
ABSTRACT
The class III histone deacetylase sirtuin 1 (SIRT1) plays a pivotal role in numerous biological and physiological functions, including inflammation. An association between SIRT1 and proinflammatory cytokines might exist. In addition to their important role in inflammation associated with rheumatoid arthritis (RA), proinflammatory cytokines mediate the development of systemic effects. Here, we evaluated systemic SIRT1 expression and enzymatic activity, in peripheral blood mononuclear cells (PBMCs) and in liver isolated from rats with adjuvant-induced arthritis (AIA), treated or not with low or high doses of glucocorticoids (GCs). We also measured the production of tumour necrosis factor alpha (TNF) and interleukin-1 beta (IL-1 beta) in PBMCs and liver. We found that SIRT1 expression and activity increased in PBMCs of AIA rats compared to healthy controls and decreased under GC treatment. Similarly, we observed an increased SIRT1 activity in the liver of AIA rats compared to healthy controls which decreased under high doses of GCs. We also found an increase in IL-1 beta and TNF levels in the liver of AIA rats compared to healthy controls, which decreased under high doses of GC. We did not observe a significant correlation between SIRT1 activity and proinflammatory cytokine production in PBMC or liver. In contrast, a strong positive correlation was found between the liver levels of TNF and IL-1 beta (rho=0.9503, p=7.5x10−21). Our results indicate that increased inflammation in AIA rats compared to healthy control is accompanied by an increased SIRT1 activity in both PBMCs and liver, which could be decreased under GC treatment.
Introduction
Rheumatoid arthritis (RA) is an autoimmune rheumatic disease characterized by a state of chronic inflammation and progressive joint destruction, along with systemic manifestations and severe disability [Citation1]. Although the etiopathogenesis of RA is not copiously delineated, it is well known that the deregulation in the cytokine network towards the activation of proinflammatory cytokines can fuel and promote the induction of autoimmunity and chronic inflammation, engendering thus joint damage [Citation2]. In this context, distinct proinflammatory cytokines are recognized to be key players in the synovial inflammation of RA [Citation3]. For instance, tumour necrosis factor (TNF) alpha and interleukin-1 beta (IL-1 beta) play a central role in mediating the inflammatory response in the rheumatoid synovial tissue. This is mediated by their pivotal role not only through aberrantly activating osteoclasts and chondrocytes that prompt bone and cartilage destruction, but also by acting as autocrine stimulators and paracrine inducers of other inflammatory players [Citation4,Citation5]. In addition to their local implication in inflammation and articular destruction in RA, proinflammatory cytokines promote the development of a multitude of systemic effects, including anaemia, cardiovascular diseases, osteoporosis, fatigue and others [Citation6]. In addition, there is increasing evidence for the critical role of the liver in modulating the immune response in autoimmune and chronic inflammatory diseases [Citation7]. Interestingly, cytokines are well thought out to be a potential powerful diagnostic tool as biomarkers of RA as well as an efficient therapeutic target, thus highlighting their crucial role in autoimmune diseases [Citation8].
Sirtuin 1 (SIRT1) is an NAD+-dependent class III histone/protein deacetylase. It is the most studied among the seven sirtuins (Silent Information Regulator proteins) family members [Citation9–11]. SIRT1 is linked to a diversity of pathophysiological processes in mammalian cells comprising the regulation of cell survival, apoptosis, gluconeogenesis, adipogenesis and lipolysis [Citation12–15]. This is ensured by the wide array of downstream targets including for instance transcription factors or chromatin-remodelling proteins such as peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1 alpha, p53 and nuclear factor-kappa B (NF-kappaB) [Citation16,Citation17]. Intriguingly, the presence of SIRT1 activity in animal mesenchymal cells has been highlighted [Citation18] and its activation promotes osteoblast differentiation via its action on p53 in a mouse model of mesenchymal stem cells [Citation19]. Besides, SIRT1 has been revealed to be a pivotal player in the regulation of local and systemic inflammation, along with bone and cartilage remodelling [Citation20–24]. Proinflammatory mediators were shown to be regulated by SIRT1 [Citation12,Citation25]. SIRT1 activators, including resveratrol, usually attenuate the release of inflammatory cytokines [Citation26,Citation27] and hinder the synthesis of TNF and IL-8 [Citation12,Citation28] possibly through the deacetylation of NF-κB RelA/p65 subunit at lysine 310 [Citation12]. In line with this, myeloid-specific SIRT1 knockout in mice resulted in a significant increase in TNF, IL-6, and IL-1 beta production by macrophages in response to infection and inflammation [Citation29]. Conversely, NF-kB activation increases both SIRT1 and TNF gene expressions [Citation30] and TNF can inactivate SIRT1, thus blocking its ‘protective’ role [Citation26,Citation27,Citation31,Citation32].
