
ABSTRACT
Although more and more children are born by Assisted Reproductive Technologies (ART), ART safety has not fully been demonstrated. Notably, ART could disturb the delicate step of implantation, and trigger placenta-related adverse outcomes with potential long-term effects, through disrupted epigenetic regulation. We have previously demonstrated that placental DNA methylation was significantly lower after IVF/ICSI than following natural conception at two differentially methylated regions (DMRs) associated with imprinted genes (IGs): H19/IGF2 and KCNQ1OT1. As histone modifications are critical for placental physiology, the aim of this study was to profile permissive and repressive histone marks in placenta biopsies to reveal a better understanding of the epigenetic changes in the context of ART. Utilizing chromatin immunoprecipitation (ChIP) coupled with quantitative PCR, permissive (H3K4me3, H3K4me2, and H3K9ac) and repressive (H3K9me3 and H3K9me2) post-translational histone modifications were quantified. The analyses revealed a significantly higher quantity of H3K4me2 precipitation in the IVF/ICSI group than in the natural conception group for H19/IGF2 and KCNQ1OT1 DMRs (P = 0.016 and 0.003, respectively). Conversely, the quantity of both repressive marks at H19/IGF2 and SNURF DMRs was significantly lower in the IVF/ICSI group than in the natural conception group (P = 0.011 and 0.027 for H19/IGF2; and P = 0.010 and 0.035 for SNURF). These novel findings highlight that DNA hypomethylation at imprinted DMRs following ART is linked with increased permissive/decreased repressive histone marks, altogether promoting a more permissive chromatin conformation. This concomitant change in epigenetic state at IGs at birth might be an important developmental event because of ART manipulations.
Introduction
It is estimated that more than six million children have been born by Assisted Reproductive Technologies (ART) worldwide, representing ~4% of all births [Citation1,Citation2]. However, the safety of these techniques has not fully been demonstrated. ART has been associated with an increased risk of placenta-related adverse pregnancy, perinatal outcomes, and imprinting disorders [Citation3–6]. As ART take place during the epigenetic-sensitive period of preimplantation when genome-wide erasure and selective reprogramming occur, these techniques could affect the implantation step, when the dialogue between endometrium and embryo establishes the placental invasion into the uterine wall [Citation7]. Together, these data raise the concern of the potential epigenetic vulnerability associated with ART.
Epigenetic mechanisms have been demonstrated to have a fundamental role in regulating placental function [Citation7]. Notably, imprinted genes (IGs) are known to modulate foetal and placental growth, for example, by regulating nutrient transfer, cell cycle, and insulin metabolism [Citation8–10]. Among imprinting mechanisms, DNA methylation in human placenta has been extensively studied, but literature about histone modifications after ART is relatively scarce. Modifications of basic histone amino (N)-terminal tail lead to changes in the overall chromatin structure and in the binding of effector molecules [Citation11] and thus changes regulation of DNA transcription, replication, recombination, and repair. For example, acetylation of the lysine 9 of histone H3 (H3K9ac) neutralizes the positive charge of histone H3, decreasing the histone’s affinity to bind DNA, resulting in a more ‘relaxed’ chromatin state which is permissive to gene expression. More complex than acetylation, histone methylation can be either a permissive or a repressive mark, according to its location on the histone tail. Though tri-methylation of lysine 4 on histone H3 (H3K4me3) is permissive, tri-methylation of lysine 9 on histone H3 is repressive when located in the promoter regions [Citation12]. A wealth of data have underlined that histone modifications are critical for trophoblast establishment [Citation13] and placental physiology [Citation14]. Notably, chronic ischaemia in the rodent placenta was linked to decreased histone H3 acetylation levels [Citation15]. In human, abnormal histone methylation at some imprinted DMRs was linked with the development of placental disorders such as preeclampsia and molar pregnancy [Citation16]. Moreover, the interest of studying histone modifications in the context of ART is reinforced by the fact that histone marks could be disturbed by environmental stressors [Citation17] and thus could mediate long-term health effects of ART.
We previously demonstrated that DNA methylation in the placenta was significantly lower after IVF/ICSI than following natural conception at two imprinted DMRs: H19/IGF2 and KCNQ1OT1 [Citation18]. The aim of this study was to determine whether DNA hypomethylation could be associated with specific histone profiles, to reveal a better understanding of the epigenetic modifications in the context of ART.
