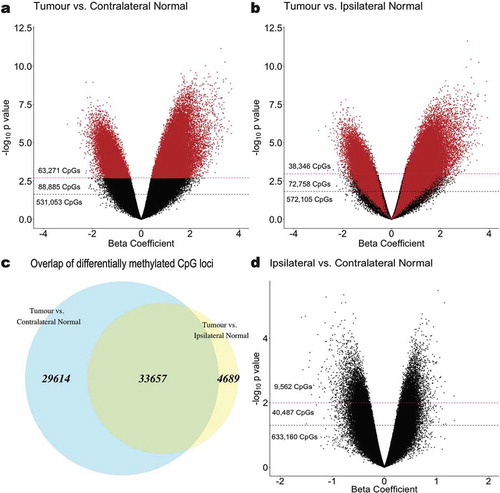
In a sensitivity analysis removing the contralateral normal-sample from the subject whose ipsilateral-normal sample was removed from analysis during QC due to > 10% of probes being above the specified detection p value (Subject 1) to allow for matched sample size across comparisons, a modest decrease was observed in the number of loci identified as differentially methylated (Additional file 2: Figure S2). However, there remained a 23% increase in the number of differentially methylated loci observed relative to the comparison using ipsilateral-normal tissue as the referent tissue.
To further assess whether the observed increase in differentially methylated CpGs were functionally redundant and reflected more loci from the same genes being identified as differentially methylated, we investigated the overlap in genes and gene sets. The 38,346 differentially methylated CpG loci identified when comparing tumour to ipsilateral-normal tissue mapped to 9,800 unique genes. Conversely, the 63,271 differentially methylated CpG loci identified when using contralateral-normal as the referent tissue mapped to 12,887 genes, 9,283 of which were overlapping with those identified in the tumour relative to ipsilateral-normal comparison.
We used the gometh function from the missMethyl package in R, which takes into account the differing number of probes per gene present on the EPIC array, to identify gene sets that were enriched among the 29,614 differentially methylated CpG loci in tumour relative to contralateral-normal tissue that were not identified when using ipsilateral normal tissue as the referent tissue. These 29,614 were compared to the background set of all 63,271 differentially methylated loci identified between tumour and contralateral-normal tissue to investigate GO terms that may be uniquely implicated in early carcinogenesis. We identified significant enrichment of (q < 0.01) 83 GO terms with the most significantly enriched GO terms relating to biological processes such as cell death, metabolic processes, and tissue development (Additional file 3: Table S1).
Hypomethylated loci in ipsilateral-normal relative to contralateral-normal tissue are enriched for transcription factor binding sites implicated in breast carcinogenesis
In analyses comparing ipsilateral-normal to contralateral-normal tissue, 9,562 CpG loci were identified as differentially methylated between the two histologically normal tissue types at a nominal significance threshold of p < 0.01 ()). Given differences in the biological interpretation of hypomethylation and hypermethylation, respectively, subsequent analyses were stratified by the direction of the identified association.
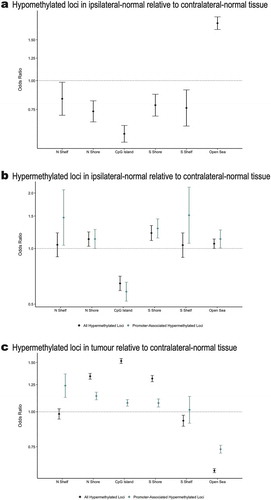
Among the 9,562 differentially methylated loci, 4,942 were identified as hypomethylated in ipsilateral-normal relative to contralateral-normal tissue (Additional file 4: Table S2). These hypomethylated loci demonstrated significant enrichment for CpG sparse open sea regions (OR = 1.77; 95% CI: 1.66, 1.88; p = 5.1 x 10−63) and significant depletion for all other CpG island associated genomic contexts ()).