Genuinely, the homoeostasis of SIRT1 expression has been reported to favour a long-lasting healthy life [Citation33–35]. In the context of RA, the function of SIRT1 as protective or proinflammatory inducer remains controversial and limited. It has been shown that RA patients exhibit a decreased SIRT1 activity and down-regulated SIRT1 protein expression compared to healthy subjects [Citation36,Citation37]. Inhibition of synovial angiogenesis through increased SIRT1 activity has been described to alleviate arthritis [Citation29]. Contrariwise, SIRT1 overexpression induced by TNF was shown to contribute to chronic inflammation by constraining apoptosis in RA synovial cells and increasing proinflammatory cytokines levels [Citation13]. In light of what was mentioned, a complex interrelation might exist between SIRT1 expression and the control of proinflammatory cytokines such as TNF, IL-1 beta and IL-6 in the context of RA. Such linkage could be possibly considered as a relevant marker of a sustained control of the inflammatory process [Citation38] and as emerging cell and cytokine targets in RA [Citation39,Citation40]. To study the impact of glucocorticoid (GC) treatment on SIRT1 expression and the production of proinflammatory cytokines, we assessed the effect of prednisolone treatment in an animal model of arthritis, specifically rats with adjuvant-induced arthritis (AIA). We fixed two main objectives for our study that focuses on the systemic effect of SIRT1 and proinflammatory cytokines. First, we assessed the expression and enzymatic activity of SIRT1 in PBMCs and liver of AIA rats compared to healthy controls and following treatment with low or high concentrations of prednisolone. Second, we analysed the relationship between SIRT1 activity and markers of inflammation, specifically TNF and IL-1 beta, in the AIA rat liver and PBMCs in the presence and absence of prednisolone treatment.
Materials and methods
Animals
Animals were studied as previously reported [Citation41]. Six-week-old male Lewis rats (n = 40) were purchased from Janvier (Le Genest-Saint-Isle, France). Animals were kept under a 12 h–12 h light–dark cycle and allowed free access to food and water. The experimental procedures were approved by the local committee for ethics in animal experimentation no. 2015–001-CD-5PR of Franche-Comte University (Besançon, France), and complied with the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
Induction and clinical evaluation of the arthritis model
Induction and clinical evaluation of the AIA rat model were done as described previously [Citation41]. Briefly, arthritis was induced in male Lewis rats (6 weeks old) by a single intradermal injection at the base of the tail of 120 microl of 1 mg of heat-killed Mycobacterium butyricum (Difco, Detroit, MI, USA) suspended in 0.1 ml of mineral oil (Freund’s incomplete adjuvant; Difco). A group of saline-infused non-arthritic rats was used as control. The AIA model is characterized by rapid onset and progression of a robust and easily measurable polyarthritis (characterized by severe erythema and diffuse soft swelling with complete ankyloses and malformations in the paws), reduced locomotor activity, and is frequently associated with ear and tail inflammation, weight loss, anorexia and diarrhea. All rats were daily monitored for clinical signs of arthritis. The scoring system was employed as follows [Citation42]: arthritis of one finger scores 0.1, weak and moderate arthritis of one big joint (ankle or wrist) scores 0.5 and intense arthritis of one big joint scores 1. Tarsus and ankle were considered as the same joint. Sum of joint scores of four limbs leads to an arthritis score of maximum 6 to each rat. Ankle diameter was measured with digital callipers (Vernier Stainless).
