
ABSTRACT
Background: Epigenetic markers are often quantified and related to disease in stored samples. While, effects of storage on stability of these markers have not been thoroughly examined. In this longitudinal study, we investigated the influence of storage time, material, temperature, and freeze-thaw cycles on stability of global DNA (hydroxy)methylation. Methods: EDTA blood was collected from 90 individuals. Blood (n = 30, group 1) and extracted DNA (n = 30, group 2) were stored at 4°C, −20°C and −80°C for 0, 1 (endpoint blood 4°C), 6, 12 or 18 months. Additionally, freeze-thaw cycles of blood and DNA samples (n = 30, group 3) were performed over three days. Global DNA methylation and hydroxymethylation (mean ± SD) were quantified using liquid chromatography–electrospray ionization–tandem mass spectrometry (LC-ESI-MS/MS) with between-run precision of 2.8% (methylation) and 6.3% (hydroxymethylation). Effects on stability were assessed using linear mixed models. Results: global DNA methylation was stable over 18 months in blood at −20°C and −80°C and DNA at 4°C and −80°C. However, at 18 months DNA methylation from DNA stored at −20°C relatively decreased −6.1% compared to baseline. Global DNA hydroxymethylation was more stable in DNA samples compared to blood, independent of temperature (p = 0.0131). Stability of global DNA methylation and hydroxymethylation was not affected up to three freeze – thaw cycles. Conclusion: Global DNA methylation and hydroxymethylation stored as blood and DNA can be used for epigenetic studies. The relevance of small differences occuring during storage depend on the expected effect size and research question.
Introduction
With the introduction of the term ‘epigenetics’ and the development of more accurate techniques to quantify epigenetic changes, this field has gained more interest over the last thirty years [Citation1]. Epigenetics comprises the reversible process of DNA and histone modifications that alter gene expression without changing the DNA sequence. DNA methylation is the most studied epigenetic modification, which involves the addition of a CH3 group to the DNA, which is in humans predominantly found on cytosines (5-methylcytosine; 5mC) [Citation2]. DNA methylation can be actively de-methylated through the oxidation by Ten-eleven-Translocation (TET) enzymes to 5-hydroxymethylcytosine (5hmC) which can also act as an epigenetic modifier itself [Citation3,Citation4]. DNA methylation and hydroxymethylation have previously been associated with several diseases like cancer, heart disease, autoimmune diseases, and neurological disorders [Citation5–10], and are therefore potential biomarkers for disease onset, progression, or response to medication [Citation11]. DNA is required for epigenetic studies, which is mainly extracted from blood samples that have been collected from patients over several years and has therefore been stored for different time periods prior to the start of a study. Despite the increasing number of studies that relate differences in DNA (hydroxy)methylation to disease, only a handful of studies have examined the effect of storage material and temperature on the stability of global DNA methylation over longer storage duration. These studies, focused either at DNA methylation of targeted regions for specific research questions or only included a few subjects [Citation12–18]. Stability of DNA hydroxymethylation was examined in even fewer studies. An increase in DNA hydroxymethylation was observed at two different sites in brain tissue of Alzheimer patients compared to age-matched controls [Citation19], which was not influenced by tissue storage time. Unfortunately, storage duration and temperature were not specified in this study.
In this longitudinal study, we examined the stability of global DNA methylation and DNA hydroxymethylation in EDTA blood and extracted DNA samples at three different temperatures (4°C, −20°C, −80°C) for the duration of 0, 1, 6, 12 and 18 months. Additionally, we investigated the effect of freeze-thaw cycles on the stability of global DNA (hydroxy)methylation.