Figure 3. (a) Enrichment/depletion of CpG island contexts from a Cochran-Mantel-Haenszel test comparing identified CpG loci to all other CpG loci included in the analysis, adjusting for Illumina probe type (a) among the 4,942 CpG loci found to be significantly (p < 0.01) hypomethylated in ipsilateral-normal relative to contralateral-normal tissue, (b) among the 4,620 CpG loci found to be significantly (p < 0.01) hypermethylated in ipsilateral-normal relative to contralateral-normal tissue as well as the subset of 1,421 hypermethylated loci annotated to gene promoter regions, (c) among the 71,407CpG loci found to be significantly (p < 0.01) hypermethylated in tumour relative to contralateral-normal tissue as well as the subset of 30,455 hypermethylated loci annotated to gene promoter regions.
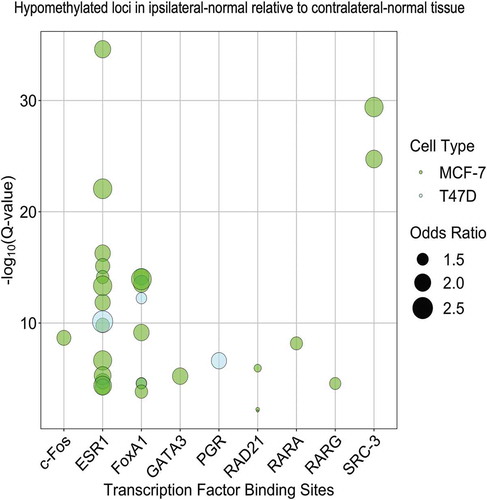
To further explore possible regulatory roles of differentially methylated CpG loci in ipsilateral-normal relative to contralateral-normal tissue we next assessed enrichment for transcription factor binding sites (TFBSs), using the Locus Overlap Analysis (LOLA) software tool. Using the LOLA core database for hg19, we assessed enrichment for these loci at TFBSs using Cistrome data for the breast cancer cell lines MCF-7 and T47D. Comparing the identified hypomethylated loci (p < 0.01) in ipsilateral-normal relative to contralateral-normal tissue to the background set of all 683,209 loci included in our epigenome-wide association study, we identified statistically significant enrichment of TFBSs for c-Fos, ESR1, FoxA1, GATA3, RAD21, RARA, RARG, and SRC-3 using MCF-7 data and ESR1, FoxA1, and PGR using T47D data (all q < 0.05, ). This suggests that cellular programmes regulated by transcription factors known to be dysregulated in invasive breast cancer also contribute to early events in breast carcinogenesis.
Figure 4. Enrichment of transcription factor binding sites in MCF-7 and T47D cell lines as assessed in LOLA using Cistrome data among the 4,942 CpG loci found to be significantly (p < 0.01) hypomethylated in ipsilateral-normal relative to contralateral-normal tissue.
Hypermethylated loci in ipsilateral-normal relative to contralateral-normal tissue are depleted for CpG islands but enriched for adjacent shore regions
4,620 loci were identified as hypermethylated in ipsilateral-normal relative to contralateral-normal tissue (Additional file 5: Table S3). Using the LOLA core database for hg19, no statistically significant enrichments for TFBSs were identified among the hypermethylated loci in ipsilateral-normal relative to contralateral-normal tissue. However, these 4,620 loci demonstrated modest enrichment for open sea regions (OR = 1.06; 95% CI: 1.00, 1.13, p = 0.044), significant depletion for CpG island regions (OR = 0.65; 95% CI: 0.59, 0.71, p = 6.8 x 10−20), and significant enrichment for CpG island shore regions (north shores OR = 1.13; 95% CI: 1.03, 1.23; p = 0.010 and south shores OR = 1.21; 95% CI: 1.11, 1.33; p = 5.7 x 10−5; )). As not all CpG islands track to promoter regions, we restricted our enrichment analyses to CpG islands in gene promoter regions which demonstrated further depletion for CpG island regions (OR = 0.58; 95% CI: 0.52, 0.66; p = 8.2 x 10−18) and further enrichment for CpG island south shore regions (OR = 1.29; 95% CI: 1.14, 1.45, p = 4.8 x 10−5; )), among CpGs hypermethylated in ipsilateral relative to contralateral normal tissue. Importantly, when assessing the genomic context distribution among the 71,407 hypermethylated loci in tumour relative to contralateral-normal tissue (p < 0.01), we observed strong enrichment for both CpG shore regions (north shore OR = 1.34; 95% CI: 1.31, 1.37; 1.7 x 10−110, south shore OR = 1.31; 95% CI: 1.28, 1.35, p = 5.2 x 10−85; )), and CpG islands (OR = 1.52; 95% CI: 1.49, 1.55; p = 1.9 x 10−319). These data provide evidence that the earliest alterations to DNA methylation occur in the shore regions bordering CpG islands and that altered DNA methylation spreads to the island regions during tumorigenesis.