Drug treatment
AIA rats were treated with GCs according to a previous protocol described in [Citation41]. Briefly, AIA rats were randomized in three groups at the start of arthritis (i.e., at days 11–12 post-immunization). Rats in the first group were injected intraperitoneally (i.p.) with prednisolone phosphate sodium salt at a dose of 13.4 mg/kg/day (equivalent to 10 mg/kg/day prednisolone) [GC high dose (GC-HD), n = 10] for 3 weeks (subchronic treatment). This dose was shown previously to result in a significant reduction in arthritis severity [Citation43]. Rats in the second group were injected intraperitoneally (i.p.) with prednisolone phosphate sodium salt at a dose of 0.134 mg/kg/day (equivalent to 0.1 mg/kg/day prednisolone) [GC low dose (GC-LD), n = 10] for 3 weeks (subchronic treatment). This low dose is a subtherapeutic dose [Citation41] used here as a pharmacological tool to determine whether a SIRT1 effect of GC might occur independently of the reduction of arthritis severity. The third group received saline at 1 ml/kg/day (i.p.) for 3 weeks (vehicle, n = 10).
Tissue collection
Twenty-one days after the initiation of treatment, rats were anaesthetized with pentobarbital (60 mg/kg, i.p.). Blood was then withdrawn from the abdominal artery and divided into two parts. One part was used for PBMC isolation. The second one was centrifuged to obtain plasma that was stored at −80°C until analysis. Livers were removed, frozen in liquid nitrogen, then stored at −80°C.
PBMC isolation from whole blood
PBMCs were isolated by Ficoll gradient centrifugation. Blood from rats was diluted with equal amounts of PBS, overlaid on Ficoll medium (Eurobio) and centrifuged at 900 X g for 30 min at 25°C. The PBMC band was removed and washed twice with PBS.
Measurement of SIRT1 expression and enzymatic activity
SIRT1 expression in PBMCs was assessed using western blotting analysis, as described previously [Citation37]. SIRT1 enzymatic activity was measured in PBMC and liver nuclear extracts using a colorimetric assay (Epigenase Universal SIRT Activity/Inhibition Assay Kit, Epigentek, Farmingdale, NY, USA), according to the manufacturer’s recommendations.
Measurement of proinflammatory cytokines
The levels of IL-1 beta and TNF were quantified in liver tissue and PBMC lysates by using an ELISA assay according to the manufacturer’s recommendations (Promokine GmbH, Heidelberg, Germany).
Quantification of proinflammatory cytokine transcripts in the liver tissue by RT-PCR
TNF and IL-1 beta mRNA levels were assessed in rat liver by RT-qPCR. Briefly, total RNA was extracted from rat liver using EZNA. RNA extraction kit according to the manufacturer’s instruction (Omega Bio-Tek). RNA was reverse transcribed into cDNA using Superscript IV (Life Technologies). The reverse transcription product was amplified using primers targeting TNF, IL-1 beta and beta-actin with KiCqStart SybrGreen mix (Sigma) according to manufacturer’s instructions on a Stratagene Mx3005P thermocycler (Agilent). The sequences of the primers used were as follows: IL-1 beta: forward 5ʹ-CACCTCTCAAGCAGAGCACAG-3ʹ, reverse 5ʹ-GGGTTCCATGGTGAAGTCAAC-3ʹ; TNF: forward 5ʹ-AAATGGGCTCCCTCTCATCAGTTC-3ʹ, reverse 5ʹ-TCTGCTTGGTGGTTTGCTACGAC-3ʹ; beta-actin: forward 5ʹ-AAGTCCCTCACCCTCCCAAAAG-3ʹ, reverse 5ʹ-AAGCAATGCTGTCACCTTCCC-3ʹ.
Data and statistical analysis
Data are expressed as the mean ± standard error of the mean (s.e.m). Comparison between two values was assessed by an unpaired Student’s t-test, or a Mann–Whitney test when the data were not normally distributed. The comparison of evolution curves of arthritis score and paw diameter was made between two groups by two-way analysis of variance (ANOVA) for repeated measures. Pearson correlation coefficient was used to identify and test the strength of a relationship between two sets of data. The significance level used was p < 0.05.
Results
Effects of GCs treatment on the arthritis severity
We assessed the effect of GCs treatment on the arthritis severity. As expected, the high dose of prednisolone significantly reduced the mean arthritis scores, whereas the low dose had no effect ( b)). The clinical effect of prednisolone was not associated with a decrease in paw diameter ( d)).