Results
Stability over time and under different storage conditions
Global DNA methylation
The effects of storage time, temperature, and material were assessed in the same model showing that global DNA methylation significantly changed over time, as the model including the time variable (Akaike information criterion; AIC = −819.06) significantly better fit the data (p < 0.0001) than the model without time variable (AIC = −685.23). To assess whether this significant change over time was affected by the type of material or storage temperature a three-way interaction between time, material, and temperature was performed, which was significant (p < 0.0001; Supplementary Table S1), indicating that DNA methylation evolved significantly different over time between blood and DNA samples and between samples stored at different temperatures. Global DNA methylation was not significantly different in blood and DNA samples stored at 4°C (when compared over 1 month), nor between blood and DNA samples stored at −80°C (compared over 18 months) (; Supplementary Table S2). Global DNA methylation in blood and DNA samples was stable at −20°C up to 12 months (). However, after 12 months mean global DNA methylation in DNA samples stored at −20°C decreased and slightly deviated from the slope of EDTA blood samples (; Supplementary Table S2). After 18 months, mean global DNA methylation in these DNA samples was 4.15 ± 0.23, which is a relative decrease of 6.1% compared to baseline (4.42 ± 0.11). In all other conditions, relative differences fluctuated between 0.5% and 4.1% compared to baseline (Supplementary Table S2), which is more comparable to the precision of the method (coefficient of variation; CV% = 2.3%).
Global DNA hydroxymethylation
The influence of time, material, and temperature on the stability of global DNA hydroxymethylation was assessed in a linear mixed model. Global DNA hydroxymethylation changed significantly over time as the AIC of the model including time variable (AIC = −5944.98) fit the data significantly better (p < 0.0001) compared to the model without time variable (AIC = −5920.03). Changes over time were significantly different for blood and DNA samples, which is described by the significant two-way interaction between time and material (p = 0.0131; Supplementary Table S1), but independent of storage temperature, as a three-way interaction between time, material, and storage was not significant and therefore excluded from the model (Supplementary Table S1). Up to 6 months of storage, there was no difference in global DNA hydroxymethylation between blood and DNA samples (). However, global DNA hydroxymethylation slightly increased in blood samples after 6 months but did not change in DNA samples (). Over 12 months, mean global DNA hydroxymethylation in blood samples stored at −20°C and −80°C increased to 0.040 ± 0.007 and 0.039 ± 0.008, respectively, which are relative changes of 17.6% and 14.7% compared to baseline (0.034 ± 0.005). Global DNA hydroxymethylation stored at −20°C also increased relatively with 12.5% at 12 months compared to baseline. Under all other conditions, differences over time were smaller and fluctuated between 0.0% and 9.4% relative to baseline. After 12 months, global DNA hydroxymethylation in blood and DNA samples stored at these temperatures decreased again to a relative difference of ≤6.3% compared to baseline, which is within the precision of the method (CV = 7.2%). At 18 months, fewer global DNA hydroxymethylation samples could be quantified from blood due to lower DNA yield upon extraction, especially from blood samples stored at −20°C (Supplementary Table S2).
Stability after repetitive freezing and thawing
DNA methylation assessed in stored blood or extracted DNA upon freezing and thawing fluctuated between 4.23 ± 0.20 and 4.29 ± 0.17 over three cycles ( and supplementary Table S3), which was no significant change (p = 0.7904). Changes compared to baseline were all <1%, which is within the within-run precision of the method (CV = 1.7%).
For global DNA hydroxymethylation in stored blood and extracted DNA samples, mean global DNA hydroxymethylation fluctuated between 0.034 ± 0.005 and 0.036 ± 0.008 upon freezing and thawing. Relative differences compared to baseline were all <6%. Although, this change was significant (p = 0.0169; Supplementary Table S3), these differences were within the within-run precision of our method (CV = 6.3%).
Discussion
For the use of stored samples in epigenetic studies it is important to know whether the stability of epigenetic markers is affected by storage conditions like time, material and temperature. In this study, we therefore examined the effect of these storage conditions on the stability of DNA methylation and hydroxymethylation. We showed that overall global DNA methylation and global DNA hydroxymethylation are stable when stored as blood or extracted DNA over a period of 18 months. However, storage of DNA at −20°C for longer than 12 months led to a significant but small decrease of 6.1% in global DNA methylation relative to baseline. Global DNA hydroxymethylation was more stable in stored DNA compared to blood after 6 months, which was independent of storage temperature. Furthermore, freeze-thaw cycles did not influence the stability of global DNA methylation and hydroxymethylation. Considering all this, we would recommend to store samples as extracted DNA at −80°C.