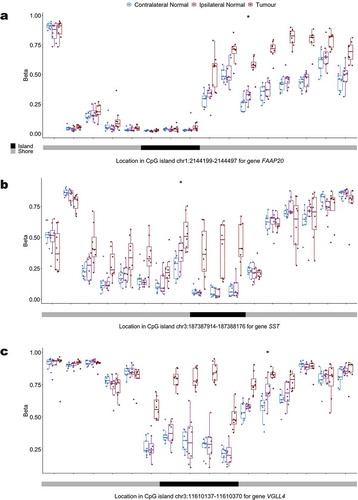
To more explicitly investigate progression of hypermethylation in tumours, we compared tumour DNA methylation data to ipsilateral normal tissue in our matched samples. Among the 4,620 hypermethylated loci in ipsilateral relative to contralateral normal tissue, 24.2% (n CpGs = 1,118), had increased methylation in tumour relative to ipsilateral normal. Of the 978 hypermethylated loci in ipsilateral relative to contralateral normal that tracked in CpG island shore regions, 32.3% (n CpGs = 316), had increased methylation in tumour relative to ipsilateral normal tissue. Among these 316 shore loci we selected the top 20 most statistically significant hypermethylated shore CpG loci in tumour relative to ipsilateral normal and investigated the potential for spreading of hypermethylation from the shore to neighbouring CpG island loci (Additional file 6: Table S4). We plotted the distribution of methylation at interrogated loci mapping to the CpG island shore of interest and the adjacent CpG island at each of these loci (, Additional file 7). For previously documented tumour suppressor genes FAAP20, SST, and VGLL4, hypermethylation can be observed spreading across the shore region in all genes as well as into the associated promoter CpG island for SST and VGLL4 in tumour tissue ().
Figure 5. Distribution of the methylation status at interrogated CpG loci in the promoter CpG island and surrounding CpG island shores across contralateral normal, ipsilateral normal, and tumour tissue for (a) FAAP20 (b) SST and (c) VGLL4.
Discussion
Our findings suggest that within the same patient, histologically normal breast tissue from the same breast as a tumour is epigenetically distinct from histologically normal tissue from the opposite breast. Furthermore, histologically normal tissue collected from the breast without the tumour appears to be more epigenetically distinct from tumour tissue than histologically normal tissue collected from the same breast as the tumour. This is suggestive of a field cancerization effect of the tumour on surrounding histologically normal tissue. These findings are consistent with previous work that demonstrated differential variance of DNA methylation in histologically normal tissue adjacent to a breast tumour relative to normal breast tissue from cancer-free women [Citation7] as an increase in the variability of DNA methylation profiles in so-called ‘adjacent normal’ tissue would decrease the power to identify differences in DNA methylation relative to tumour. While our study is limited by sample size, it presents a unique and valuable resource due to the paired nature of our samples.
While differences between ipsilateral-normal and contralateral-normal tissue were modest and did not achieve statistical significance at an FDR threshold of q < 0.05, differentially methylated CpG loci at a nominal significance threshold of p < 0.01 were significantly enriched for regions of functional significance including CpG island shore regions and tissue specific regulatory regions. This suggests that these differences may have important implications for the regulation of gene expression between the two tissue types.
