
ABSTRACT
The epitranscriptomic writer Alkylation Repair Homolog 8 (ALKBH8) is a transfer RNA (tRNA) methyltransferase that modifies the wobble uridine of selenocysteine tRNA to promote the specialized translation of selenoproteins. Using Alkbh8 deficient (Alkbh8def) mice, we have investigated the importance of epitranscriptomic systems in the response to naphthalene, an abundant polycyclic aromatic hydrocarbon and environmental toxicant. We performed basal lung analysis and naphthalene exposure studies using wild type (WT), Alkbh8def and Cyp2abfgs-null mice, the latter of which lack the cytochrome P450 enzymes required for naphthalene bioactivation. Under basal conditions, lungs from Alkbh8def mice have increased markers of oxidative stress and decreased thioredoxin reductase protein levels, and have reprogrammed gene expression to differentially regulate stress response transcripts. Alkbh8def mice are more sensitive to naphthalene induced death than WT, showing higher susceptibility to lung damage at the cellular and molecular levels. Further, WT mice develop a tolerance to naphthalene after 3 days, defined as resistance to a high challenging dose after repeated exposures, which is absent in Alkbh8def mice. We conclude that the epitranscriptomic writer ALKBH8 plays a protective role against naphthalene-induced lung dysfunction and promotes naphthalene tolerance. Our work provides an early example of how epitranscriptomic systems can regulate the response to environmental stress in vivo.
Introduction
Epitranscriptomic marks in the form of RNA modifications are catalysed by specific enzymes or RNA-protein complexes on the base or ribose sugar of the canonical adenosine, cytosine, guanosine and uridine nucleosides. RNA modifications can include simple changes such as methylation or acetylation, or hyper-modifications involving multiple enzymes and cofactors. Transfer RNA (tRNA) is the most heavily modified RNA species, with an average of 13 RNA modifications comprising ~17% of the 70–90 ribonucleosides. Epitranscriptomic marks can be located throughout tRNA molecules and can affect tRNA stability, folding, localization, transport, processing, and function [Citation1]. Modifications in the anticodon loop of tRNA can function as regulators of translation by promoting or preventing codon-anticodon interactions, which can be achieved by altering charge or base-pairing potential. Mammalian Alkylation Repair Homolog 8 (ALKBH8) is a writer enzyme that is required for the formation of modified wobble uridines on tRNASec to promote the translation of specific selenoproteins [Citation2–Citation4]. Global studies using cell systems have demonstrated that anticodon specific epitranscriptomic marks, as well as others, are dynamically regulated during cellular stress. Further it has been shown that the collection of epitranscriptomic marks change depending on the mechanism of action of the toxicants [Citation5,Citation6].
Epitranscriptomic marks reprogrammed in the anticodon of tRNA have been shown to promote the translation of codon-biased mRNAs, in order to increase the synthesis of critical survival proteins. Translational regulation by reprogrammed epitranscriptomic marks is detailed in bacteria [Citation7], yeast [Citation8–Citation11], mouse embryonic fibroblasts [Citation2] and human melanoma cells [Citation12]. A specific example of epitranscriptomic marks driving codon biased translation is found with the ALKBH8 dependent RNA modifications 5-methoxycarbonylmethyluridine (mcm5U) and 5-methoxycarbonylmethyluridine-2ʹ-O-methyluridine (mcm5Um) housed on the wobble uridine of selenocysteine tRNA (tRNASec) in mice and humans ()[Citation3,Citation4]. The epitranscriptomic marks mcm5U and mcm5Um are an integral part of the specialized addition of the selenocysteine (Sec) amino acid into proteins. The resulting selenoproteins are made by stop codon recoding [Citation2,Citation4,Citation13–Citation15], where mcm5 U and mcm5 Um promote binding of the tRNASec anticodon to the UGA stop codon on mRNA. Corresponding selenoprotein transcripts are inherently codon-biased because of this unusual in-frame UGA stop codon as well as the terminal stop codon [Citation16]. Importantly though, other core elements are required for stop codon recoding and they include a stem-loop structure in the 3ʹ untranslated region (3ʹUTR) of the transcript termed the Sec Insertion Sequence (SECIS) element, a SECIS Binding Protein (SBP2), and SEC-specific elongation factors (EFSec) [Citation17].
Figure 1. ALKBH8 domain structure, modification chemistry and the translation of selenoproteins.
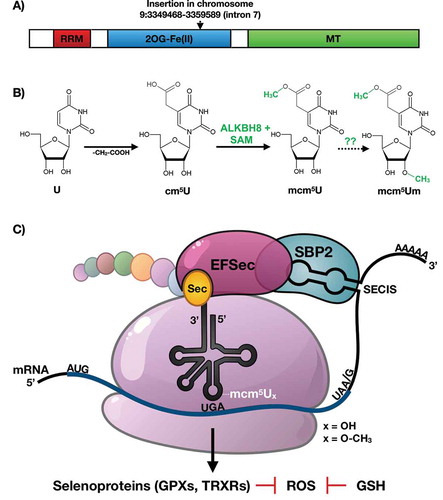
ALKBH8, mcm5U and mcm5Um modifications on tRNASec and other cis and trans accessory factors () help translationally regulate the production of selenoproteins, which comprise an important group of activities involved in development, reproduction and protection against oxidative stress. Selenoproteins such as glutathione peroxidase (GPX) family members catalyze the reduction of hydrogen peroxide and lipid peroxides by glutathione (GSH) and play an essential role in protecting cells against oxidative damage. Thioredoxin reductases (TRXRs) are involved in maintaining the reduced state of thioredoxins (TRXs), which function to reduce oxidative stress and play critical roles in development. We have previously shown that a deficiency in ALKBH8 prevents a H2O2 induced increase in mcm5Um and Alkbh8 deficient mouse embryonic fibroblasts (MEFs) have decreased selenoprotein expression levels, increased ROS and show sensitivity to the ROS-producing agent H2O2 [Citation2,Citation18] ALKBH8 deficient (Alkbh8def) mice are viable under normal care conditions which include a 12-hour light/dark cycle, 68-72°F temperature controlled environment, a standardized and balanced diet with free access to water, and minimal stress-inducing noise, odours and handling. Their ability to thrive in a normal environment, as well as produce pups that survive to adulthood, suggested that Alkbh8def mice adapt to the epitranscriptomic deficiency. Similar to other important stress and selenoprotein deficiency [Citation19], the Alkbh8def mice may need to experience an external stress to display observable phenotypes.
Naphthalene is a polyaromatic hydrocarbon (PAH) and exposure to this environmental toxicant can promote ROS, DNA damage and disrupt cellular glutathione levels [Citation20–Citation25]. Naphthalene is found in cigarette smoke, gasoline, and mothballs [Citation26–Citation28] as well as being derived in a number of industrial processes. In mouse, rat and human respiratory tissue, cytochrome P450 (CYP) enzymes bioactivate naphthalene to reactive and toxic metabolites. Naphthalene exposure is highly prevalent in the human population as detectable metabolites (1- and 2-hydroxynaphthalene) were found in urine of all 2,749 human civilians tested across 30 U.S. cities. In addition to its 100% detection rate in urine, naphthalene contributes approximately 75% to the total concentration of all PAHs tested in the study [Citation29]. Naphthalene is currently classified as a possible human carcinogen [Citation30] and GSH depletion is a major determinant in naphthalene-induced lung toxicity [Citation31–Citation33]. Because of the naphthalene connection to GSH and stress, we postulated that ALKBH8 could play an important role in response to this toxicant. Notably there are few animal studies testing the importance of epitranscriptomic systems in response to PAHs.