Materials and methods
Study population
Patients were prospectively included from 1 January, 2013, to 30 April, 2015, in the Department of Obstetrics, Gynaecology, and Reproductive Biology at Dijon University Hospital, France. ‘Natural conception’ group included singleton pregnancies of women that had conceived spontaneously within 1 year after stopping contraception. ‘IVF/ICSI’ group included singleton pregnancies achieved following fresh embryo transfer after 2 days of in vitro culture. This cohort has previously been described [Citation18] and used to compare the DNA methylation, by pyrosequencing, of 51 IVF/ICSI vs. 48 placentas from natural conception for three imprinted DMRs associated with the H19/IGF2:IG-DMR, KCNQ1OT1:TSS-DMR, and SNURF:TSS-DMR, named according to the recommendations for nomenclature [Citation19]. For the present study, to determine whether DNA hypomethylation could be associated with specific histone profiles, 16 placentas from the IVF/ICSI group who presented with below 5th percentile for methylation for at least one of these DMRs were selected (). They were compared with 16 controls matched for parity, newborn's sex, and gestational age at delivery. The controls were selected among the 48 women with natural pregnancy from the previous study.
Figure 1. Flowchart. From our precedent study [Citation18], we selected the patients from IVF/ICSI group who presented with below the 5th percentile of percentage methylation for at least one of the studied DMRs (H19 DMR, KCNQ1OT1 DMR and SNURF). The 16 selected patients were then matched for 16 controls from the natural conception group for parity, newborn's sex, and gestational age at delivery
![Figure 1. Flowchart. From our precedent study [Citation18], we selected the patients from IVF/ICSI group who presented with below the 5th percentile of percentage methylation for at least one of the studied DMRs (H19 DMR, KCNQ1OT1 DMR and SNURF). The 16 selected patients were then matched for 16 controls from the natural conception group for parity, newborn's sex, and gestational age at delivery](/cms/asset/d6509ece-f202-4025-b571-658c584353ab/kepi_a_1783168_f0001_oc.jpg)
Sample preparation
Placenta samples (1 cm3) were extracted from the foetal side within 15 min after delivery, washed twice in 0.9% NaCl before being snap-frozen in liquid nitrogen and conserved at −80°C.
DNA methylation and expression
Data for expression and DNA methylation experiments were obtained from our previous publication using real-time PCR and pyrosequencing, respectively [Citation18], and analysed on this new cohort of 32 samples.
Histone modifications analyses by Chromatin ImmunoPrecipitation (ChIP)
For the three imprinted DMRs previously analysed three permissive histone marks (di/trimethylation of lysine 4 of histone H3, H3K4me2/3; acetylation of lysine 9 of histone H3, H3K9ac) [Citation20] and two repressive histone marks associated with heterochromatic states (di/trimethylation of lysine 9 of histone H3, H3K9me2/3) [Citation21,Citation22] were studied.
Preparation of chromatin from placenta samples
Approximately 2 g of frozen placenta was rinsed two times in cold PBS and placed in lysis tubes (Zymo Research BashingBeads Lysis Tubes – 0.5 mm) containing 1 mL buffer I (0.5 M Tris-HCl pH 7.5, 0.5 M KCl, 2.5 M NaCl, 0.5 M MgCl2, 25 mM EGTA, 0.3 M sucrose, 0.5 mM DTT, 0.1 mM PMSF, 3.6 ng/mL aprotinin, 5 mM sodium butyrate) and subject to three intervals of agitation (90 sec, 5000 rpm) using a Precellys24 homogenizer (Bertin technologies) with 5 minutes on ice between each agitation cycle. The cell suspension was then placed in 7 mL of buffer II (buffer I with NP40 at a final concentration of 0.2%) to purify nuclei by centrifugation at 8500 rpm/12,720 g for 20 minutes with low acceleration and low deceleration on a sucrose gradient (8 mL from the previous step carefully placed on 25 mL of buffer III) (0.5 M Tris-HCl pH 7.5, 0.5 M KCl, 2.5 M NaCl, 0.5 M MgCl2, 25 mM EGTA, 1.2 M sucrose, 0.5 mM DTT, 0.1 mM PMSF, 3.6 ng/mL aprotinin, 5 mM sodium butyrate) in SorvallTM RC 6 Plus Centrifuge (ThermoScientificTM). The nuclear pellet was resuspended in digestion buffer (0.32 M sucrose, 50 mM Tris-HCl pH 7.5, 4 mM MgCl2, 1 mM CaCl2, 0.1 mM PMSF, 5 mM sodium butyrate) to 0.4 mg DNA/mL (Quantification by absorbance). Aliquots of 500 µL were distributed in 1.5 mL tubes. Micrococcal nuclease (Nuclease S7 15 IU/µL, Roche; final concentration 30 mIU/µL) was used to digest the chromatin to yield fragments one to five nucleosomes in length, which typically presented an incubation time of 3 minutes at 37°C. Digestion was stopped by adding 0.5 M EDTA at a final concentration of 20 mM and cooling on ice. After centrifugation (10 min, 15,800 g, 4°C), the supernatant was designated fraction S1. The pellet was resuspended in 500 µL lysis buffer (1 mM Tris-HCl pH 7.5, 0.2 mM EDTA, 0.2 mM PMSF, 5 mM sodium butyrate) and left 20–30 minutes on ice and subject to a second centrifugation step (10 min, 15,800 g, 4°C), the supernatant of which was designated fraction S2. The size of the nucleosomes was determined following Nucleospin gel and PCR clean-up (Macherey-Nagel) of ~100 µL of each fraction, to ensure the S1 chromatin fraction mainly comprised of mono and dinucleosomes and the S2 poly-nucleosomes of 2 to 5 nucleosomes (Supplemental Figure 1).