Furthermore, enrichment of CpG island shore regions but depletion of CpG island regions among hypermethylated loci in ipsilateral-normal relative to contralateral-normal tissue, with greater magnitudes of enrichment and depletion observed when restricting to gene promoter regions, suggests that the earliest molecular alterations in carcinogenesis might appear as hypermethylation of CpG island shore regions and that these early alterations may also be present in histologically normal tissue surrounding the tumour. These findings are further supported by existing work demonstrating methylation encroachment at the borders of CpG islands in breast cancer and prostate cancer cells [Citation8], suggesting that hypermethylation of promoter regions over the course of carcinogenesis might begin at CpG island shores and progress into the islands.
Inspection of the promoter CpG island shore methylation profile for genes FAAP20, SST, and VGLL4, showed that while there was hypermethylation of a single shore locus in ipsilateral relative to contralateral normal tissue, hypermethylation spread in tumour tissue was consistently observed across the shore and, for SST and VGLL4, into the island itself. This finding lends additional support for the hypothesis that initial seeding events for the widely observed hypermethylation of promoter CpG islands in tumour tissue occur in the bordering shore regions [Citation8]. Furthermore, the hypermethylation of promoter CpG islands for the interrogated genes have important functional implications for breast carcinogenesis. FAAP20 plays an important role in DNA repair via the Fanconi anaemia-BRCA pathway [Citation9] and prior work has suggested that epigenetic alterations to genes in this pathway may play a role in the development of cervical cancer [Citation10] and ovarian cancer [Citation11]. SST is a neuropeptide that has been shown in models of breast cancer to both inhibit growth factors and hormones known to promote tumour growth, as well as prevent receptor signalling that can trigger apoptosis [Citation12]. Additionally, the role of epigenetic silencing as a means of suppressing SST expression in tumour cells has previously been suggested by the observed hypermethylation of the SST promoter CpG island in multiple tumour types including cervical cancer [Citation13], colorectal cancer [Citation14,Citation15], gastric cancer [Citation16], and renal cell carcinoma [Citation17]. VGLL4 has also been identified as a potential tumour suppressor gene across multiple tumour types including breast cancer [Citation18], gastric cancer [Citation19], and lung cancer [Citation20] and overexpression of VGLL4 has been found to suppress breast tumour growth in vivo [Citation18]. Therefore, the potential for suppression of VGLL4 expression through promoter hypermethylation could contribute to breast carcinogenesis.
Histologically normal tissue in the breast harbouring tumour also shows evidence of early molecular alterations that may be implicated in carcinogenesis among loci that are hypomethylated in this tissue relative to contralateral-normal tissue. These loci demonstrate enrichment for binding sites for transcription factors previously implicated in breast cancer, suggesting that hypomethylation of these loci may play a role in transcriptional regulation. It is also important to note that the cross-sectional nature of the study design precludes us from assessing the temporality of these alterations. Therefore, it is also possible that molecular alterations observed in ipsilateral-normal relative to contralateral-normal tissue reflect paracrine effects of the tumour on surrounding tissue [Citation21,Citation22] rather than precursor events to tumorigenesis. However, loci that demonstrate a directionally consistent association between both tumour and ipsilateral normal tissue relative to contralateral normal tissue, as well as CpG islands demonstrating spreading from shore hypermethylation in ipsilateral normal to island hypermethylation in tumour tissue relative to contralateral normal tissue suggest progression of local DNA methylation alterations in carcinogenesis. Prior work assessing the DNA methylation profiles of pre-invasive ductal carcinoma in situ lesions has also demonstrated that alterations to DNA methylation occur early on in breast carcinogenesis [Citation5]. Nonetheless, the observed differences in the epigenetic profile of ipsilateral-normal and contralateral-normal tissue suggest that histologically normal tissue collected from the affected breast is an imperfect referent tissue for the tumour.