In the following study we have tested the hypothesis that the ALKBH8 epitranscriptomic writer plays a protective role against naphthalene-induced lung toxicity in vivo. We have characterized the lungs of Alkbh8def mice under normal conditions to determine how these mice adapt to an epitranscriptomic defect. We have shown that lungs from the writer deficient mice have increased markers of oxidative stress and have reprogrammed at the molecular level to display increased stress response transcripts. We have also performed naphthalene exposure studies on WT, Alkbh8def, and Cyp2abfgs-null mice. The Cyp2abfgs-null mice are a total-body knockout of a 12-gene cluster of lung preferential CYPs needed for naphthalene bioactivation in the lung [Citation34]. Naphthalene bioactivation is a well-documented requirement of naphthalene toxicity, thus the Cyp2abfgs-null mice served as our negative controls [Citation35,Citation36]. Alkbh8def mice are significantly more sensitive to naphthalene than WT, showing higher susceptibility to lung damage both at the cellular and molecular levels. Tolerance to naphthalene is defined as resistance to a high challenging dose after repeated exposures. WT mice develop tolerance to naphthalene after 3 days while Alkbh8def mice fail to develop tolerance. Our data supports a model in which the ALKBH8 writer, epitranscriptomic marks and some selenoproteins allow cells and tissues to develop tolerance and adapt to chronic naphthalene stress. Our work highlights that epitranscriptomic systems can regulate the response to environmental stress in vivo.
Materials and methods
Animal experiments
This study was carried out in accordance with the recommendations in the guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Albany, SUNY (Albany, NY) protocol #17-016 for breeding and basal characterization and #18-011 for naphthalene challenge. Additionally, it was approved by the Wadsworth Centre IACUC committee protocol 14–331 (Albany, NY) and The University of Arizona IACUC, protocol 17–335 (Tucson, AZ). Four homozygous strains of mice were bred and used in our study. They included WT, Alkbh8 deficient (Alkbh8def) and Cyp2abfgs-null, all of which were on the C57BL/6 background. A double mutant strain was also established via an initial cross of our homozygous Cyp2abfgs-null x Alkbh8def mice with offspring used to generate homozygous Cyp2abfgs-null/Alkbh8def double mutants (denoted as DM in this manuscript). Alkbh8def mice were originally generated using an insertional mutagenesis approach targeting the Alkbh8 gene in the parental E14Tg2a.4 129P2 embryonic stem cell line using a vector containing a splice acceptor sequence upstream of a β-geo cassette (β-galactosidase/neomycin phosphotransferase fusion). The construct was inserted into intron 7 at chromosome position 9:3,349,468–3,359,589, creating a fusion transcript of Alkbh8-β-geo [Citation37], as previously described [Citation2]. The resulting mutant mice were backcrossed 10 generations to C57BL/6 to make a congenic strain, and crossed with WT C57BL/6 mice every 10 generations thereafter to prevent genetic drift. Multiplex qRT-PCR and relative cycle threshold analysis (ΔΔ CT) on genomic DNA derived from tail biopsies was used to determine animal zygosity for Alkbh8def with TaqMan primers specific for neomycin (Neo, target allele) and T-cell receptor delta (Tcrd, endogenous control). The following primers were used: Neo forward (5ʹ-CCA TTC GAC CAC CAA GCG-3ʹ), Neo reverse (5ʹ-AAG ACC GGC TTC CAT CCG-3ʹ), Neo probe (5ʹ-FAM – AAC ATC GCA TCG AGC GAG CAC GT – TAMRA-3ʹ), Tcrd forward (5ʹ-CAG ACT GGT TAT CTG CAA AGC AA-3ʹ), Tcrd reverse (5ʹ-TCT ATG CAA GTT CCA AAA AAC ATC-3ʹ), and Tcrd probe (5ʹ-VIC – ATT ATA ACG TGC TCC TGG GAC ACC C – TAMRA −3ʹ). Cyp2abfgs-null mice were generated using the in vivo Cre-mediated gene deletion approach as described [Citation34], in which a 1.4-megabase pair genomic fragment containing 12 Cyp genes was deleted, including Cyp2a, Cyp2b, Cyp2f, Cyp2g, and Cyp2s subfamilies. Genotyping of Cyp2abfgs-null mice was performed as described [Citation34]. Male mice in age between 8–12 weeks were used in all experiments. Mice were sacrificed via CO2 asphyxiation and lungs excised, flash-frozen and stored at −80°C, or immediately processed for downstream experiments.
Naphthalene solution preparation and injection
Naphthalene (99% pure) and corn oil were purchased from Sigma-Aldrich (St. Louis, MO). Due to the volatile nature of naphthalene, stock solutions were stored at −80°C and dilutions were made fresh at the start of each experiment. In the case of multiple injections, diluted solutions were stored at −80°C and discarded after 5 days. For injections, mice were weighed, placed under brief anaesthesia (~1 minute) using 5% isoflurane gas, manually restrained, injection site wiped with 70% ethanol, and the corn oil or naphthalene solution injected into the intraperitoneal cavity in the lower left abdominal quadrant using Coviden™ Monoject™ 27 g, ½ inch standard hypodermic needles (Fisher Scientific, Franklin, MA) and sterile 1 ml Coviden™ Monoject™ tuberculin syringes (Fisher Scientific). 50–200 mg/kg of naphthalene or an equal volume of corn oil proportional to body weight was used for each injection. In case of multiple doses, the injections were administered between 10 and 11 AM (US EST) 24 h apart. Mice were monitored for signs of excess stress and weight loss. Mice were sacrificed 24 h after injection, or 24 h after last injection in case of multiple doses.
RNA extraction and Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from mouse lungs under basal and naphthalene-challenged conditions. Immediately after excision, fresh lungs were placed in Trizol reagent (Invitrogen, CA), cut into small pieces with scissors, and homogenized in 2 ml Trizol with a Tissue-Tearor homogenizer (BioSpec, Bartlesville, OK). Samples were left at room temperature for 10 minutes then placed on dry ice and transferred to −80°C for temporary storage. RNA isolation was performed using the standard Trizol reagent manufacturer’s protocol (http://tools.thermofisher.com/content/sfs/manuals/trizol_reagent.pdf). Alkbh8 transcript levels were measured in the lung using a Taqman Gene Expression assay. All qRT-PCR was carried out using a Thermo Fisher Scientific QuantStudio™ 3 Real-Time PCR system (Waltham, MA). Each sample was tested in triplicate and normalized to a β-Actin endogenous control. Statistically significant differences between the ΔΔCT values were determined using an unpaired Student’s T-test. Error bars denote standard error of the mean.
mRNA sequencing
Basal, 24-hour corn oil, and 24-hour naphthalene treated WT and Alkbh8def mice were sacrificed and lungs were harvested and RNA was extracted using the QIAGEN RNease mini kit (Qiagen, Germantown, MD). Samples were sent to Qiagen Genomic Services (Frederick, MD) for quality control (QC), library prep and targeted RNA sequencing. RNA integrity and concentration were evaluated using Agilent TapeStation 4200 and Thermo Scientific Nanodrop spectrophotometer, respectively. The starting RNA input for library prep was 500 ng. QIAseq Targeted RNA with a custom panel of 350 stress response transcripts specific to DNA damage, oxidative stress, protein damage, inflammation, metabolism and signalling pathways were targeted for library prep and sequencing. The targeted libraries were pooled together and sequenced using miSeq™ 500 system (Illumina, San Diego, CA). See Supplemental Table 1 for complete gene list. FASTQ files for each sample were obtained from BaseSpace and were further analysed using the QIAGEN GeneGlobe Data Analysis Centre (QIAGEN.com). Linear fold-change values were reported after normalization and assembled into a heat map using Java TreeView.
Measurements of Oxidation Reduction Potential (ORP)
Lung tissue was solubilized in extraction buffer (0.1 g per 1 mL phosphate buffered saline + 0.1% v/v Triton-X-100) using a rotor/stator tissue homogenizer. 30 mL of lysate was then applied to a filter-based sensor inserted into the RedoxSYS® galvanostat (RedoxSYS, Aytu BioScience, Inc., Englewood, CO). An electrochemical circuit was made when the sample (drawn by liquid flow) entered the sensor cell, after which an initial ORP measurement was made followed by the application of a current sweep through the sample, which exhausts the antioxidants present in the sample; the amount of current required to do this was determined over a 3-minute period. The primary output was a measurement of static ORP (mV), also known as the redox potential, representing the potential for electrons to move from one chemical species to another.