Immunoprecipitation of fresh chromatin
For ChIP, we used antibodies directed against H3K4me3 (Diagenode C15410003-50), H3K4me2 (Millipore 07–030), H3K9ac (Cell Signalling 9649 S), H3K9me3 (Abcam AB8898), H3K9me2 (Diagenode C15410060), and a negative control (mock precipitation with mouse IgG Millipore 12–371).
Chromatin was quantified by absorbance. For each condition, 4 µg of chromatin was used (constituted of 75% S1 and 25% S2) and suspended in incubation buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 20 mM sodium butyrate, 5 mM EDTA, 0.1 mM PMSF) in a total volume of 500 µL for each condition. We precleared chromatin by agitating overnight at 4°C with 4% Dynabeads ® Protein G for immunoprecipitation (Invitrogen) washed three times in PBS-BSA 5%. In parallel, antibodies were combined to Dynabeads ® Protein G for immunoprecipitation (Invitrogen), each antibody being agitated overnight in 250 µL of PBS-BSA 5% containing 16% of beads previously washed three times in PBS-BSA 5%.
The following day, beads were removed from precleared chromatin and antibodies-beads complexes were washed two times in PBS-BSA 5%. ChIP was then carried out for 4 h at 4°C
The antibody-chromatin complexes were subsequently washed three times with each buffer A (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 5 mM sodium butyrate, 75 mM NaCl), B (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 5 mM sodium butyrate, 125 mM NaCl), and C (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 5 mM sodium butyrate, 175 mM NaCl) to ensure only the fraction of chromatin linked to the antibodies was retained. Elution was performed in 400 µL of elution buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM EDTA, 1% SDS). After digestion with proteinase K (100 µg/mL) for 1 h at 65°C, DNA was obtained from the input and bound fractions with Nucleospin gel and PCR clean-up (Macherey-Nagel), according to the manufacturer’s protocol (protocol for samples SDS rich for bound fractions) with a final elution with 40 µL of water.
Quantification of immunoprecipitated chromatin
For an initial check of precipitated DNA quality, allelic specificity PCR assays were performed on all heterozygous samples. The PCR regions incorporated a Single Nucleotide Polymorphisms (SNPs) to allow both alleles to be discriminated. PCR and direct sequencing as used interrogate sequence traces, using Sequencher v4.6 (Gene Codes Corporation, MI). Primers, PCR mix, and conditions are available in Supplemental Table S1.
Levels of immunoprecipitated chromatin at each region of interest were determined by quantitative real-time PCR amplification with the QuantStudioTM 5 Real-Time PCR system (Applied BiosystemsTM), using the SYBRTM Green PCR Master Mix (Applied) (see supplemental Table S2). Data were analysed with QuantStudioTM Design & Analysis Software (v1.3.1). Each PCR was run in triplicate and level of immunoprecipitation was quantified as a percentage of total input material as follows: % of input = 2(-ΔCt) where ΔCt is the difference in mean Ct triplicate between the DNA of interest and the DNA of the input.
To overcome the inherent variability of different immunoprecipitations, precipitation levels obtained at the region of interest were normalized to the level obtained for positive control intervals. Interrogation of placenta ChIP-seq datasets in the Genome Data viewer function in the GEO data repository revealed that the promoter of KLF10 was enriched for the permissive histone marks H3K4me3, H3K4me2, and H3K9ac and was selected as a control region. For a control of repressed chromatin, we selected a heterochromatic satellite region on chromosome 4, which is ubiquitously associated with both H3K9me3 and H3K9me2.
Methylation-sensitive genotyping
Approximately 500 ng of heterozygous placenta DNA was digested with 10 units of HpaII and BstU1 restriction endonuclease for 6 hours at 37°C. The digested DNA was subject to ethanol precipitation and resuspended in a final volume of 20 μl TE. Approximately, 50 ng of digested DNA was used in each amplification reaction. The resulting amplicons were sequenced, and the sequence traces were compared to those obtained for the corresponding undigested DNA template.