We did not identify significant differences between the methylation status of Alu and LINE-1 repetitive elements across tissue types. While previous work has identified hypomethylation of these repetitive elements over the course of carcinogenesis [Citation23], these findings were subtype-specific. Therefore, given our limited sample size, it is unclear whether such associations might be observed given the lack of power to stratify analyses by molecular subtype.
Observed differences have important implications for molecular research in which histologically normal tissue adjacent to a tumour is used as a comparison group when evaluating for molecular alterations in tumour tissue. Given that our study collected ipsilateral-normal tissue at a distance of 3 cm from the tumour, 1 cm more distal from the tumour than the TCGA specified 2 cm minimum distance for the collection of adjacent normal tissue, this is suggestive that adjacent normal tissue may not be an adequate referent tissue. The observed effect of the tumour/tumour microenvironment on adjacent normal tissue may attenuate results when adjacent normal tissue is used as a comparison. In such cases, research may fail to identify significant differences at certain loci between tumour tissue and benign breast tissue within the same individual due to an unquantified field cancerization effect on the referent tissue. In addition, these loci may reflect the earliest alterations to DNA methylation in breast carcinogenesis and, therefore, failing to identify alterations at these loci may limit our understanding of early carcinogenesis. Our findings suggest that additional research is needed to identify molecular differences between breast tumours and normal breast tissue that previous research utilizing histologically normal adjacent tissue may have been unable to capture. While outside of the scope of the present work, extending the present findings to histologically normal breast tissue from women without breast cancer and histologically normal tissue adjacent to breast tumours could further elucidate our understanding of how DNA methylation mediates the association between known breast cancer risk factors and subsequent breast cancer development, building upon the existing body of literature in this space [Citation7,Citation24].
Our results indicate that histologically normal tissue collected from the same breast as the tumour harbours field cancerization effects and is not an ideal referent tissue for identifying breast tumour DNA methylation alterations, particularly early events in carcinogenesis. Our results also suggest that early hypermethylation events in breast carcinogenesis are more likely to occur in the regions immediately surrounding CpG islands than within CpG islands. This work offers an opportunity to focus investigations of early DNA methylation alterations in breast carcinogenesis and potentially develop epigenetic biomarkers of disease risk.
Methods
Study population
This was a retrospective study using formalin-fixed, paraffin-embedded (FFPE) archival tissue blocks from treatment-naïve women who had undergone elective bilateral mastectomies (tumour mastectomy and prophylactic contralateral mastectomy) due to patient choice for aggressive therapy at Baystate Medical Center, Springfield, MA and who were enrolled in the Rays of Hope Center for Breast Research Registry (IRB Baystate Health, Springfield, MA protocol number 568088).
Laser-capture microdissection and DNA isolation
Breast tumour, ipsilateral-normal, and contralateral-normal tissue blocks were identified and pulled for each of 9 women. H&E stained sections of the tumour and benign samples were used to estimate the total area for microdissection; a minimum of 5 × 106 μm2 was required to ensure sufficient material for adequate DNA yield (~800 ng minimum). There was insufficient tumour tissue available for one woman. Consecutive tissue sections (6-μm thick) were cut and mounted on membrane slides (MMI, Rockledge, FL). For tumour blocks, every 4th section was H&E stained and the tumour tissue was marked by a breast pathologist (GMC). For ipsilateral-normal samples, benign glandular areas were selected from blocks that were at least 3 cm from the tumour tissue. For contralateral-normal samples, benign glandular areas were selected. For both ipsilateral and contralateral samples, glandular tissue was marked on an H&E slide by a pathologist (GMC). For each sample the 6-μm sections mounted on membrane slides were deparaffinized in 3 changes of Xylene and allowed to air dry under vacuum in a desiccator for 30 min prior to the laser-capture microdissection. The unstained sections were oriented for microdissection aided by landmarks defined on the H&E stained slides. Areas to be microdissected were circumscribed using MMI Cell Tools software (Version Celltools-4.4 #261, Rockledge FL). Microdissected tumour, ipsilateral-normal, and contralateral-normal samples from each patient were collected separately onto caps. The collected tissue was placed in 150 μL digestion buffer containing 10 μL proteinase K, kept overnight at 55°C, and stored at −80°C until further processing. DNA was isolated using the manufacturer’s protocol for the BiOstic DNA FFPE Tissue Kit (MoBio).