Histopathology
WT, Alkbh8def, Cyp2abfgs-null and Alkbh8def/Cyp2abfgs-null double mutant mice were exposed to naphthalene as described above and sacrificed 24 h after the last injection by CO2 asphyxiation. Lungs and trachea were exposed, a small incision was placed in the trachea, and the lungs were slowly inflated with 0.8 mL Z-Fix formalin based fixative (Anatech LTD, Battle Creek, MI) using a sterile, disposable blunt end needle (22 gauge, 0.5 in, Part #B22-50, SAI Infusion Technologies, Lake Villa, IL). The lungs were placed into a 50 ml microcentrifuge tube containing an additional 20 ml of Z-fix. After 24 h, the fixative was removed and replaced with 70% ethanol. Samples were sent to either the Wadsworth histopathology core (Albany, NY) or Mass Histology Service, Inc (Worcester, MA) for processing, paraffin embedding, sectioning and haematoxylin and eosin (H & E) staining. Mass Histology Service, Inc. captured 40X slide scan images of lung airways and alveolar regions. H & E stained slides were analysed by board-certified pathologist at Mass Histology Service, Lawrence McGill, DVM, PhD, DACVP, and assessed for pathologies and extent of damage, with specific focus on club cells.
The 8-isoprostane ROS assay
8-isoprostane was measured as an indicator of ROS in mouse lungs using an ELISA kit (Catalogue # ab175819, Abcam). All mouse treatments were performed in triplicate. Additional reagents used: 2 N sulphuric acid stop solution (catalogue # DY994, R&D Systems, Minneapolis, MN), triphenylphosphine (TPP) (Catalogue # T84409-1 G, Sigma-Aldrich), and ethyl acetate (Catalogue # 270,989, Sigma-Aldrich). Mice were sacrificed via CO2 asphyxiation at basal conditions and after challenge with 200 mg/kg naphthalene or corn oil carrier for 24 h. Lungs were excised and divided among 2 tubes with each containing ½ a lung, flash frozen and stored at −80°C. The right lobes (on the right as observed with the mouse ventral-side up) were processed for 8-isoprostane measurements as outlined in the manufacturer protocol and described below. The left lobes were processed for glutathione quantification (Section 2.7).
Lungs were weighed and then homogenized in 500 μL of H2O containing 0.00125 mg TPP. 1 μL of glacial acetic acid was added to acidify the samples, and pH measured using Hydrion S/R pH test paper (Micro Essential Labs, Inc., Brooklyn, NY) to ensure a pH between 3–4. 500 μL ethyl acetate was added, the sample was vortexed, spun down at 5000 rpm for 5 minutes, and the organic phase decanted into a new tube. The ethyl acetate extraction was repeated 2 more times and the organic phases were pooled and then dried using nitrogen gas. 5 μL of ethanol (200 Proof) (Catalogue #BP2818100, Fisher Scientific) was added to each sample to dissolve the dried residue. 300 μL of 1X sample dilution buffer (provided in the kit) was added to each sample. Samples were spun at 10,000 rpm for 5 min, and the supernatant was removed and placed in a new 1.5 ml microcentrifuge tube. The supernatant was used for ELISA analysis per manufacturer’s instructions.
Glutathione quantification
Sample preparation
Mice were left untreated, or exposed to corn oil or naphthalene for 24 h. The mice were sacrificed via CO2 asphyxiation and the lungs removed and immediately flash frozen. Samples were stored on dry ice and GSH & GSSG quantification was determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Tissue samples were homogenized in 19x volumes of Acetate buffer (100 mM Tris-base, 1.0 mM ethylenediaminetetraacetic acid (EDTA), 150 mM potassium chloride (KCl), pH 7.4) using a Polytron PT 10-35-GT powered homogenizer (Kinematica, Bohemia, NY). Tissue homogenates were processed using protein precipitation and centrifugation as follows: 40 μL internal standard, GSH-13C2,15N (3 µg/mL in 10% acetonitrile), was added to 40 μL of tissue homogenate, followed by the addition of 200 μL methanol and 200 μL water. The samples were mixed and centrifuged at 4°C and 14,000 rpm for 10 minutes. The supernatant was collected. 300 μL 0.1 N HCL was added to 100 μL supernatant and vortexed for 30 seconds and centrifuged at 4°C and 14,000 rpm for 10 minutes. The supernatant was collected and 3 μL was injected into the LC-MS system.
LC-MS conditions for analysis of GSH and GSSG
The LC-MS system consisted of an Agilent HPLC-1290 (Agilent, USA), and a Qtrap 6500 plus mass spectrometer (Sciex, Ontario, Canada) equipped with a Turbo IonSpray source. For data acquisition and processing, Analyst Software 1.6.3 was used.
Chromatographic separation was performed on a reversed-phase ZORBAX SB-C8 column (2.1 × 100 mm, 1.8 µm) using a mobile phase that consisted of 0.1% formic acid (A) and methanol (B) at a flow rate of 0.25 mL/min. The following mobile phase gradient was used: at 1%B, 0–3.5 min; 1%-50%B change, 3.5–4.0 min; 50%-95%B change, 4.0–4.5 min; at 95%B, 4.5–6.0 min; 95%-1%B change, 6.0–6.1 min; re-equilibration of the column at 1%, 6.1–9.5 min. The mass spectrometer was operated in positive ionization mode with an electrospray ionization probe and multiple reaction monitoring (MRM) was used. The MRM transitions used for detection of GSH and GSSG were 308.0/179.0 and 613.1/355.1, respectively. Isotopically labelled GSH (GSH-13C2,15N) was detected with an MRM transition of 311.0/182.0.
Calibration curves
Calibration standards for GSH and GSSG were prepared by diluting the stock solutions with water:acetonitrile (9:1,v/v) to the final concentrations of 1, 2, 10, 20, 50, 100 µg/mL. Calibration standards were processed using protein precipitation and centrifugation as follows: 40 μL internal standard, GSH-13C2,15 N (3 µg/mL in 10% acetonitrile), was added to 40 μL of calibration standards and followed by the addition of 200 μL methanol and 200 μL water. The samples were mixed and centrifuged at 4°C and 14,000 rpm for 10 minutes. The supernatant was collected. 300 μL 0.1 N HCL was added to 100 μL supernatant and vortexed for 30 s and centrifuged at 4°C and 14,000 rpm for 10 minutes. The supernatant was collected and 3 μL injected into the LC-MS system.
Protein assays
A Simple Western (Wes) protein analysis tool was used to quantify protein levels following manufacturer protocols (Protein Simple, Kanata, ON, Canada). Commercial antibodies used were as follows: GPX1 (Catalogue #AF3798, R&D Systems, Minneapolis, MN), GPX3 (Catalogue #AF4199, R&D Systems), TRXR1 (Catalogue #MAB7428), TRXR2 (Catalogue #ab180493, Abcam), TRX1 (Catalogue #MAB19701), TRX2 (Catalogue #MAB5765), SELS (Catalogue #HPA010025, Sigma) and GAPDH (Catalogue # CB1001, Millipore Sigma). Lungs were homogenized in 1–1.5 ml RIPA Buffer (50 mM Tris-HCl, pH 7.4, with 150 mM NaCl, 1% TritonX-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulphate) supplemented with an EDTA-free phosphatase inhibitor cocktail 2 (Catalogue #P5726, Sigma) and phosphatase inhibitor cocktail 3 (Catalogue #P0044, Sigma). Extracted protein was quantified using either Bradford assay (BioRad, Portland, OR) or Pierce BCA Protein Assay Kit (Thermo Fisher) according to manufacturer’s instructions, then diluted to equal concentrations and, due to Wes’ sensitivity, measured a second time using the BCA assay, and adjusted if necessary. The final loading concentration of lysate into each capillary was 3 μL of 1.5 mg/ml lung protein.