Statistical analyses
Categorical variables were expressed as numbers (percentages) and compared using the Chi-2 test or Fisher exact test when appropriate. Continuous variables were expressed as means ± standard deviations (SD) or medians and interquartile ranges [IQR] and compared using the Student or Mann–Whitney test, as appropriate. Birth weights were normalized by conversion to z-scores calculated using normal birthweight curves of our population accounting for gestational age and newborn's sex [Citation23]. Placental weights were also converted into z-scores according to gestational age and newborn's sex [Citation24]. All statistical analyses were performed with SAS software, version 9.4 (SAS Institute Inc, USA). A two-tailed P < 0.05 was considered significant.
Results
Confirmation of in-silico histone modifications profiles in term placentas
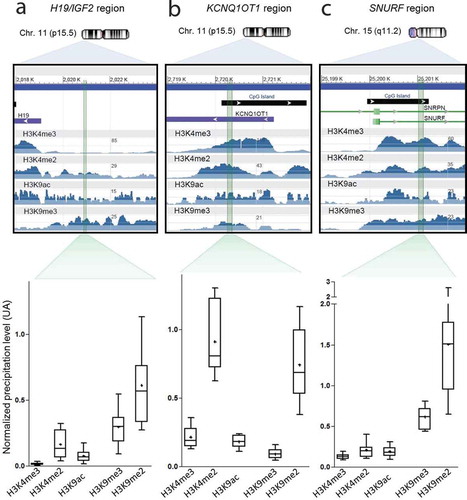
To ensure the PCR amplified intervals mapping to the imprinted DMRs were enriched for the histone modification of interest, we performed an in-silico analysis to ensure primer design coincided with the largest peaks in placenta-derived ChIP-seq datasets (GEO accession numbers GSM1160199 for H3K4me3; GSM753439 for H3K4me2, GSM818049 for H3K9ac and GSM1160204 for H3K9me3). Following primer optimization, amplicons of ~120–200 bp, which would allow for amplification of dinucleosome fragments and larger, were used to quantify the precipitation levels for the three imprinted loci of interest: H19/IGF2 ()), KCNQ1OT1 ()), and SNURF ()) DMRs.
Figure 2. Mapping of histone marks in placenta for each region of interest. For each region of interest (H19 DMR (a), KCNQ1OT1 DMR (b) and SNURF DMR (c)), we marked in green the sequence amplified by the qRT-PCR ChIP primers. To ensure the PCR amplified intervals mapping to the imprinted DMRs were enriched for the histone modification of interest, we performed an in-silico analysis to ensure primer design coincided with the largest peaks in placenta-derived ChIP-seq datasets. We used the Gene Expression Omnibus (GEO) application, available at https://www.ncbi.nlm.nih.gov/geo/. The GEO accession numbers for H3K4me3, H3K4me2, H3K9ac, and H3K9me3 were GSM1160199 (Histone H3K4me3 ChIP-Seq of Foetal Placenta), GSM753439 (ChIP-Seq Analysis of H3K4me2 in BMP4 Trophoblast Cells), GSM818049 (ChIP-Seq Analysis of H3K9ac in BMP4 Trophoblast Cells) and GSM1160204 (Histone H3K9me3 ChIP-Seq of Foetal Placenta), respectively. In parallel, normalized precipitation levels obtained in the 16 control samples of our cohort are displayed for each studied region. Precipitation levels of permissive marks H3K4me3, H3K4me2 and H3K9ac were normalized on precipitation levels of the KLF10 gene whereas repressive marks H3K9me3 and H3K9me2 were normalized on the satellite region SAT4
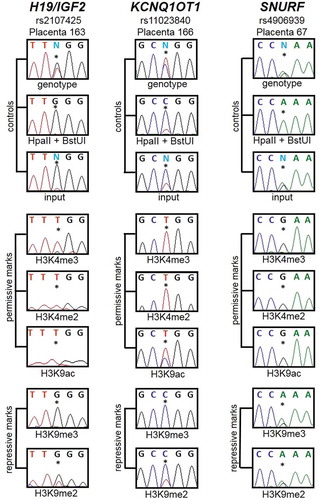
Subsequently, we analysed the allelic precipitation of the ChIP material in the 16 naturally conceived control samples since we anticipated that permissive and repressive histone marks should be on opposite parental alleles at these imprinted DMRs. PCR was performed using primers that flanked highly informative SNPs and the resulting amplicons sequenced. In total nine samples were heterozygous for H19/IGF2 (rs2107425), nine for KCNQ1OT1 (rs11023840) and nine for rs4906939 within the SNURF DMR. The allelic precipitation levels were compared to methylation-sensitive genotyping, which revealed that the permissive marks were solely on the unmethylated allele and the repressive marks preferentially on the opposite allele ().