DNA methylation profiling
DNA samples were sent to the Molecular Genomics/Methylation Core at the University of Southern California. Bisulphite treatment with the EZ DNA methylation kit (Zymo) was performed and bisulphite-converted DNA was processed as described in the Infinium FFPE Restoration guide (Illumina, San Diego, CA, USA). Modified DNA was assessed with four PCR reactions to estimate quantity and test for complete bisulphite conversion [Citation25]. DNA samples that passed the PCR quality controls were amplified at 37°C for 20–24 hours after treatment with 0.1 N NaOH. The DNA was fragmented at 37°C for 1 hour and subsequently precipitated in 100% 2-propanol at 4°C for 30 minutes followed by centrifugation at 3000xg at 4°C for 20 minutes. Dried DNA-pellets were resuspended in hybridization buffer at 48°C for 1 hour, denatured for 20 minutes at 95°C and loaded onto the Human MethylationEPIC BeadChip, and hybridized at 48°C for 16–24 hours. Unhybridized and non-specific DNA was removed using wash buffers. After a single base extension of the hybridized primers using labelled nucleotides, the BeadChip was stained with Cy-3 and Cy-5 fluorescent dyes and read using the Illumina iScan Reader.
DNA methylation data processing
Sample intensity data (IDAT) files were processed with the R package minfi using the ‘Funnorm’ normalization method on the full dataset. CpGs with a detection p value > 1E-06 in more than 5% of samples, CpGs with high frequency SNP(s) in the probe, probes previously described to be potentially cross-hybridizing, and sex-specific probes were filtered. Samples with more than 10% of probes above the detection p value or with bisulphite conversion intensity less than 3 standard deviations of the mean were removed.
One ipsilateral-normal sample was removed due to poor sample quality (>10% of probes above the detection p value). From an original set of 853,307 measured CpG loci on the Illumina HumanMethylationEPIC array (EPIC), our filtering steps removed 96,564 probes exceeding the detection p value limit, 71,478 probes that were SNP associated or cross-hybridizing, and 15,585 sex-specific probes. The final data for analysis resulted in 683,209 CpG loci measured in 25 matched samples from nine women () from tumour tissue (n = 8), ipsilateral-normal tissue (n = 8), and contralateral-normal tissue (n = 9).
Statistical analysis
Unsupervised hierarchical clustering was conducted based on Manhattan distance and using the 10,000 CpG loci that demonstrated the most variability in methylation beta value across all samples.
To quantify differential methylation, pairwise comparisons were run on logit-transformed methylation beta values (M-values) between contralateral-normal and tumour tissue, ipsilateral-normal and tumour tissue, and contralateral-normal and ipsilateral-normal tissue. Linear mixed-effect models were performed to model the relation between tissue type (tumour relative to contralateral-normal, tumour relative to ipsilateral-normal, and ipsilateral-normal relative to contralateral-normal, respectively) and CpG methylation M-value, controlling for age and parity and allowing a random effect for each subject. Due to the limited sample size of the study, we utilized nominal significance levels of p < 0.01 for subsequent analyses and interpretations.
To classify the genomic context of identified differentially methylated CpG loci, annotation data for the Illumina HumanMethylationEPIC array was used to identify the relation to CpG islands for each probe. The UCSC Genome Browser UCSC_hg19_refGene file was used to define promoters, exons, introns, and intergenic regions and the R package GenomicRanges was used to map these regions to all CpG loci on the Illumina HumanMethylationEPIC array. In cases where CpG loci mapped to more than one genomic region, loci were preferentially assigned to promoters, then exons, and then introns such that each locus only had one associated genomic region.