Bronchoalveolar lavage
Mice were euthanized using CO2 asphyxiation, the lungs and trachea were exposed, a small incision was placed in the trachea, intubated using a sterile, disposable blunt end needle, 22 gauge, 0.5 in (Part #B22-50, SAI Infusion Technologies, Lake Villa, IL) secured with a piece of thread. Next, the lungs were slowly inflated with 0.8 ml PBS, using a 1-ml Coviden™ Monoject™ tuberculin syringe (Fisher Scientific, Franklin, MA) and the fluid removed and re-injected 2 additional times over the course of ~1 minute to draw out the maximum number of inflammatory cells. The PBS containing cells was then transferred to a 2-ml microcentrifuge tube. The PBS lavage step was repeated 2 more times with 0.5 ml of PBS. The pooled Bronchoalveolar Lavage Fluid (BALF) was then mixed by gentle pipetting, dissolved 1:1 with trypan blue (Thermo Fisher), and live cells were counted on a haemocytometer.
Results
Alkbh8def lungs have adapted by molecular reprogramming
Under standard care conditions 12-week-old WT and Alkbh8def mice and lungs have no observable differences in tissue appearance or morphology (). Similarly, H & E stained lung sections of the bronchioles and alveolar regions do not show any noticeable difference (). Lung sections from WT and Alkbh8def mice show healthy epithelial cells, comprised primarily of club cells and ciliated cells, lining the bronchioles. Epithelial cells maintain a cuboidal structure and line the perimeter of the bronchiole, with club cells distinguishable from ciliated cells by the absence of cilia on the surface facing the bronchiolar lumen. No inflammation (i.e., macrophages present) or swelling in the alveolar regions was observed in either genotype. Together, these results support that even though there is a deficiency in Alkbh8 () mouse development and viability appear normal under normal growth conditions.
Figure 2. Alkbh8def lungs have decreased levels of specific selenoproteins and transcriptionally reprogram stress response systems.
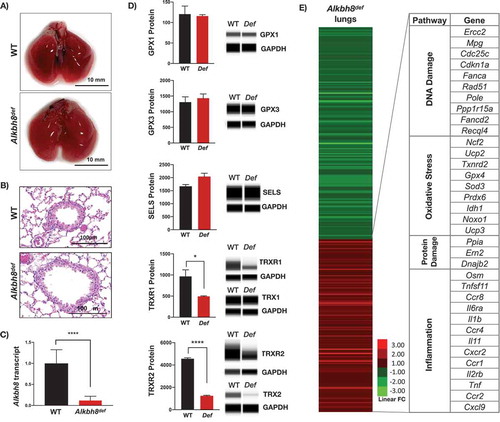
We also investigated molecular changes in the lungs of Alkbh8def mice by measuring selenoprotein and TRX1 and TRX2 protein levels (). Under normal growth conditions, we observed little difference in GPX1, GPX3, and Selenoprotein S (SELS) protein levels. In contrast we observed a significant decrease in TRXR1 and TRXR2 protein levels in the lungs of Alkbh8def mice. TRXRs are Sec-containing proteins critical for all signalling pathways in which thioredoxin (TRX) is involved as a reducing agent, as TRXRs maintain TRX in its reduced and active state. Due to significant deficiency in TRXRs, we also measured the associated thioredoxin (TRX) levels, and note a significant decrease in the mitochondrial form (TRX2), while the cytoplasmic form (TRX1) was unchanged.
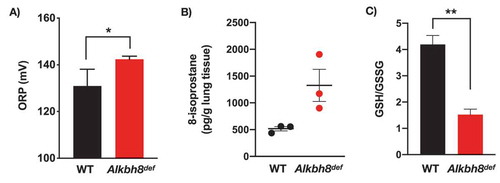
Next we used targeted mRNA sequencing () to determine if specific stress response pathways are transcriptionally regulated to adapt to the Alkbh8 deficiency, as well as TRXR1, TRXR2 and GPX deficiencies. mRNA seq data was submitted to the Gene Expression Omniubus (https://www.ncbi.nlm.nih.gov/gds). Transcripts belonging to inflammatory pathways were upregulated in the lungs of Alkbh8def mice, relative to WT. In addition, we observed increased levels of transcripts for protein damage response proteins, as well as those specific to some DNA damage and oxidative stress response systems in the lungs of Alkbh8def mice, relative to WT. Together these mRNA results ( and Suppplemental Figure 1) support the idea that Alkbh8def lungs alter their gene expression to adapt to stress. Oxidation-reduction potential (ORP) is a direct measure of the balance between ROS and antioxidants in a system. The more positive the ORP, the higher the oxidant activity or alternately, the lower the antioxidant activity. ORP is considered a new marker of ROS and has previously been used clinically as a measure of oxidative stress in blood of patients with traumatic brain injury, stroke, sepsis, metabolic disorder and liver toxicity [Citation38–Citation43]. We observed a significant increase in oxidation reduction potential (ORP) in the Alkbh8def lungs (). As a secondary measure of increased oxidants, we also analysed isoprostane levels. Isoprostanes are a family of eicosanoids produced by the random oxidation of tissue phospholipids by ROS and provide one of the most reliable measures of oxidative stress in vivo [Citation44,Citation45]. We observed a modest increase in 8-isoprostane levels () in the lungs of Alkbh8def mice, but this was just above the threshold to be significant. The Alkbh8def mice also have significantly decreased GSH/GSSG ratio under basal conditions (; Supplemental Figure 1). Together, our ORP, 8-isoprostane and GSH/GSSG findings support the idea that there is modest increase in ROS in the lungs of Alkbh8def mice and that GSH levels are dysregulated, relative to WT.
Figure 3. Markers of oxidative stress and increased DNA damage response observed in Alkbh8def lungs.
Alkbh8def mice show hypersensitivity to naphthalene and have increased lung airway inflammation after naphthalene exposure
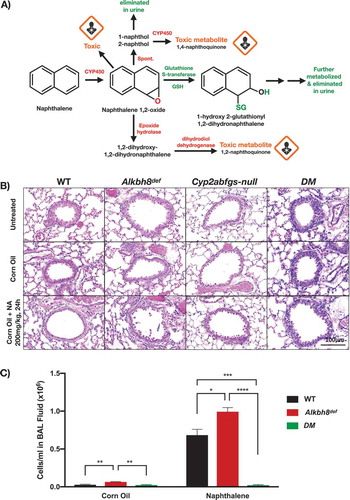
Our data from Alkbh8def mice grown under normal unstressed conditions supports the idea that the lungs have adapted to the writer defect and have minor signs of oxidative stress. We next asked whether the Alkbh8def mice would be sensitive to a challenge by the environmental toxicant naphthalene. We treated WT, Alkbh8def, Cyp2abfgs-null, and DM (double mutant Cyp2abfgs-null/Alkbh8def) mice to a single 200 mg/kg dose of naphthalene. The 200 mg/kg dose via I.P. injection used in our study is well established in the literature and the amount of naphthalene delivered to the lung is relevant to Occupational Safety and Health Administration (OSHA) standard for naphthalene exposure in the workplace (10 ppm, or 15 ppm short-term). Major steps in naphthalene bioactivation and metabolism are highlighted in . Alkbh8def mice have an observable difference in appearance after naphthalene exposure compared to WT or DM mice (data not shown). They display more fluffed fur, which is a classic marker of stress in mice. DM mice, despite Alkbh8 deficiency, have a smooth coat of fur, supporting previous studies that naphthalene bioactivation by Cyp2abfgs is required for toxicity [Citation46,Citation47]. shows representative lung airway images of all four strains of mice, either untreated or treated with corn oil or naphthalene (200 mg/kg) after 24 h. The H & E stained lung sections were focused on the bronchioles (round structures) and alveolar regions (perimeter) as these are prime spots for naphthalene damage due to the proximity of bioactivating enzymes. Healthy lung airways were observed under untreated and corn oil conditions in all 4 strains. After naphthalene exposure, the Cyp2abfgs-null and DM mice are resistant to naphthalene-induced pulmonary damage. WT mice show a flattening and elongating of epithelial cells, an event that occurs after early naphthalene-induced necrosis of club cells [Citation25]. Club cells participate in the biotransformation of many harmful substances introduced into the lung through inhalation, including naphthalene, via CYP-mediated bioactivation. The lung airway of Alkbh8def mice appear altered relative to WT, as what appears to be necrotic cells were observed in the airway epithelium. Lungs were sectioned, H & E stained, and analysed by a board-certified pathologist as described in the methods. Pathologies associated with naphthalene-induced lung damage include free floating necrotic cells in the airway lumen, flattened and elongated club cells in the airway epithelium, absent club cells in parts of the airway, and increased presence of macrophages. The pathologies were more extreme in Alkbh8def mice, which was further increased at prolonged exposure durations (discussed below). The single dose of naphthalene (200 mg/kg) resulted in elongated club cells, a classic response to naphthalene, in most WT and Alkbh8def mice. However, the Alkbh8def mice exhibit necrosis of epithelial cells in more bronchioles. We next used bronchoalveolar lavage to measure the number of cells in the recovered fluid, so as to gauge inflammation in WT, Alkbh8def and DM mice (). Alkbh8def mice have a significantly increased number of cells in the bronchoalveolar lavage fluid (BALF) compared to WT mice, under both corn oil and naphthalene treated conditions.