Figure 3. Histone post-translational modifications are imprinted in the placenta. For each region of interest, an informative SNP was selected, the control DNA was genotyped, and heterozygous samples were studied. DNA was digested by HpaII and BstUI before sequencing to evidence the methylated allele. Then the ChIP products were also sequenced to assess which allele was the most represented in either permissive or repressive marks. It appears that the unmethylated allele is mostly represented in the permissive marks H3K4me3, H3K4me2 and H3K9ac. On the contrary, the methylated allele is mostly represented in the repressive marks H3K9me3 and H3K9me2. Thus methylated regions are associated with repressive histone marks whereas unmethylated regions are associated with permissive histone marks
Comparison between IVF/ICSI and natural conception groups
The two groups were comparable in terms of parental and new-born characteristics (). As expected, the mean group DNA methylation of H19/IGF2, KCNQ1OT1, and SNURF DMRs was significantly lower in the IVF/ICSI group (45.1% [43.2–48.9]; 32.8% [31.7–35.7] and 38.3% [35.5–40.5], respectively) compared to those conceived naturally (53.5% [49.6–59.3], P = 0.004; 39.4% [34.8–41.9], P = 0.001, and 41.2% [38.4–42.1], P = 0.036, respectively; , )). Relative expression was not different between groups ().
Table 1. Population characteristics
Table 2. DNA methylation and expression according to the mode of conception
Figure 4. Comparisons between IVF/ICSI group and controls. DNA methylation levels were lower in the IVF/ICSI group than in the natural conception group (a), some repressive and permissive marks normalized precipitation levels were lower and higher, respectively, in the IVF/ICSI group compared to the natural conception group (b and c, respectively). Each box represents the interquartile range (IQR). Lines inside the boxes are the median. Whiskers represent the 10th and 90th percentiles. Crosses represent the mean. For the histone marks profiling, the figures present the ratio between the % of input obtained at the region of interest and the % of input obtained at the control region. IVF: In Vitro Fertilization, ICSI: IVF with Intra Cytoplasmic Sperm Injection, ns: non-significant. Nat.: natural conception group
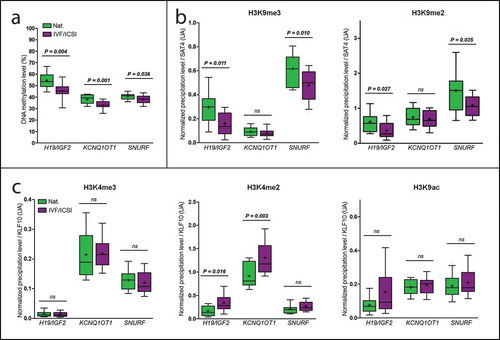
Quantitative PCR targeting H19/IGF2 and SNURF DMRs in the H3K9me3 and H3K9me2 precipitated material revealed significantly lower quantities of H3K9me3 and H3K9me2 in the IVF/ICSI group than in the natural conception group (P = 0.011 and 0.027 for H19/IGF2, respectively; and P = 0.010 and 0.035 for SNURF, respectively; )). There was no significant difference for either repressive mark at KCNQ1OT1 DMR ()).
The quantity of H3K4me2 at H19/IGF2 and KCNQ1OT1 DMRs was significantly higher in the IVF/ICSI group than in the natural conception group (P = 0.016 and 0.003, respectively; )). There was no significant difference for H3K4me2 for SNURF or for the other two permissive marks (H3K4me3, H3K9ac; )).
When the eight conventional IVF cases were compared with the eight IVF with ICSI, it showed that there was no methylation difference between both groups (Supplemental Figure 2(a)). However, the quantity of H3K9me3 at KCNQ1OT1 DMR was significantly lower in the ICSI than in the IVF group (P = 0.032; Supplemental Figure 2(b)), while the quantity of H3K4me2 in the same interval was significantly higher in the ICSI than in the IVF group (P = 0.003; Supplemental Figure 2(c)). There was no significant difference for the other permissive marks H3K4me3 and H3K9ac, repressive mark H3K9me2, nor for the SNRPN and H19/IGF2 DMRs (Supplemental Figure 2).