Gene set enrichment analyses of identified differentially methylated CpG loci were conducted using the gometh function from the missMethyl package in R [Citation26]. In brief, this method assesses enrichment of represented gene ontology (GO) terms among a set of differentially methylated CpG loci relative to a specified background set while adjusting for potential bias introduced by differences in the number of probes that map to each gene leading to altered probability of identifying a differentially methylated locus across different genes.
Cochran-Mantel-Haenszel tests were used to calculate odds ratios (ORs) and p values for the enrichment of CpG island contexts, adjusting for Illumina probe type, among CpG loci that were significantly hypomethylated or hypermethylated (p < 0.01) in ipsilateral-normal relative to contralateral-normal tissue, compared to all other CpG loci included in the analysis.
To assess possible enrichment of regulatory elements among hypomethylated and hypermethylated (p < 0.01) loci, respectively, in ipsilateral-normal relative to contralateral-normal tissue, we utilized Locus Overlap Analysis (LOLA) [Citation27] using Cistrome data for MCF-7 and T47D breast cancer cell lines from the LOLA core database for hg19. In brief, LOLA conducts a Fisher’s Exact Test for the enrichment of the identified differentially methylated loci relative to a specified background set (here specified as the 683,209 loci included in the analysis) overlapping with specified regulatory elements.
Methylation status of Alu and long interspersed nucleotide element-1 (LINE-1) repetitive elements were predicted using the REMP package in R [Citation28]. Median value of Alu and LINE-1 were subsequently calculated across all reported regions for each sample. Median Alu and LINE-1 methylation status was then compared between tissue types using a Wilcoxon rank-sum test.
Authors’ Contributions
MEM conceived and designed the analytical approach, performed statistical analyses, interpreted the results, and wrote and revised the manuscript. AJT conceived and designed the analytical approach, performed statistical analyses, interpreted the results, and wrote and revised the manuscript. LAS designed the analytical approach, interpreted the results, and revised the manuscript. OMW designed the analytical approach, interpreted the results, and revised the manuscript. CM assisted with microdissection, isolated DNA, interpreted the results, and revised the manuscript. KJG sectioned tissue, performed microdissections, interpreted results, and revised the manuscript. SSS conceived and designed the approach, interpreted the results, and revised the manuscript. GMC reviewed normal and tumor tissue, marked areas for microdissection, interpreted the results, and revised the manuscript. RMJ reviewed normal and tumor tissue, interpreted the results, and revised the manuscript. CNO conceived and designed the approach, interpreted the results, and revised the manuscript. BCC conceived and designed the approach, oversaw project development, interpreted the results, and revised the manuscript. KFA conceived and designed the approach, oversaw project development, interpreted the results, and revised the manuscript. All authors read and approved the final manuscript.
Availability of Data and Materials
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus under the accession number GSE133985 (https://www.ncbi.nlm.nih.gov/geo/).
Ethics Approval and Consent to Participate
Written informed consent was obtained from all subjects. Approval for this research was obtained from the Baystate Health Institutional Review Board (protocol number 568088).
Supplemental Material
Download MS Word (12.6 KB)Supplemental Material
Download Zip (1.7 MB)Acknowledgments
The authors thank all the women who donated tissue to this study.
Disclosure Statement
The authors declare that they have no competing interests.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. American Cancer Society; 2019;69:7–34.
- Koboldt DC, Fulton RS, McLellan MD, et al. Comprehensive molecular portraits of human breast tumours. Nature. Nature Publishing Group; 2012;490:61–70.
- Christensen BC, Kelsey KT, Zheng S, et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. United States; 2010;6:e1001043.
- McPherson K, Steel CM, Dixon JM. Breast cancer—epidemiology, risk factors, and genetics. BMJ Br Med J. British Medical Journal; 2000;321:624–628.