Figure 4. Alkbh8def mice and lungs are more sensitive to damage from naphthalene.
Alkbh8def mice show an increase in molecular markers of stress and fail to increase some protein levels compared to WT mice under NA challenge conditions
Next we investigated the redox and transcriptional states of WT, Alkbh8def and DM mice 24 hours after a single naphthalene exposure. ORP measures highlight a significant naphthalene-induced increase in oxidants, or a deficiency in antioxidants, in Alkbh8def mice relative to treated WT mice (). We observed an increase in 8-isoprostane levels in both WT and Alkbh8def mice treated with naphthalene, relative to the untreated sample (). WT naphthalene-treated mice had a significant increase (P ≤ 0.05) in 8-isoprostane levels relative to the corn-oil treated WT group, while naphthalene-treated Alkbh8def mice show a more significant increase (P ≤ 0.001) relative to corn oil-treated mice. We note significant difference in the corn oil condition between WT and Alkbh8def, supporting the idea of metabolic differences between the mice. We measured the GSH and GSSG in corn oil and 200 mg/kg naphthalene-treated mouse lungs after 24 hour exposure. While GSH depletion is considered a critical step in naphthalene-induced cytotoxicity occurring maximally between 1–3 hours after exposure and generally restored by 24 hours [Citation25,Citation31], we predicted that selenoprotein deficiency may cause extended disruption of the GSH system. We observe a decreased GSH/GSSG ratio in Alkbh8def mice under both conditions ().
Figure 5. Altered stress response to naphthalene observed in Alkbh8def lungs.
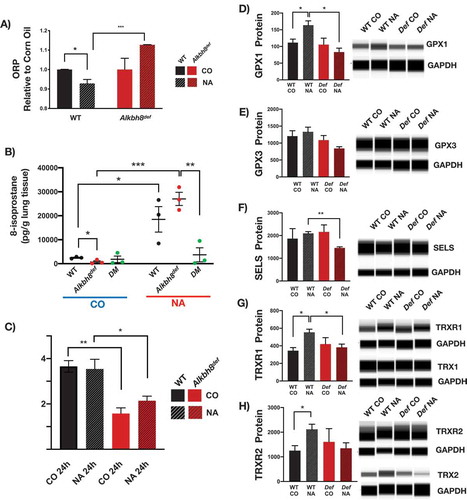
We also measured protein levels to determine if specific selenoproteins are upregulated in response to naphthalene and whether this was corrupted in the Alkbh8def lungs. WT mice upregulate the selenoproteins GPX1, TRXR1 and TRXR2, and non-selenoprotein TRX2 in response to naphthalene stress (-h). In contrast the Alkbh8def mice fail to upregulate GPX1, TRXR1, TRXR2 and TRX2 (-h; Supplemental Figure 3). Together the data support a model in which there are increased ROS levels and decreased ROS detoxifiers in the Alkbh8def lungs after naphthalene treatment, relative to WT.
Alkbh8def mice are sensitized to naphthalene and fail to develop tolerance
We next used daily 200 mg/kg naphthalene exposures to determine whether there were differences in survival between WT and Alkbh8def mice. The resulting Kaplan-Meier plot () provides evidence that Alkbh8def mice have increased sensitivity to naphthalene, relative to WT. We observed that over 50% of the WT mice survive and start gaining weight around day 3–4 with no deaths observed after day 5 (-b). In contrast, all Alkbh8def mice die by day 7 and do not show marked recovery in weight. As the turnaround time for WT mice was around day 3, we analysed lung tissues after 3-days of exposure by H & E (Supplemental Figure 4). While WT mice had the early classic response of flattened and elongated club cells and some necrosis, they begin to recover the cuboidal structure of their airway epithelial cells around day 3, a sign of tolerance. Meanwhile, Alkbh8def lungs remain severely damaged with complete absence of epithelial cells in many parts of the bronchioles and injury and necrosis extending to ciliated cells to a moderate degree. The Alkbh8def mice also had blood clots in the alveoli suggesting an accumulation of sloughed cells or possibly a greater incidence of haemolytic anaemia, which naphthalene is known to cause and which can contribute to intravascular coagulation [Citation20,Citation48].
Figure 6. Alkbh8 deficient mice show increased mortality and fail to develop tolerance to naphthalene exposure.
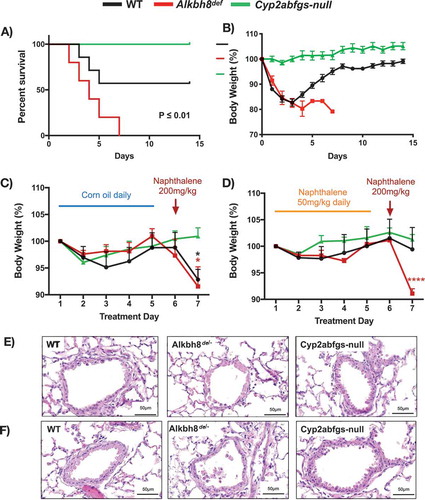
Based on these data WT mice appear to be developing tolerance to naphthalene by day 3, and we performed a dedicated tolerance study to investigate. As a control (non-tolerant) experiment we treated mice with 5 daily doses of corn oil, followed by a naphthalene dose of 200 mg/kg on day 6. Both WT and Alkbh8def mice lost significant amounts of weight and show lung airway damage 24 hours after naphthalene treatment (,e), with WT and Alkbh8def lung airways showing a similar response to a single 24-hour naphthalene treatment, as expected (). Next, we made the mice tolerant with 5 daily doses of 50 mg/kg naphthalene, followed by a high dose of 200 mg/kg on day 6 (). As opposed to non-tolerant mice (5 days of corn oil), naphthalene tolerant WT mice (5 days of naphthalene treatment of 50 mg/kg) maintain their weight after exposure to 200 mg/kg of naphthalene on day 6. In contrast Alkbh8def mice lost a similar amount of weight as in the non-tolerant study. H & E stained lung sections from the tolerant mice provide evidence that the Alkbh8def mice have a marked increase in lung airway damage compared to WT and Cyp2abfgs-null mice after exposure to 200 mg/kg of naphthalene, with continued necrosis and exfoliation of epithelial cells to the airway lumen in most bronchioles, as compared to WT mice ().