Figure 1. Evolution of arthritis severity in vehicle- and prednisolone-treated rats. Arthritis scores were plotted over time after adjuvant-induced arthritis induction and were compared to AIA left untreated (AIA) in low dose of prednisolone group (a, LD-GCs) and high dose of prednisolone group (b, LD-GCs). The evolution of paw diameter was represented (c, d). Values are the mean (S.E.M.) (n = 10/group). ***P < 0.001
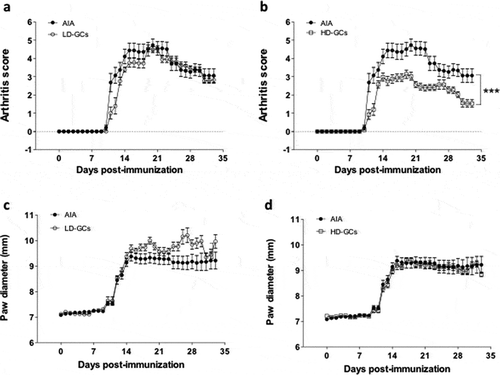
Activity of SIRT1 in PBMCs isolated from rats with AIA and treated with GCs
Nuclear extracts of PBMCs were isolated from healthy rats, rats with AIA left untreated or treated with LD and HD of GCs. Enzymatic activity of SIRT1 in those extracts was then measured using a colorimetric enzymatic assay. As expected, SIRT1 activity was increased by 68% (p = 0.008) in PBMCs isolated from AIA rats compared to healthy rats ()). Alternatively, treatment of AIA rats with LD-GCs and HD-GCs reduced SIRT1 activity by 23% (non-significant) and 33% (p = 0.039), respectively compared to AIA rats ()). To note that AIA rats with LD-GCs still show a significant increase by 30% (p = 0.027) in the enzymatic activity compared to healthy rats, with an increase of 14% (non-significant) in the AIA rats with HD-GCs ()).
Figure 2. SIRT1 activity and expression in PBMCs isolated from healthy control rats and rats with AIA left untreated or treated with GCs. (a) Nuclear SIRT1 activity in PBMCs isolated from healthy rats (n = 10) and rats with AIA treated with low (n = 10) and high (n = 10) doses of prednisolone or left untreated (n = 10) was measured as indicated in Materials and Methods. Boxes show mean values, with Q1-Q3 range. (b) Western-bot analysis for SIRT1 expression in PBMCs isolated from healthy rats (n = 4) and rats with AIA treated with low (n = 4) and high (n = 4) doses of prednisolone or left untreated (n = 4). Beta-actin was used as an internal control. Representative data are shown. (c) Ratio of SIRT1/beta-actin expression as measured by arbitrary units (n = 16 rats). Boxes show mean values, with Q1-Q3 range
Expression of SIRT1 in PBMCs isolated from rats with AIA and treated with GCs
Next, the expression (amount) of SIRT1 in PBMCs isolated from healthy rats, rats with AIA left untreated or treated with LD and HD of GCs was assessed using western blotting. Data were then analysed by densitometry quantification using ImageJ software ()). In line with the enzymatic activity, SIRT1 expression was increased by 2.3-fold (p = 0.029) in PBMCs isolated from AIA rats compared to healthy rats ()). Conversely, treatment of AIA rats with LD-GCs and HD-GCs reduced SIRT1 activity by 31% and 28%, respectively compared to AIA rats but not in a significant manner ()). Expression of SIRT1 in AIA rats treated with LD-GCs or HD-GCs still show an increase by 66% (non-significant) compared to healthy rats ()).
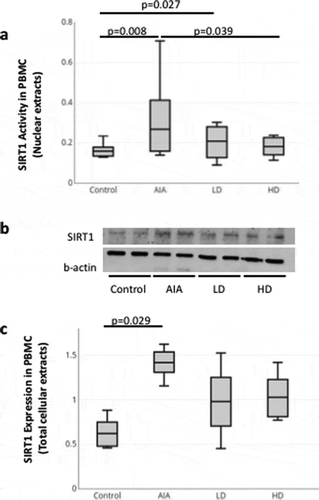
Activity of SIRT1 in livers isolated from rats with AIA and treated with GCs
Nuclear extracts of livers were isolated from healthy rats, rats with AIA left untreated or treated with LD and HD of GCs. Enzymatic activity of SIRT1 in those extracts was then measured using a colorimetric enzymatic assay. As expected, SIRT1 activity was increased by 2.6-fold (p = 0.015) in livers isolated from AIA rats compared to healthy rats (). Alternatively, treatment of AIA rats with HD-GCs reduced SIRT1 activity by 33% (non-significant, p = 0.151) compared to AIA rats (). To note that AIA rats with LD-GCs still show a non-significant increase (p = 0.310) in the enzymatic activity compared to healthy rats ().