We have chosen storage temperatures and time according to daily practice. Therefore, EDTA blood at 4°C was stored for 1 month as in most laboratories longer storage takes place at −20°C or −80°C. Although global DNA methylation in blood samples stored at 4°C for 1 month slightly decreased, this decrease was similar to that in blood stored at −20°C and −80°C and to DNA samples stored at 4°C during the first month, where after mean methylation in all these conditions increased again towards the mean of baseline measurements. These changes lay within the precision of the method, indicating that these are probably due to between-run variances.
Mean DNA methylation levels were comparable to previously reported levels quantified by liquid chromatography – electrospray ionization – tandem mass spectrometry (LC-ESI-MS/MS) with mean DNA methylation between ~2 − 5% and hydroxymethylation of ~0.03% [Citation20–22]. These DNA methylation levels are also comparable to quantification using enzyme-linked immunosorbent assay (ELISA) – based methods with mean levels in the same range [Citation23]. However, LC-MS has been shown to be more specific compared to ELISA-based methods [Citation24]. Other methods that are used as a proxy for global DNA methylation usually measure highly repetitive and densely methylated regions, which is the case for long interspersed nuclear elements (LINE-1) and short interspersed nuclear elements (SINE), together constituting of ~30% of the genome. Because these regions are highly methylated, quantification of DNA methylation results in higher percentages (>50%). For the quantification of actual global DNA methylation and hydroxymethylation percentages LC-MS/MS is the gold standard, while for other research questions other methods could be considered [Citation24,Citation25].
Few other groups investigated the stability of global DNA methylation. One study observed a strong decrease in global DNA methylation upon three days of whole blood storage at room temperature, 4°C and −80°C, assessed using a dot-blot assay [Citation16]. They observed a strong decrease in DNA methylation, however, this was a small study, including 10 subjects only and the decrease was accompanied by a decrease in DNA concentration due to white blood cell lysis. In another study, the authors examined mean methylation of multiple CpG sites grouped within specific genomic regions (transcription start site, promoter, gene body, CpG islands, and shores) assessed by bisulphite conversion followed by pyrosequencing. In line with our results, they observed similar methylation patterns over time for DNA extracted from stored blood (4°C, -80°C) for 20 years and fresh samples. Like in our study, small differences over time were observed; however, their study design was cross-sectional and ours longitudinal. They observed that the variation between fresh samples and stored samples was similar, indicating that there was no real effect of storage [Citation15]. In our study, we observed small differences over time that were comparable to the precision of the method, with the exception of stored DNA at −20°C after 12 months and blood after 12 months, here we saw a relative decrease in global DNA methylation and a relative increase in global DNA hydroxymethylation respectively that exceeded the precision of the method.
In contrast to our study, most studies focused on the stability of DNA methylation at candidate genes. These results depended on the targeted site, the technique used and the material stored. Schröder et al. for instance observed a strong increase in mean DNA methylation in the intron 1 of HIF3A gene of blood stored at room temperature, 2–8°C, −20°C and −70°C, after 10 months of storage, assessed using bisulphite sequencing [Citation17]. While, others did not find significant differences in the hypermethylated promoter region of testis-specific histone 2B (TSH2B) and the hypomethylated transcription start site of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) quantified using methylated DNA immunoprecipitation coupled with next-generation sequencing (MeDIP-seq) in dried blood spots (DBS) stored at room temperature for 16 years compared to freshly extracted material [Citation26]. Thus, global DNA methylation is mainly stable upon storage, however, changes may occur at gene-specific sites.