Discussion
We have previously shown that under normal cell culture growth conditions of 21% O2 mouse embryonic fibroblasts deficient in the epitranscriptomic writer ALKBH8 have increased ROS and DNA damage but adapt to these stresses by molecular reprogramming of stress-response transcript levels [Citation2]. We have also recently shown that the decreased selenoproteins in Alkbh8def is linked to senescence and mitochondrial dysfunction, with extensive reprogramming of associated transcripts under conditions of high oxygen [Citation18]. Our current study supports the idea that adaptive gene expression reprogramming can be extended to the lungs of Alkbh8def mice, as they have adjusted to the epitranscriptomic deficiency and corresponding decrease in selenoprotein levels by upregulating the expression of some inflammation, DNA repair and protein stress-response transcripts and markers. Molecular reprogramming of stress response systems in response to an epitranscriptomic writer deficiency has also been reported in lower organisms. For example, deficiencies in wobble uridine modification systems in budding yeast have been linked to upregulation of heat shock and unfolded protein response pathways [Citation49]. Molecular reprogramming due to cellular stress, including oxidative stress, protein stress and DNA damage, is also a well-documented response and has implications in development, ageing and disease susceptibility. For example, the nuclear factor erythroid 2-related factor 2 (NRF2) is a master regulator of cellular resistance to oxidative stress [Citation50,Citation51]. In response to oxidative stress, NRF2 dissociates from its negative regulator KEAP1, translocates to the nucleus, and binds to specific antioxidant response elements (AREs) in the promoter region of antioxidant and cytoprotective protein genes, including enzymes that catalyse glutathione synthesis [Citation50,Citation52]. Knockout of Nrf2 in mice results in increased sensitivity to oxidative stress-inducing agents including environmental toxicants, cigarette smoke, drugs and diseases [Citation53–Citation59], while boosting NRF2 activity has a protective effect against oxidative damage [Citation60]. Nevertheless, mice lacking NRF2 have been shown to compensate for the deficiency by upregulating endothelial nitric oxide synthase (eNOS), which mediates cardioprotection against myocardia ischaemia in conditions of decreased antioxidant capacity [Citation61]. eNOS synthesizes nitric oxide, which has potent antioxidant effects [Citation61,Citation62]. Despite severely decreased glutathione levels and general antioxidant capacity, Nrf2−/- mice were able to preserve their redox status, vascular function, and resist myocardial ischaemia/perfusion damage [Citation61]. NRF2 deficiency has also been shown to play a protective role against cardiomyopathy with molecular reprogramming of antioxidants at the transcript and protein levels playing a contributing role, with a noted increase in GPX1 in NRF2 deficient mice with cardiomyopathy [Citation63]. NRF2 and selenoproteins have been shown to work in conjunction to protect against oxidative damage [Citation64].
There are 25 Sec-containing proteins in humans, and 24 in rodents, with many serving as antioxidants in various thiol-dependent reactions [Citation65,Citation66]. Mice deficient in the selenoproteins TRXR1, Selenoprotein R (SELR), or Selenoprotein P (SELP) have growth delays and display markers of oxidative stress and metabolic deficiencies [Citation67-Citation69]. There are strong links between selenoprotein defects and inflammation. For example, selenoprotein S is an important regulator of the inflammatory response [Citation70,Citation71], with polymorphisms in the promoter region of SelS linked to decreased SELS expression and increased IL-6, IL-1β, and TNF-α levels [Citation70]. A deficiency or mutation in SELS is also associated with inflammation-related disorders and has been linked to gastric cancer [Citation72] and inflammatory bowel disease [Citation73]. In our study, we found a significant difference in SELS protein levels between WT and Alkbh8def naphthalene treated mice, which may also explain the dramatic increase in IL-6 and other inflammatory markers at the transcript level in the Alkbh8def lungs. The increase in inflammation markers in writer deficient lungs can be also linked to increased ROS and decreased antioxidant enzymes in the form of specific seleno- and other proteins. In support of this idea, we observed deficiencies in selenoproteins TRXR1 and TRXR2 and non-selenoprotein TRX2 in Alkbh8def mice under untreated conditions, and associated increase in oxidative stress markers, ORP and 8-isoprostanes. TRXs primarily reduce oxidized protein cysteine residues to form reduced disulfide bonds, with TRX2 localized to the mitochondria. TRXs are maintained in their reduced and active state by TRXRs using NADPH as a reducing equivalent, with TRXR1 located in the cytoplasm and TRXR2 in the mitochondria. This system is critical for signaling pathways involved in the protection from oxidants [Citation74]. TRXRs are also required for development as complete knockout of TRXR1 and TRXR2 in mice result in severe growth abnormality and embryonic death at day 8.5 and 13, respectively [Citation67,Citation75,Citation76].
GPX family members catalyze the reduction of harmful peroxides by glutathione and play an essential role in protecting cells against oxidative damage. GPX1 is the most abundant selenoprotein in mice, and its importance in naphthalene stress response is demonstrated by WT mice significantly upregulating SELS, TRXR1, TRXR2, TRX2 and GPX1 protein levels after naphthalene exposure, whereas Alkbh8def mice failed to do so. Notably Alkbh8def mice have decreased GPX1 levels compared to their carrier-treated counterparts. Previous studies have shown that glutathione (GSH) protects cells against naphthalene metabolites (i.e., 1,2-naphthoquinone). GSH is a key detoxification pathway and its depletion is a major determinant of naphthalene respiratory toxicity [Citation25,Citation31]. Many of the selenoproteins linked to ALKBH8 use GSH, including GPXs 1-6 in humans, and 1-5 in rodents, to transform H2O2 to water and lipid peroxides to alcohols. While the writer deficient lungs reprogram and upregulate other stress response systems under normal growth conditions, they are ill equipped to handle environmental challenge. These observations support the idea that dysregulated epitranscriptomic systems sensitizes lungs to naphthalene-induced injury.
In general, cells and tissues respond to environmental stress by regulating stress response pathways. Lungs respond to toxicant challenge via recruitment of inflammatory cells and secretion of inflammatory cytokines, as well oxidative modification of biomolecules (in part due to high oxygen exposure in the lung and high surface contact with the environment). Oxidatively modified compounds, as well as bioactivated components of some environmental toxicants, are able to enter the blood stream and other organs and produce systemic oxidative stress and inflammation [Citation77,Citation78]. Thus, inhaled environmental toxicants often have an effect that exceeds that of the lung. We have observed this phenomenon in our Alkbh8def mice, as weight loss is increased and animal survival decreased after naphthalene-exposure, which is likely due to systemic issues. Our study demonstrates that the epitranscriptomic writer ALKBH8 is required to mitigate the effects of naphthalene. It represents one of the first examples of how a mammalian RNA modification system is vital after environmental challenge. As there are over twenty epitranscriptomic writers in mammals [Citation79], there are most likely other RNA modifications, writers and perhaps erasers essential to the response to common environmental toxicants.
The highest occupational exposure to naphthalene occurs in industries dealing with wood treatment, coal tar production, mothball production and jet fuel industries. Naphthalene bioactivation by CYP enzymes is a well-documented requirement of naphthalene toxicity [Citation20,Citation35]. Bioactivation and now regulators of selenoproteins, in this case ALKBH8-dependent epitranscriptomic control, should also be considered modifiers of naphthalene exposure effects. Age-associated decline in GSH synthesis and activity has been reported in mice and rats [Citation80–Citation82]. Decreased ALKBH8 has been identified in aged fibroblasts, relative to young, with deficiencies also linked to early senescence [Citation18]. Due to age induced changes in transcription and epitranscriptomic activity, naphthalene sensitivity may be amplified in older people, but that is speculation that needs to be further studied. In support though, age related decreases have been established for drug and xenobiotic metabolism capacity in ageing mice [Citation83] and humans [Citation84].
Tolerance is defined as resistance to a high challenging dose after repeated exposures. Upregulation of glutathione synthesis has been reported as a tolerance mechanism in lung airway epithelial club cells [Citation25,Citation33,Citation85]. Glutathione depletion is a hallmark of naphthalene toxicity [Citation31]. γ-GCS is the rate-limiting enzyme in GSH synthesis, and treatment of mice with buthionine sulfoximine, a γ-GCS inhibitor, eliminates the tolerant phenotype in a majority of mice post- naphthalene exposure [Citation32,Citation33]. Selenoprotein H (SELH) has been shown to be an important transcription factor promoting expression of genes involved in GSH synthesis, including γ-GCS [Citation86]. Decreased SELH may explain our finding that ALKBH8-deficient mice fail to develop tolerance to naphthalene, but we were unable to identify suitable antibodies to measure it. Our study highlights the importance of ALKBH8 in promoting tolerance, most likely via translational regulation of selenoproteins and maintenance of GSH levels. The molecular reprogramming that occurs in response to the epitranscriptomic deficiency may also prevent tolerance in the Alkbh8def mice. It is notable though that tolerance to a high dose chemical challenge has been demonstrated for a homolog of ALKBH8, namely E. coli AlkB [Citation87]. The adaptive response to alkylating agents was characterized under conditions that mimic tolerance, as E.coli cells pretreated with a low dose of the alkylating agent N-methyl-N’-nitro-nitrosoguanidine (MNNG) were able to survive a high dose challenge, relative to naive cells. The adaptive response includes up-regulation of members of the Ada operon, the DNA glycosylase AlkA and the demethylase AlkB. A major finding of our study is the determination that the epitranscriptomic writer ALKBH8 is a key driver of tolerance, which highlights a novel role for tRNA modifications and translational regulatory mechanisms in response to environmental pollutants.