Figure 3. SIRT1 activity in livers isolated from healthy control rats and rats with AIA left untreated or treated with GCs. Nuclear SIRT1 activity in livers isolated from healthy rats (n = 5) and rats with AIA treated with low (n = 5) and high (n = 5) doses of prednisolone or left untreated (n = 5) was measured as indicated in Materials and Methods. Boxes show mean values, with Q1-Q3 range
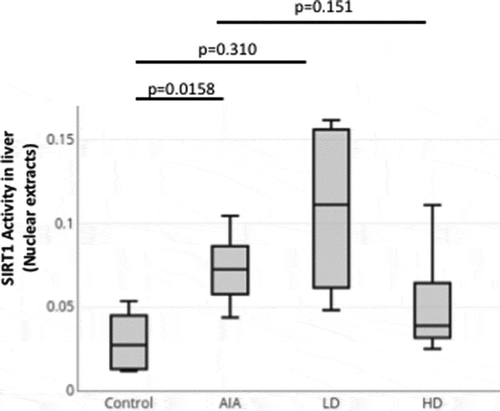
Measurement of the levels of proinflammatory cytokines in liver and PBMCs of rats with AIA treated with GCs
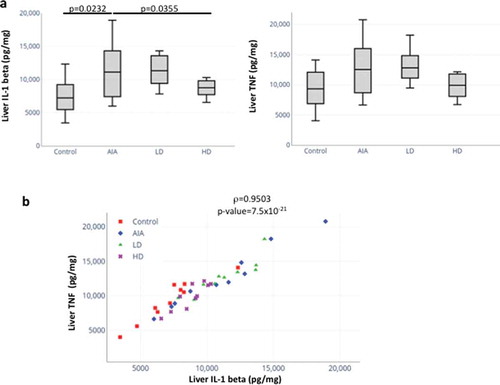
The levels of proinflammatory cytokines TNF and IL-1 beta in cell lysates from liver isolated from healthy rats, rats with AIA untreated or treated with LD-GCs and HD-GCs were measured ()). A statistically significant increase in IL-1 beta levels (p = 0.0232) was found between AIA rats compared to healthy control rats. In addition, a statistically significant decrease in IL-1 beta levels (p = 0.0355) was recorded in AIA rats treated with HD-GCs compared with AIA rats ()). For AIA rats treated with LD-GCs, the level of liver IL-1 beta did not change significantly compared to untreated rats ()). On the other hand, a non-significant increase for TNF in the liver from rats with AIA compared to HC and a non-significant decrease in the expression of TNF in AIA rats treated with HD-GCs compared to untreated AIA rats were found (). For AIA rats treated with LD-GCs, the level of liver TNF did not change significantly compared to untreated AIA rats ()). Furthermore, by using Pearson correlation, we were able to show a positive trend between the liver levels of TNF and IL-1 beta (rho = 0.9503, p = 7.5x10−21) ()).