- Johnson KC, Koestler DC, Fleischer T, et al. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin Epigenetics. BioMed Central; 2015;7:75.
- Fleischer T, Frigessi A, Johnson KC, et al. Genome-wide DNA methylation profiles in progression to in situand invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. BioMed Central; 2014;15:435.
- Teschendorff AE, Gao Y, Jones A, et al. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun. 2016;7. DOI:10.1038/ncomms10478
- Skvortsova K, Masle-Farquhar E, Luu P-L, et al. DNA hypermethylation encroachment at CpG Island borders in cancer is predisposed by H3K4 monomethylation patterns. Cancer Cell. Elsevier; 2019;35:297–314.e8.
- Leung JWC, Wang Y, Fong KW, et al. Fanconi anemia (FA) binding protein FAAP20 stabilizes FA complementation group A (FANCA) and participates in interstrand cross-link repair. Proc Natl Acad Sci U S A. 2012;109(12):4491–4496.
- Narayan G, Arias-Pulido H, Nandula SV, et al. Promoter hypermethylation of FANCF. Cancer Res. 2004;64(9):2994–2997.
- Wang Z, Li M, Lu S, et al. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting fanconi anemia-BRCA pathway. Cancer Biol Ther. 2006;5(3):256–260.
- Watt HL, Kharmate G, Kumar U. Biology of somatostatin in breast cancer. Mol Cell Endocrinol. 2008;286(1–2):251–261.
- Verlaat W, Snijders PJF, Novianti PW, et al. Genome-wide DNA methylation profiling reveals methylation markers associated with 3q gain for detection of cervical precancer and cancer. Clin Cancer Res. 2017;23(14):3813–3822.
- Leiszter K, Sipos F, Galamb O, et al. Promoter hypermethylation-related reduced somatostatin production promotes uncontrolled cell proliferation in colorectal cancer. PLoS One. 2015;10(2):e0118332.
- Mori Y, Cai K, Cheng Y, et al. A genome-wide search identifies epigenetic silencing of somatostatin, Tachykinin-1, and 5 other genes in colon cancer. Gastroenterology. 2006;131(3):797–808.
- Jackson K, Soutto M, Peng D, et al. Epigenetic silencing of somatostatin in gastric cancer. Dig Dis Sci. 2011;56(1):125–130.
- Ricketts CJ, Morris MR, Gentle D, et al. Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics. 2012;7(3):278–290.
- Zhang Y, Shen H, Withers HG, et al. VGLL4 selectively represses YAP-dependent gene induction and tumorigenic phenotypes in breast cancer. Sci Rep. 2017;7:1–14.
- Jiao S, Wang H, Shi Z, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25(2):166–180.
- Zhang W, Gao Y, Li P, et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014;24(3):331–343.
- Maia J, Caja S, Strano Moraes MC, et al. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. Frontiers; 2018;6:18.
- Syn N, Wang L, Sethi G, et al. Exosome-mediated metastasis: from epithelial–mesenchymal transition to escape from immunosurveillance. Trends Pharmacol Sci. Elsevier Current Trends; 2016;37:606–617.
- Park SY, Seo AN, Jung HY, et al. Alu and LINE-1 hypomethylation is associated with HER2 enriched subtype of breast cancer. El-Maarri O, editor. PLoS One. Public Library of Science; 2014;9:e100429.
- Johnson KC, Houseman EA, King JE, et al. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. BioMed Central; 2017;19:81.
- Campan M, Weisenberger DJ, Trinh B, et al. Methylight. In: Tost J, editor. DNA Methylation. Methods in Molecular Biology. Humana Press; 2009. p. 325–337.
- Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2015;32:btv560.
- Sheffield NC, Bock C. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and bioconductor. Bioinformatics. Oxford University Press; 2016;32:587–589.
- Zheng Y, Joyce BT, Liu L, et al. Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res. Oxford University Press; 2017;45:8697–8711.