Conclusions
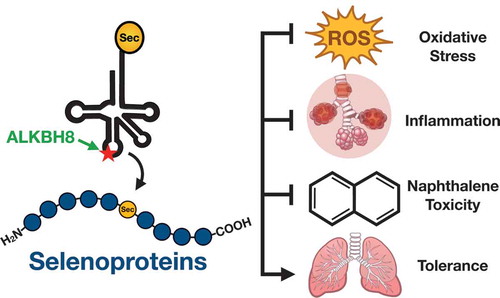
Our study supports a model () in which ALKBH8-catalysed epitranscriptomic marks promote the translation of selenoproteins important for handling stress, preventing naphthalene toxicity and driving tolerance. Efforts to identify other epitranscriptomic writers and marks, and translational control systems that mammals use to survive and tolerate naphthalene and other exposures, are needed to further understand the roles of RNA modifications in environmental exposure induced pathobiological changes.
Figure 7. Model for the epitranscriptomic writer based response to naphthalene. Our study supports a model in which ALKBH8-catalysed epitranscriptomic marks promote the translation of selenoproteins important for handling stress, preventing naphthalene toxicity and driving tolerance.
Supplemental Material
Download Zip (5.1 MB)Acknowledgments
We would like to thank our colleagues who have provided helpful suggestions. We would also like to thank the dedicated animal care staff at the University and Albany, Wadsworth Institute, and University of Arizona for their helpful assistance in all issues related to our animal colonies and Ms. Weizhu Yang for assistance with mouse breeding.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data of this article can be accessed here.
Additional information
Funding
References
- Jackman JE, Alfonzo JD. Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip Rev RNA. 2013;4(1):35–48.
- Endres L, Begley U, Clark R, et al. Alkbh8 regulates selenocysteine-protein expression to protect against reactive oxygen species damage. PLoS ONE. 2015;10(7):1–24.
- Fu D, Brophy JA, Chan CT, et al. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol. 2010;30(10):2449–2459.
- Songe-Moller L, van den Born E, Leihne V, et al. Mammalian ALKBH8 possesses trna methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol. 2010;30(7):1814–1827.
- Chan CTY, Deng W, Li F, et al. Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon-biased genes. Chem Res Toxicol. 2015;28(5):978–988.
- Chan CTY, Dyavaiah M, DeMott MS, et al. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6(12):e1001247.
- Chionh YH, McBee M, Babu IR, et al. tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat Commun. 2016;7:1–12.
- Bauer F, Hermand D. A coordinated codon-dependent regulation of translation by Elongator. Cell Cycle. 2012;11(24):4524–4529.
- Bauer F, Matsuyama A, Candiracci J, et al. Translational control of cell division by elongator. Cell Rep. 2012;1(5):424–433.
- Chan CTY, Pang YLJ, Deng W, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3(1):937.
- Deng W, Babu IR, Su D, et al. Trm9-catalyzed tRNA modifications regulate global protein expression by codon-biased translation. PLoS Genet. 2015;11(12):1–24.
- Rapino F, Delaunay S, Rambow F, et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature. 2018;558(7711):605–609.
- Copeland PR, Fletcher JE, Carlson BA, et al. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. Embo J. 2000;19(2):306–314.
- Copeland PR. Regulation of gene expression by stop codon recoding: selenocysteine. Gene. 2003;312:17–25.
- Low SC, Berry MJ. Knowing when not to stop: selenocysteine incorporation in eukaryotes. Trends Biochem Sci. 1996;21(6):203–208.
- Doyle F, Leonardi A, Endres L, et al. Gene- and genome-based analysis of significant codon patterns in yeast, rat and mice genomes with the CUT Codon UTilization tool. Methods. 2016;107:98–109.
- Driscoll DM, Copeland PR. Mechanism and regulation of selenoprotein synthesis. Annu Rev Nutr. 2003;23(1):17–40.
- Lee MY, Leonardi A, Shapiro R, et al. (2019). Loss of epitranscriptomic control of selenocysteine utilization engages senescence and metabolic reprogramming. Redox Bio. 28:10137. Available from: https://www.sciencedirect.com/science/article/pii/S2213231719313692
- Haan D, Bladier C, Griffiths P, et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem. 1998;273(35):22528–22536.
- Agency for Toxic Substances and Disease Registry (ATSDR). TToxicological profile for Naphthalene, 1-Methylnaphthalene, and 2-Methylnaphthalene. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service. 2005.U.S. Environmental Protection Agency (EPA).
- Bagchi D, Bagchi M, Balmoori J, et al. Induction of oxidative stress and DNA damage by chronic administration of naphthalene to rats. Res Commun Mol Pathol PharmacolResearch. 1998a;101(3):249–257.
- Bagchi M, Bagchi D, Balmoori J, et al. Naphthalene-induced oxidative stress and DNA damage in cultured macrophage J774A.1 cells. Free Radic Biol Med. 1998b;25(2):137–143. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9667488
- Carratt SA, Hartog M, Buchholz BA, et al. Naphthalene genotoxicity: DNA adducts in primate and mouse airway explants. Toxicol Lett. 2019;305(January):103–109.
- Lin PH, Pan WC, Kang YW, et al. Effects of naphthalene quinonoids on the induction of oxidative DNA damage and cytotoxicity in calf thymus DNA and in human cultured cells. Chem Res Toxicol. 2005;18(8):1262–1270.
- Plopper CG, Malburg SRC, Nishio SJ, et al. Early events in naphthalene-induced acute clara cell toxicity. Am J Respir Cell Mol Biol. 2013;24(3):272–281.
- Jia C, Batterman S. A critical review of naphthalene sources and exposures relevant to indoor and outdoor air. Int J Environ Res Public Health. 2010;7(7):2903–2939.
- Kakareka SV, Kukharchyk TI. PAH emission from the open burning of agricultural debris. SciTotal Environ. 2003;308(1–3):257–261.
- Sudakin DL, Stone DL, Power L. Naphthalene mothballs: emerging and recurring issues and their relevance to environmental health. Curr Topics Toxicol. 2011;7(541):13–19.
- Li Z, Sandau CD, Romanoff LC, et al. Concentration and profile of 22 urinary polycyclic aromatic hydrocarbon metabolites in the US population. Environ Res. 2008;107(3):320–331.
- IARC. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr Eval Carcinogenic Risks Humans. 2002;82:1–556. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12687954
- Phimister AJ, Lee MG, Morin D, et al. Glutathione depletion is a major determinant of inhaled naphthalene respiratory toxicity and naphthalene metabolism in mice. Toxicol Sci. 2004;82(1):268–278.
- West JAA, Williams KJ, Toskala E, et al. Induction of tolerance to naphthalene in Clara cells is dependent on a stable phenotypic adaptation favoring maintenance of the glutathione pool. Am J Pathol. 2002;160(3):1115–1127.
- West JAA, Van Winkle LS, Morin D, et al. Repeated inhalation exposures to the bioactivated cytotoxicant naphthalene (NA) produce airway-specific clara cell tolerance in mice. Toxicol Sci. 2003;75(1):161–168.
- Li L, Megaraj V, Wei Y, et al. Identification of cytochrome P450 enzymes critical for lung tumorigenesis by the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK): insights from a novel Cyp2abfgs-null mouse. Carcinogenesis. 2014;35(11):2584–2591.
- Li L, Wei Y, Van Winkle L, et al. Generation and characterization of a Cyp2f2-null mouse and studies on the role of CYP2F2 in naphthalene-induced toxicity in the lung and nasal olfactory mucosa. J Pharmacol Exp Ther. 2011;339(1):62–71.