Finally, stability of global DNA methylation and hydroxymethylation were not affected by freezing and thawing of blood or DNA samples for at least three freeze-thaw cycles at −80°C, which is in accordance with Li and colleagues who observed no effect of freeze-thaw cycles on DNA methylation [Citation15]. Additionally, to our knowledge we were the first to assess the stability of global DNA hydroxymethylation, thus more studies are required to confirm our findings.
Strengths of this study are that this is a longitudinal study, excluding potential differences due to inter-subject variability over time. Relative to other studies, our study groups were large, as most studies assessing DNA methylation stability only included 6–10 subjects per condition. Additionally, for all subjects, fresh material was collected and stored at the same time, so there was no time-to storage variability between the samples and samples from similar materials were quantified at the same time to reduce between-run variability. Additionally, global DNA methylation and hydroxymethylation were quantified using a global LC-MS/MS method, which is the gold standard method for analysis of global DNA methylation and enables to assess mean stability of methylated and hydroxymethylated cytosines all over the genome [Citation27]. An advantage of our method over other global DNA methylation methods is that no bisulphite conversion nor whole genome amplification is required to distinguish methylated from unmethylated cytosines, which minimizes the risk to introduce biases. Also, in our study DNA was digested into single nucleotides representing cytosines from the whole genome instead of a surrogate measure for global DNA methylation, like repetitive elements. Additionally, DNA hydroxymethylation was quantified simultaneously, which allows us to compare DNA methylation and hydroxymethylation within the same samples. A limitation of the method is that we cannot draw conclusions from stability at targeted CpG sites, while methylation at some sites have been shown to be more stable than others.
Other limitations of our study are that we did not account for cell type ratios. From literature, it is known that different cell types have different (hydroxy)methylomes [Citation28]. Previously, it was suggested that some cell types are more stable than others upon storage, which may lead to different cell type ratios upon cell lysis and consequently in different DNA methylation percentages [Citation17]. Although, in our study, this seems unlikely as we then would expect to find changes in blood samples over time and not in stored DNA samples, which was not the case.
Conclusion
In conclusion, overall global DNA (hydroxy)methylation was stable. However, small significant differences over time were observed in global DNA methylation and global DNA hydroxymethylation upon storage. Therefore, when samples are stored for extended periods of time or when samples are stored for different time durations, small differences in relation to disease should be interpreted with caution. Subsequently, for the assessment of global DNA (hydroxy)methylation, samples can best be stored at −80°C as extracted DNA.
Methods
Subjects and study design
EDTA whole blood was collected from leftover blood samples of the department of clinical chemistry at the Erasmus MC University medical centre of 90 individuals and divided into three groups of 30 individuals. Blood from patients from the oncology department was excluded due to the chance of decreased white blood cell concentrations, required for DNA extraction. None of the subjects included in the study objected for the use of their material for scientific research (opt in-opt out procedure) and all samples were anonymized after collection and the study follows the principles of the Declaration of Helsinki of 1975. 4 mL EDTA blood of the first 30 subjects (group 1) was stored in aliquots of 375 μL. From the second group, 4 mL EDTA blood was used to extract DNA, where after DNA samples were stored in aliquots of 25 μL. Blood and DNA aliquots were stored at three different temperatures (4°C, −20°C, −80°C) for 1, 6, 12 or 18 months prior to (hydroxy)methylation quantification (). Blood samples stored at 4°C were stored for 1 month, as blood storage at this temperature is usually short term. From all subjects, one sample was processed and quantified immediately, which was used as baseline measurement. From the third group, 1.6 mL EDTA blood was collected of which half was used to store blood samples and half was used to extract and store DNA. Blood and DNA samples of this group were used to assess the effect of freeze-thaw cycles over three consecutive days after blood collection (0, 1, 2, 3 times) on the stability of DNA (hydroxy)methylation. DNA and blood samples were frozen at −80°C and thawed at room temperature on a laboratory tube roller for 2 h (=1 cycle) before they were refrozen or further processed for DNA (hydroxy)methylation quantification. DNA (hydroxy)methylation was quantified within the same day in all samples.