Figure 4. Expression and correlation of proinflammatory cytokines in liver from healthy rats, rats with AIA left untreated or treated with GCs. (a) The levels of proinflammatory cytokines TNF and IL-1 beta were measured in cell lysates from livers isolated from healthy rats (n = 10), rats with AIA untreated (n = 10) or treated with LD-GCs (n = 10) and HD-GCs (n = 10) using an ELISA assay (n = 2). Boxes show mean values, with Q1-Q3 range. p-values were calculated using a Mann-Whitney test between two groups. (b) Significant positive correlation between TNF and IL-1 beta expression in liver of rats using Pearson correlation (rho = 0.95, p = 7.5x10−21)
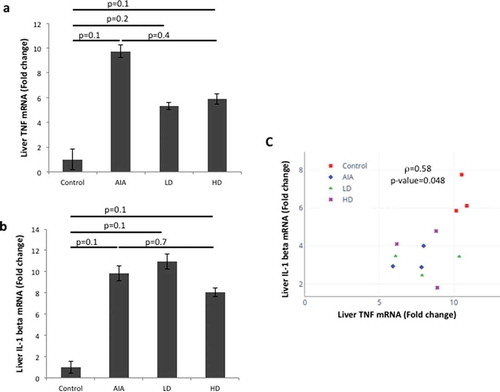
We then measured TNF and IL-1 beta transcripts in cell lysates from liver isolated from healthy rats, rats with AIA untreated or treated with LD-GCs and HD-GCs (). We observed an increase of nine-fold in the levels of TNF transcripts in AIA rats compared to control rats ()). Levels of TNF transcripts were decreased by 45% and 40% respectively, in AIA rats treated with LD-GCs and HD-GCs compared to untreated AIA rats ()). An increase of nine-fold in the levels of IL-1 beta transcripts in AIA rats compared to control rats was also found ()). Levels of IL-1 beta transcripts were decreased by 19% in AIA rats treated with HD-GCs compared to untreated AIA rats ()). A positive correlation was found between the levels of TNF and IL-1 beta transcripts in liver (rho = 0.58, p = 0.048) ()). Although SIRT1 activity and TNF and IL-1 beta levels in liver showed similar trends, the levels of TNF and IL-1 beta and the SIRT1 activity in the liver did not show a significant correlation (rho = 0.1314, p = 0.581 and rho = 0.2174, p = 0.357, respectively) (Suppl. Table 1). Similarly, SIRT1 activity in PBMC and TNF and IL-1 beta levels in PBMC did not show a significant correlation (rho = −0.1835, p = 0.299 and rho = 0.1765, p = 0.626, respectively) (Suppl. Table 1).
Figure 5. Expression levels of TNF and IL-1 beta transcripts in liver from healthy rats, rats with AIA left untreated or treated with GCs. The expression level of TNF (a) and IL-1 beta (b) was assessed by RT-qPCR on RNA extracted from livers isolated from healthy rats (n = 3), rats with AIA untreated (n = 3) or treated with LD-GCs (n = 3) and HD-GCs (n = 3). Histograms show the mean (±SD) fold-change in expression as compared to healthy rats. (c). Positive correlation between TNF and IL-1 beta transcripts levels in liver using Pearson correlation (rho = 0.58, p = 0.048)
Discussion
In this study, the activity and expression of SIRT1 in PBMCs and liver isolated from rats with AIA compared to healthy rats, as well as the effect of GC on SIRT1 were assessed. In parallel, levels of proinflammatory cytokines in liver and PBMCs were measured. Our results show an increase in SIRT1 activity and expression in PBMCs isolated from rats with AIA compared to healthy rats, with a noted decrease under GC treatment. Similarly, increased SIRT1 activity was observed in liver isolated from rats with AIA compared to healthy rats, with a remarkable reduction under high dose GC treatment. In the liver of AIA rats, both SIRT1 activity and levels of proinflammatory cytokines were increased compared to control rats, and diminished under GC treatment. However, and independently of the tissue studied (PBMCs, liver), we did not observe a significant correlation between SIRT1 activity and proinflammatory cytokines production.
As expected [Citation39], a high dose of prednisolone induced a significant decrease in the arthritis score in the AIA model. This effect was not associated with a decrease in paw diameter, suggesting that a dissociation may exist between the effect on the synovitis and on the bone tissue. In line with this, radiographic analysis of ankle and foot in rats treated by the same dose of prednisolone showed a better effect on erosion than in bone formation [Citation42].
In addition to their local effects in RA, proinflammatory cytokines mediate the development of some systemic effects such as anemia and cardiovascular diseases [Citation6]. In agreement with the reported systemic effects in RA, we observed an increased SIRT1 expression in PBMCs isolated from AIA rats compared to healthy rats. This is in line with the SIRT1 overexpression reported in the RA synovium [Citation13]. This was also consistent with a previous report by Engler et al. that showed a positive correlation between amplified synovitis and enhanced SIRT1 expression in RA synovial tissues [Citation44]. Given that we cannot rule out that the measure of SIRT1 activity rather than SIRT1 expression is a more reliable marker of SIRT1 biological effect, we therefore analysed SIRT1 activity. In consort with the increased expression, we also observed an increase in SIRT1 activity in PBMCs of AIA rats compared to healthy rats. We reported previously that SIRT1 activity is present both in the cytoplasm and in the nucleus of human PBMCs [Citation9,Citation36]. In addition, a nucleo-cytoplasmic shuttling of SIRT1 has been reported [Citation16]. In the present study, we measured the activity of SIRT1 in nuclear lysates. In fact, several factors including age, diabetes and obesity have been reported to modulate SIRT1 activity [Citation14]. In contrast to the increased nuclear SIRT1 activity in PBMCs from rats with AIA compared to healthy controls, our group did not find previously any significant difference in the cytoplasmic SIRT1 activity in PBMCs of RA patients compared to healthy subjects [Citation37]. This apparent discrepancy could result not only from distinct SIRT1 regulation in rats and in humans, but also from the study of nuclear SIRT1 activity versus cytoplasmic SIRT1 activity. Similar to PBMC biological findings, we observed an increase in SIRT1 activity in livers of AIA rats compared to healthy rats, indicating that upregulated SIRT1 activity might be a more general biological feature of AIA irrespective of the tissue studied, which is in line with the systemic effects observed in RA.