- Li L, Carratt S, Hartog M, et al. Human CYP2A13 and CYP2F1 mediate naphthalene toxicity in the lung and nasal mucosa of CYP2A13/2F1-humanized mice. Environ Health Perspect. 2017;125(6):067004.
- Stryke D, Kawamoto M, Huang CC, et al. BayGenomics: A resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31(1):278–281.
- Bar-Or R. Raman spectral signatures of human liver perfusates correlate with oxidation reduction potential. Mol Med Rep. 2009;2(2):175–180.
- Bjugstad KB, Fanale C. A 24 h delay in the redox response distinguishes the most severe stroke patients from less severe stroke patients. J Neurol Neurophysiol. 2016;07:05.
- Bjugstad KB, Rael LT, Levy S, et al. Oxidation-reduction potential as a biomarker for severity and acute outcome in traumatic brain injury. Oxid Med Cell Longev. 2016;(2016:1–9.
- Bobe G, Cobb TJ, Leonard SW, et al. Increased static and decreased capacity oxidation-reduction potentials in plasma are predictive of metabolic syndrome. Redox Biol. 2017;12(February):121–128.
- Rael LT, Bar-Or R, Aumann RM, et al. Oxidation–reduction potential and paraoxonase–arylesterase activity in trauma patients. Biochem Biophys Res Commun. 2007;361(2):561–565.
- Spanidis Y, Goutzourelas N, Stagos D, et al. Assessment of oxidative stress in septic and obese patients using markers of oxidation-reduction potential. In Vivo. 2015;29(5):595–600. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26359419
- Gross M, Steffes M, Jacobs DR, et al. Plasma F2-isoprostanes and coronary artery calcification: the CARDIA study. Clin Chem. 2005;51(1):125–131.
- Morrow JD. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler Thromb Vasc Biol. 2005;25(2):279–286.
- Buckpitt AR, Warren DL. Evidence for hepatic formation, export and covalent binding of reactive naphthalene metabolites in extrahepatic tissues in vivo. J Pharmacol Exp Ther. 1983;225(1):8–16. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6834280
- Warren DL, Brown DL, Buckpitt AR. Evidence for cytochrome P-450 mediated metabolism in the bronchiolar damage by naphthalene. Chem Biol Interact. 1982;40(3):287–303. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7083396
- Cappellini MD. Coagulation in the pathophysiology of hemolytic anemias. Hematol Educ Program Am Soc Hematol. 2007;74–78. DOI:10.1182/asheducation-2007.1.74
- Patil A, Chan CTY, Dyavaiah M, et al. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol. 2012;9(7):990–1001.
- Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53(1):401–426.
- Vomund S, Schäfer A, Parnham MJ, et al. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. 2017;18(12):1–19.
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009. DOI:10.1074/jbc.R900010200
- Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Nat Acad Sci. 2001;98(8):4611–4616.
- Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8(1–2):76–87.
- Johnson DA, Amirahmadi S, Ward C, et al. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2009;114(2):237–246.
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47(1):89–116.
- Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012;64(4):1055–1081.
- Motohashi H, Yamamoto M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10(11):549–557.
- Rangasamy T, Tuder RM, Biswal S, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke – induced emphysema in mice Find the latest version: genetic ablation of Nrf2 enhances susceptibility to cigarette smoke – induced emphysema in mice. J Clin Invest. 2004;114(9):1248–1259.
- Talalay P, Dinkova-Kostova AT, Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul. 2003;43:121–134. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12791387
- Erkens R, Suvorava T, Sutton TR, et al. Nrf2 deficiency unmasks the significance of nitric oxide synthase activity for cardioprotection. Oxid Med Cell Longev. 2018;(2018:1–15.
- Wink DA, Miranda KM, Espey MG, et al. Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal. 2001;3(2):203–213.
- Kannan S, Muthusamy VR, Whitehead KJ, et al. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc Res. 2013;100(1):63–73.
- Kawatani Y, Suzuki T, Shimizu R, et al. Nrf2 and selenoproteins are essential for maintaining oxidative homeostasis in erythrocytes and protecting against hemolytic anemia. Blood. 2011;117(3):986–996.
- Hawkes WC, Alkan Z. Regulation of redox signaling by selenoproteins. Biol Trace Elem Res. 2010;134(3):235–251.
- Reeves MA, Hoffmann PR. The human selenoproteome: recent insights into functions and regulation. Cell Mol Life Sci. 2009;66(15):2457–2478.
- Bondareva AA, Capecchi MR, Iverson SV, et al. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43(6):911–923.
- Fomenko DE, Novoselov SV, Natarajan SK, et al. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J Biol Chem. 2009;284(9):5986–5993.
- Schomburg L, Schweizer U, Holtmann B, et al. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem J. 2003;370(2):397–402.
- Curran JE, Jowett JBM, Elliott KS, et al. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37(11):1234–1241.
- Gao Y, Hannan NRF, Wanyonyi S, et al. Activation of the selenoprotein SEPS1 gene expression by pro-inflammatory cytokines in HepG2 cells. Cytokine. 2006;33(5):246–251.
- Mao H, Cui R, Wang X. Association analysis of selenoprotein S polymorphisms in Chinese Han with susceptibility to gastric cancer. Int J Clin Exp Med. 2015;8(7):10993–10999.
- Seiderer J, Dambacher J, Kühnlein B, et al. The role of the selenoprotein S (SELS) gene −105G>A promoter polymorphism in inflammatory bowel disease and regulation of SELS gene expression in intestinal inflammation. Tissue Antigens. 2007;70(3):238–246.
- Saccoccia F, Angelucci F, Boumis G, et al. Thioredoxin reductase and its inhibitors. Curr Protein Pept Sci. 2014;15(6):621–646.
- Conrad M, Jakupoglu C, Moreno G, et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. 2004;24(21):9414–9423.
- Jakupoglu C, Przemeck GKH, Schneider M, et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25(5):1980–1988.
- Dalle-Donne I, Rossi R, Colombo R, et al. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52(4):601–623.
- Gomez-Mejiba SE, Zhai Z, Akram H, et al. Inhalation of environmental stressors & chronic inflammation: autoimmunity and neurodegeneration. Mutat Res Genet Toxicol Environ Mutagen. 2009;674(1–2):62–72.
- Kadumuri RV, Janga SC. Epitranscriptomic code and its alterations in human disease. Trends Mol Med. 2018;24(10):886–903.
- Chen TS, Richie JP, Lang CA. The effect of aging on glutathione and cysteine levels in different regions of the mouse brain. Proc Soc Exp Biol Med. 1989;190(4):399–402.
- Kim HG, Hong SM, Kim SJ, et al. Age-related changes in the activity of antioxidant and redox enzymes in rats. Mol Cells. 2003;16(3):278–284. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14744015
- Liu RM. Down-regulation of gamma-glutamylcysteine synthetase regulatory subunit gene expression in rat brain tissue during aging. J Neurosci Res. 2002;68(3):344–351.
- Kwak HC, Kim HC, Oh SJ, et al. Effects of age increase on hepatic expression and activity of cytochrome P450 in male C57BL/6 mice. Arch Pharm Res. 2015;38(5):857–864.
- Sotaniemi EA, Arranto AJ, Pelkonen O, et al. Age and cytochrome P450-linked drug metabolism in humans: an analysis of 226 subjects with equal histopathologic conditions. Clin Pharmacol Ther. 1997;61(3):331–339.
- Van Winkle LS, Buckpitt AR, Nishio SJ, et al. Cellular response in naphthalene-induced clara cell injury and bronchiolar epithelial repair in mice. A J Physiol. 1995;269(6 Pt 1):L800–18.
- Panee J, Stoytcheva ZR, Liu W, et al. Selenoprotein H is a redox-sensing high mobility group family DNA-binding protein that up-regulates genes involved in glutathione synthesis and phase II detoxification. J Biol Chem. 2007;282(33):23759–23765.
- Samson L, Cairns J. A new pathway for DNA repair in escherichia coli. Nature. 1977;267(5608):281–283. Available from: http://www.ncbi.nlm.nih.gov/pubmed/325420