Figure 1. Experimental set up of material stored at different temperatures
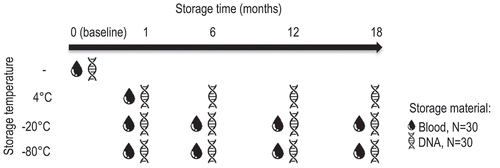
Figure 2. DNA (hydroxy)methylation over time for blood and DNA samples stored at different temperatures
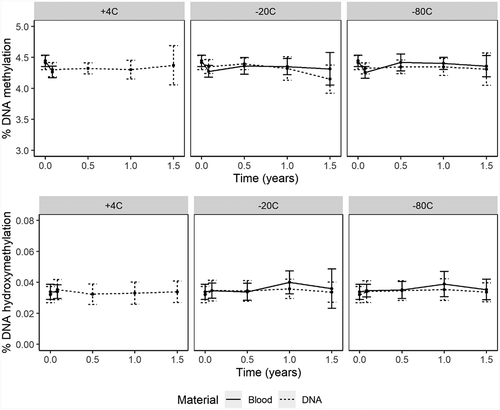
Figure 3. Effect of freezing and thawing on the stability DNA (hydroxy)methylation
DNA extraction
DNA was extracted from EDTA whole blood using the MagNA Pure Compact Nucleic Acid Isolation Kit I, following manufacturer’s protocol (cat.no: 03730964001, Roche Molecular Biochemicals®) [Citation10][. DNA concentrations were assessed using a NanoDrop ND-1000 Spectrophotometer with DNA-50 default settings (NanoDrop Technologies, Wilmington, DE, USA).
DNA (hydroxy)methylation quantification
Quantification of DNA (hydroxy)methylation was performed using a liquid chromatography – electrospray ionization – tandem mass spectrometry (LC-ESI-MS/MS) method, as previously described [Citation10][]. To reduce technical variances, a batch sample of internal standard and a batch calibration curve (stored at −80°C) were used for (hydroxy)methylation quantification at each time point. For hydroxymethylation, samples with analyte areas <50 000 were excluded, as these were too low to correctly quantify DNA hydroxymethylation. For DNA methylation, all areas were above 50 000. As an additional quality check, we assessed the ratios between areas of the quantifier and qualifier of 2ʹ-deoxyguanosine, as these should be similar each day. DNA methylation samples in which this ratio deviated >10% from the mean of that day, were excluded. A random DNA sample was stored in batches of 27 ng/µL at −80°C which was used as batch control: these samples were digested and quantified in each run together with the rest of the samples and acted as quality control. The method was previously validated. To determine precision 5-hmdC/2-dG and 5-mdC/2-dG were quantified in fourfold over 5 days in DNA samples with low (27 ng/µL) and high (54 ng/µL) concentrations. Results of the precision experiment are shown in . Samples were all diluted to 30 ng/µL, hence for this study, we compared samples to the precision of the method for the low DNA concentration (27 ng/µL). The LLOQ for DNA hydroxy(methylation) was 10 ng/mL, which was determined according to Clinical Laboratory and Standards Institute (CLSI) protocol EP-17a. To determine the dynamic range of the method, a random DNA sample of 27 ng/ul was spiked with 6 increasing concentrations, each measured in quadruple. Linearity was assessed using a lack-of-fit test using the Excel plug-in Analyse-it, where values <3.29 were considered linear. The range and corresponding lack-of-fit measure were as follows: 5-hmdC between 5.9 and 36 nM (lack-of-fit = 0.16), 5-mdC between 552 and 3553 nM (lack-of-fit = 2.47) and 2-dG between 13159 and 23159 nM (lack-of-fit = 2.22).