One hypothesis that could explain the increased of permissive histone modifications in some samples was the presence of these marks on the normally repressed allele. To address this, we focused on the allelic precipitation profiles in IVF/ICSI samples with highest precipitation levels (>75th percentile) of permissive marks at the H19/IGF2 DMR. Sequencing of samples heterozygous for SNPs revealed that the normally methylated allele was decorated with H3K4me2 and H3K9ac (Supplemental Figure 3). However, similar experiments targeting the KCNQ1OT1 and SNURF regions revealed maintained monoallelic precipitation patterns, comparable to spontaneously conceived controls (Supplemental Figures 4 and 5, respectively).
Discussion
These data demonstrate that DNA hypomethylation at imprinted DMRs could be associated with an increase in permissive histone marks and/or with a decrease in repressive histone modifications. This is consistent with a more ‘permissive’ chromatin conformation on the normally repressed allele. However, by focusing on outlier samples with highest precipitation levels of permissive marks and heterozygous for SNPs, we observed the enrichment of H3K4 methylation and H3K9 acetylation on the normally repressed and DNA methylated allele at the H19/IGF2 region. This suggests that some cells within the samples could lose their allelic methylation and subsequently gained the permissive histone modifications. Single-cell studies, possible incorporating Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) would be required to clarify this observation. In the KCNQ1OT1 and SNURF regions, i.e. maternally imprinted genes, the monoallelic imprint seemed to be preserved.
Several studies have addressed the stability of DNA methylation in placenta after IVF. The first reported lower DNA methylation levels in placentas after IVF than after natural pregnancy [Citation25], whilst other observed hypomethylation at the MEST and H19 loci [Citation25–27]. Our previous work evidenced lower DNA methylation levels of two imprinted loci (H19/IGF2 and KCNQ1OT1) and two transposable elements (LINE-1 and ERVFRD-1) in IVF placentas while there was not any statistical difference between IVF and controls for SNURF DNA methylation [Citation18]. However, not all studies have shown such clear-cut differences [Citation28]. Concerning gene expression, higher levels of expression of some IGs such as MEST and H19 have been demonstrated after IVF [Citation25,Citation26], but other studies found lower expression levels for IGF2 and H19 [Citation29].
To our knowledge, this study is the first reporting altered post-translational histone modification abundance in the human placenta after ART. Indeed, most studies focusing on histone regulation have been conducted in mouse models and mainly in pre-implantation embryos. For example, a study profiling epigenetic modifications at the Mest and H19 loci in mouse blastocysts cultured in vitro found an increased abundance of permissive histone marks and a decrease in repressive histone modifications [Citation21]. The same team confirmed these trends at the H19/Igf2 region on two cohorts of 2-cells embryos cultured in vitro until the blastocyst stage or vitrified/thawed and then cultured in vitro until the blastocyst stage [Citation30]. Similarly, altered methylation of histones and DNA at the H19/Igf2 region has also been shown in embryonic stem cells derived from mice preimplantation embryos [Citation31]. Interestingly, in an IVF cattle model, a higher expression of the imprinted gene PHLDA2 was associated with an increase in the permissive mark H3K4me2 in its promoter [Citation32]. Overall, our results are consistent with those reported in these models.
The increased in permissive and decrease of repressive histone marks observed in our study in hypomethylated samples following ART support the hypothesis that chromatin could be more permissive to transcription. However, increased expression was evidenced neither in this study nor in our previous one [Citation18]. Nevertheless, as we worked on term placentas, plasticity, and adaptability of placenta to the environment [Citation7] suggest that the altered expression could occur throughout pregnancy and no longer be visible at birth. This is well demonstrated by increased Igf2 expression after ART in superovulated mice placenta during gestation but no longer visible at birth [Citation33,Citation34] and by the observation that a positive correlation between placental IGF2 expression and birth weight is only present during the first trimester and not at term [Citation35]. Indeed, as the placenta undergoes rapid epigenomic changes during gestation, a placenta collected at birth may not reflect the changes occurring throughout pregnancy [Citation17]. However, these epigenetic changes occurring during the prenatal period, probably participating in compensation mechanisms [Citation7,Citation18], raise questions about the potential long-term effects of such modifications on children conceived by ART. As for the origin of these modifications, recent studies showed that levels of histones mRNA could be different in infertile patients’ sperm compared to controls [Citation36], and that H3K4me2 could be a molecular marker of sperm quality [Citation37], which raises questions about potential modifications in histone physiology related to infertility. Moreover, increase in permissive and decrease in repressive histone marks at KCNQ1OT1 DMR in ICSI group compared to IVF group suggest that specific ICSI protocols might influence histone regulation.
A limitation of this study could be the restricted number of IGs and histone marks studied. It would be interesting to extend analyses to other imprinted DMRs as well as imprinted genes with unmethylated promoters, regulated by neighbouring DMRs in cis, such as CDKN1 C and PHLDA2 [Citation22]. Furthermore, studying non-imprinted loci associated with early and late placental development could be revealing. It would also be interesting to decipher the proper roles of each placental cell type, although it would be technically difficult.