Table 1. Precision results of method validation
Statistics
Percentages of global DNA (hydroxy)methylation were presented as mean ± standard deviation (SD). Linear mixed models were used to assess changes in DNA (hydroxy)methylation over time. Main effects included in the models were: temperature, material, and time. To assess whether DNA (hydroxy)methylation changed differently over time for DNA and blood samples stored at different temperatures, two and three-way interaction terms between temperature, material, and time were examined. The models with two-way interaction terms and a model with a three-way interaction term between time, material, and temperature were compared using a chi-square log likelihood ratio test, where a p-value <0.05 was considered a significant difference. Additionally, overall model qualities were assessed using the Akaike information criterion (AIC), where the lower the AIC the better the model fits the data. Here, a difference of at least two AIC units was considered a significantly better fit [Citation29]. Upon significance of (higher-term) interactions, all lower-term interactions were kept in the model to keep the models hierarchically well formulated (HWF) [Citation30,Citation31]. Additionally, to assess whether DNA (hydroxy)methylation was stable over time, we completely removed time from the model and compared the model with and without time component. Upon a change of ≥2 units in AIC upon removal of the time component, time was considered significantly important for the model, meaning that there are changes over time. Random effects included in the model were: (A) random intercept for time (accounting for inter-subject variability in (hydroxy)methylation levels at baseline, which may influence stability over time) and (B) random slopes for subjects (hydroxymethylation) or for subjects grouped within storage temperature (methylation), adjusting for the fact that samples from the same subject were higher correlated over time than from different subjects. Final model for the analysis of DNA methylation over time per condition was: lme(fixed = methylation ~ material * temperature * time, random = ~time|id/temperature) and for DNA hydroxymethylation: lme(fixed = hydroxymethylation ~ temperature + material * time, random = ~time|id). For the analysis of freeze-thaw cycles on the stability of DNA (hydroxy)methylation we used a linear mixed model including fixed effects for: material and number of cycles (cycles) and the following random effects: random intercept and random slope for subjects (id). Final models for both DNA methylation and hydroxymethylation upon freezing and thawing were as followsꓽ lme(data = DF, fixed = (hydroxy)methylation ~ material * cycles, random = ~ 1 | id). All analyses were performed in R studio (R version 3.6.1 (2019–07-05)) using the linear mixed effect model (lme) function of the ‘nlme’ package and were fitted using the maximum likelihood ‘ML’ method [Citation32].
Supplemental Material
Download MS Word (73.6 KB)Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- Felsenfeld G. A brief history of epigenetics. Cold Spring Harb Perspect Biol. 2014;6. DOI:10.1101/cshperspect.a018200
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38.
- Melamed P, Yosefzon Y, David C, et al. Variants, and differential effects on function. Front Cell Dev Biol. 2018;6:22.
- Pfeifer GP, Kadam S, Jin S-G. 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics Chromatin. 2013;6(1):10.
- Henderson-Smith A, Fisch KM, Hua J, et al. DNA methylation changes associated with Parkinson’s disease progression: outcomes from the first longitudinal genome-wide methylation analysis in blood. Epigenetics. 2019;14(4):365–382.
- Jiang D, Sun M, You L, et al. DNA methylation and hydroxymethylation are associated with the degree of coronary atherosclerosis in elderly patients with coronary heart disease. Life Sci. 2019;224:241–248.
- Ciccarone F, Valentini E, Malavolta M, et al. DNA hydroxymethylation levels are altered in blood cells from down syndrome persons enrolled in the MARK-AGE project. Journals Gerontol Ser A. 2018;73:737–744.
- Zouali M. Epigenetics of autoimmune diseases. Autoimmune Dis. 2020:429–466 Elsevier. doi:10.1016/b978-0-12-812102-3.00025-7
- Skvortsova K, Stirzaker C, Taberlay P. The DNA methylation landscape in cancer. Essays Biochem. 2019;63(6):797–811.
- Gosselt HR, van Zelst BD, de Rotte MCFJ, et al. Higher baseline global leukocyte DNA methylation is associated with MTX non-response in early RA patients. Arthritis research & therapy. 2019;21(1):157. DOI:10.1186/s13075-019-1936-5
- Ciechomska, M., Roszkowski, L., & Maslinski, W. DNA methylation as a future therapeutic and diagnostic target in rheumatoid arthritis. Cells. 2019;8(9):953.