Conclusion
These novel findings highlight that DNA hypomethylation at imprinted DMRs after ART is linked with increased permissive/decreased repressive histone marks, altogether promoting a ‘permissive’ conformation of the chromatin. This concomitant change in epigenetic state at IGs at birth might be an important developmental event as a consequence of ART. To date, exact causes and consequences of these changes are not known. Better knowledge of the mechanisms at stake could enable to adapt our daily practice in order to reduce the impact of these changes.
Declarations
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
PF, DM, and CC were the principal investigators and take primary responsibility for the paper. PF, DM, PP, and CC were responsible for the study design. CC and PF recruited the patients. CC, DM, PP, MS, JH, and AM were involved in experiments. PF, DM, and CC coordinated the research. CC performed the statistical analyses. DM, PF, and CC drafted the manuscript. All authors read and approved the final manuscript.
Consent for publication
Not applicable
Ethics approval and consent to participate
All women had given written informed consent in accordance with the Declaration of Helsinki. The study was approved by the Institutional Review Board and the Ethics Committee of Dijon University Hospital (Comité de Protection des Personnes [CPP] Est I, n°2012-A01010-43).
Supplemental Material
Download MS Word (1.1 MB)Acknowledgments
We thank the midwives and nurses of Dijon University Hospital for their help in collecting samples. We thank Benjamin Tournier and Laurence Jego for their help in optimizing protocols. We thank Imprinting and Cancer group, Cancer Epigenetic and Biology Program, Bellvitge Biomedical Research Institute for their help in realizing experiments. We thank Sandrine Daniel, Marie-Laure Humbert-Asensio and Lydie Rossye (member of the “Centre d’Investigation Clinique-Epidémiologie Clinique/essais cliniques” of Dijon) for their precious help in monitoring and analysing the data. We thank Maud Carpentier of the “Direction de la Recherche Clinique et de l’Innovation” (DRCI) of Dijon University Hospital for the promotion and the management of the study. We thank Philip Bastable for his help in writing the manuscript.
Disclosure statement
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Messerlian C, Gaskins AJ. Epidemiologic approaches for studying assisted reproductive technologies: design, methods, analysis and interpretation. Curr Epidemiol Rep. 2017;4(2):124–132.
- Spaan M, van den Belt-dusebout AW, van den Heuvel-eibrink MM, et al. Risk of cancer in children and young adults conceived by assisted reproductive technology. Hum Reprod. 2019;34(4):740–750.
- Lazaraviciute G, Kauser M, Bhattacharya S, et al. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum Reprod Update. 2014;20(6):840–852.
- Pinborg A, Wennerholm UB, Romundstad LB, et al. Why do singletons conceived after assisted reproduction technology have adverse perinatal outcome? Systematic review and meta-analysis. Hum Reprod Update. 2013; 19:87–104.
- Qin J, Liu X, Sheng X, et al. Assisted reproductive technology and the risk of pregnancy-related complications and adverse pregnancy outcomes in singleton pregnancies: a meta-analysis of cohort studies. Fertil Steril. 2016;105:73–85e1-6.
- Zhu L, Zhang Y, Liu Y, et al. Maternal and live-birth outcomes of pregnancies following Assisted Reproductive Technology: a retrospective cohort study. Sci Rep. 2016; 6:35141.
- Choux C, Carmignac V, Bruno C, et al. The placenta: phenotypic and epigenetic modifications induced by Assisted Reproductive Technologies throughout pregnancy. Clin Epigenetics. 2015;7(1):87.
- Angiolini E, Fowden A, Coan P, et al. Regulation of placental efficiency for nutrient transport by imprinted genes. Placenta. 2006;27(27 Suppl A):S98–102.
- Ferguson-Smith AC, Moore T, Detmar J, et al. Epigenetics and imprinting of the trophoblast – a workshop report. Placenta. 2006; 27(Suppl A):S122–6. DOI:10.1016/j.placenta.2006.01.015
- Monk D. Genomic imprinting in the human placenta. Am J Obstet Gynecol. 2015;213(4):S152–62.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395.
- Portha B, Fournier A, Kioon MD, et al. Early environmental factors, alteration of epigenetic marks and metabolic disease susceptibility. Biochimie. 2014;97(97):1–15.
- Rugg-Gunn PJ. Epigenetic features of the mouse trophoblast. Reprod Biomed Online. 2012;25(1):21–30.