- Talens RP, Boomsma DI, Tobi EW, et al. Variation, patterns, and temporal stability of DNA methylation: considerations for epigenetic epidemiology. Faseb J. 2010;24(9):3135–3144.
- Zaimi I, Pei D, Koestler DC, et al. Variation in DNA methylation of human blood over a 1-year period using the Illumina MethylationEPIC array. Epigenetics. 2018;13(10–11):1056–1071.
- Wang Y, Zheng H, Chen J, et al. The impact of different preservation conditions and freezing-thawing cycles on quality of RNA, DNA, and proteins in cancer tissue. Biopreserv Biobank. 2015;13(5):335–347.
- Li Y, Pan X, Roberts ML, et al. Stability of global methylation profiles of whole blood and extracted DNA under different storage durations and conditions. Epigenomics. 2018;10:797–811.
- Huang LH, Lin PH, Tsai KW, et al. The effects of storage temperature and duration of blood samples on DNA and RNA qualities. PLoS One. 2017;12. DOI:10.1371/journal.pone.0184692.
- Schröder C, Steimer W. gDNA extraction yield and methylation status of blood samples are affected by long-term storage conditions. PLoS One. 2018;13. DOI:10.1371/journal.pone.0192414
- Bulla A, De Witt B, Ammerlaan W, et al. Blood DNA yield but not integrity or methylation is impacted after long-term storage. Biopreserv Biobank. 2016;14(1):29–38.
- Coppieters N, Dieriks BV, Lill C, et al. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol Aging. 2014;35:1334–1344.
- Song L, James SR, Kazim L, et al. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77(2):504–510.
- Kok, R. M., Smith, D. E., Barto, R., et al. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: analytical technique, reference values and determinants in healthy subjects in. Clinical Chemistry and Laboratory Medicine (CCLM). 2007;45(7):903–911.https://www.degruyter.com/view/journals/cclm/45/7/article-p903.xml
- Godderis L, Schouteden C, Tabish A, et al. Global methylation and hydroxymethylation in DNA from blood and saliva in healthy volunteers. Biomed Res Int. 2015;2015:1–8.
- Weiner AS, Boyarskikh UA, Voronina EN, et al. Methylenetetrahydrofolate reductase C677T and methionine synthase A2756G polymorphisms influence on leukocyte genomic DNA methylation level. Gene. 2014;533(1):168–172.
- Liu J, Hesson LB, Ward RL. Liquid chromatography tandem mass spectrometry for the measurement of global DNA methylation and hydroxymethylation J Proteomics Bioinform S. 2013;2:5–10.
- Kurdyukov S, Bullock M, Methylation Analysis: DNA Choosing the right method. Biology, 2016;5(1):3. DOI:10.3390/biology5010003
- Staunstrup NH, Starnawska A, Nyegaard M, et al. Genome-wide DNA methylation profiling with MeDIP-seq using archived dried blood spots. Clin Epigenetics. 2016;8(1):81.
- Lisanti S, Omar WAW, Tomaszewski B, et al. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS One. 2013;8:79044.
- Hohos NM, Lee K, Ji L, et al. DNA cytosine hydroxymethylation levels are distinct among non-overlapping classes of peripheral blood leukocytes. J Immunol Methods. 2016;436:1–15.
- Burnham KP, Anderson DR Multimodel inference understanding AIC and BIC in model selection. Sociological methods & research. 2004;33(2):261–304. doi:10.1177/0049124104268644.
- Zerback T. Interaction Effects. Int. Encycl. Commun. Res. Methods. Wiley; 2017. 1–9. Doi:10.1002/9781118901731.iecrm0118
- Modeling Strategy Guidelines. Logist Regres. New York: Springer-Verlag. 2006;161–189. doi:10.1007/0-387-21647-2_6
- Package “nlme”. 2020.