- Kohan-Ghadr H-R, Kadam L, Jain C, et al. Potential role of epigenetic mechanisms in regulation of trophoblast differentiation, migration, and invasion in the human placenta. Cell Adh Migr. 2016;10(1–2):126–135.
- Eddy AC, Chapman H, George EM. Acute hypoxia and chronic ischemia induce differential total changes in placental epigenetic modifications. Reprod Sci. 2019;26(6):766–773.
- Rahat B, Mahajan A, Bagga R, et al. Epigenetic modifications at DMRs of placental genes are subjected to variations in normal gestation, pathological conditions and folate supplementation. Sci Rep. 2017;7(1):40774.
- Barouki R, Melen E, Herceg Z, et al. Epigenetics as a mechanism linking developmental exposures to long-term toxicity. Environ Int. 2018;114(114):77–86.
- Choux C, Binquet C, Carmignac V, et al. The epigenetic control of transposable elements and imprinted genes in newborns is affected by the mode of conception: ART versus spontaneous conception without underlying infertility. Hum Reprod. 2018;33(2):331–340.
- Monk D, Morales J, den Dunnen JT, et al. Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics. 2018;13(2):117–121.
- Umlauf D, Goto Y, Cao R, et al. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet. 2004;36(12):1296–1300.
- Jahangiri M, Shahhoseini M, Movaghar B. H19 and MEST gene expression and histone modification in blastocysts cultured from vitrified and fresh two-cell mouse embryos. Reprod Biomed Online. 2014;29(5):559–566.
- Monk D, Arnaud P, Apostolidou S, et al. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci U S A. 2006;103(17):6623–6628.
- Rousseau T, Ferdynus C, Quantin C, et al. [Liveborn birth-weight of single and uncomplicated pregnancies between 28 and 42 weeks of gestation from Burgundy perinatal network]. J Gynecol Obstet Biol Reprod (Paris). 2008;37(6):589–596.
- Thompson JM, Irgens LM, Skjaerven R, et al. Placenta weight percentile curves for singleton deliveries. BJOG. 2007;114(6):715–720.
- Katari S, Turan N, Bibikova M, et al. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet. 2009;18(20):3769–3778.
- Nelissen EC, Dumoulin JC, Daunay A, et al. Placentas from pregnancies conceived by IVF/ICSI have a reduced DNA methylation level at the H19 and MEST differentially methylated regions. Hum Reprod. 2013;28(4):1117–1126.
- Rancourt RC, Harris HR, Michels KB. Methylation levels at imprinting control regions are not altered with ovulation induction or in vitro fertilization in a birth cohort. Hum Reprod. 2012;27(7):2208–2216.
- Camprubi C, Iglesias-Platas I, Martin-Trujillo A, et al. Stability of genomic imprinting and gestational-age dynamic methylation in complicated pregnancies conceived following assisted reproductive technologies. Biol Reprod. 2013;89:50.
- Turan N, Katari S, Gerson LF, et al. Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS Genet. 2010;6(7):e1001033.
- Jahangiri M, Shahhoseini M, Movaghar B. The effect of vitrification on expression and histone marks of Igf2 and Oct4 in blastocysts cultured from two-cell mouse embryos. Cell J. 2018;19:607–613.
- Li T, Vu TH, Ulaner GA, et al. IVF results in de novo DNA methylation and histone methylation at an Igf2-H19 imprinting epigenetic switch. Mol Hum Reprod. 2005;11(9):631–640.
- Arnold DR, Gaspar RC, da Rocha CV, et al. Nuclear transfer alters placental gene expression and associated histone modifications of the placental-specific imprinted gene pleckstrin homology-like domain, family A, member 2 (PHLDA2) in cattle. Reprod Fertil Dev. 2017; 29:458–467.
- Fortier AL, Lopes FL, Darricarrere N, et al. Superovulation alters the expression of imprinted genes in the midgestation mouse placenta. Hum Mol Genet. 2008;17:1653–1665.
- Fortier AL, McGraw S, Lopes FL, et al. Modulation of imprinted gene expression following superovulation. Mol Cell Endocrinol. 2014;388:51–57.
- Moore GE, Ishida M, Demetriou C, et al. The role and interaction of imprinted genes in human fetal growth. Philos Trans R Soc Lond B Biol Sci. 2015; 370:20140074.
- Hamad MF. Quantification of histones and protamines mRNA transcripts in sperms of infertile couples and their impact on sperm’s quality and chromatin integrity. Reprod Biol. 2019;19:6–13.
- Stiavnicka M, Garcia-Alvarez O, Ulcova-Gallova Z, et al. H3K4me2 accompanies chromatin immaturity in human spermatozoa: an epigenetic marker for sperm quality assessment. Syst Biol Reprod Med. 2020;66(1):3–11.