GCs constitute a cornerstone in the management of RA [Citation45,Citation46]. GC has been reported to inhibit SIRT1 in human tenocytes [Citation46]. SIRT1 interacts physically with the GC receptor and is a transcriptional enhancer of GR-induced transcriptional activity [Citation47]. Although the expression of SIRT1 and the regulation of proinflammatory cytokines such as TNF and IL-1 beta have been addressed previously [Citation48–50], the impact of GCs on SIRT1 expression and activity in PBMCs and liver and its interrelation with TNF and IL-1 beta have not been commonly studied so far in the context of arthritis. In a previous report from our group, we observed a decrease in the plasma levels of TNF and IL-1 beta in rats with AIA treated with GCs compared to healthy rats [Citation41]. In the present study, we decided to quantify the levels of proinflammatory cytokines in both PBMCs and liver of AIA rats treated or not with GCs. We observed a low level of IL-1 beta and TNF in the liver of AIA rats treated with GCs. We observed an increase in TNF and IL-1 beta transcript levels in the liver of AIA rats compared to healthy rats (). This is consistent with increased levels of TNF and IL-1 beta proteins measured in the liver of AIA rats ()). Parallel to the decreased levels of TNF and IL-1 beta proteins measured in the liver of AIA rats treated with a high dose of GCs compared to untreated AIA rats ()), we observed decreased levels of TNF and IL-1 beta transcripts ( b)). In addition, a positive correlation was found between the levels of TNF and IL-1 beta transcripts in the liver ()). Altogether, our results indicate that the control of TNF and IL-1 beta expression in the liver of AIA rats treated with GCs might occur at the transcriptional level.
Distinct cell types may express distinct levels of SIRT1 and could respond differently to GC treatment in vitro. SIRT1 inhibition decreased the production of IL-6, TNF and RANTES (regulated upon activation normal T cell expressed and secreted) in LPS-stimulated macrophages [Citation32], suggesting that sirtuin activation may favour proinflammatory cytokine production by the activated macrophages. It is worthy to mention that polarization of macrophages from SIRT1 transgenic (Tg)-mice towards the M2 phenotype resulted in lower TNF and IL-1 beta expressions [Citation51]. In patients with RA, the inhibition of SIRT1 enzymatic activity reduced LPS-induced levels of TNF in monocytes [Citation13]. NF-kB activation results in the enhancement of both SIRT1 and TNF gene expressions [Citation30,Citation52]. Therefore, the parallel increase in SIRT1 activity and proinflammatory cytokines such as TNF could result from the activation of a common pathway leading to NF-kB activation. Although we did not measure NF-kB activation in our study, we observed a parallel increase in SIRT1 activity and levels of proinflammatory cytokines namely TNF and IL-1beta in the liver of AIA rats compared to healthy controls.
In contrast, SIRT1 inhibition has been reported to increase IL-6 expression via caveolin in fibroblasts [Citation53]. Likewise, myeloid cell-specific deletion of SIRT1 increased IL-1 and TNF expression, thus exacerbating inflammatory arthritis in an in vivo mice model [Citation54], advocating that SIRT1 activation may be favourable in the treatment of RA. This is in line with SIRT1-mediated suppression of IL-6 release after activation by resveratrol in fibroblastic cells [Citation55]. Hence, the regulation of IL-6, IL-1 and TNF production by SIRT1 may depend on the cell type or the model involved, highlighting the importance of defining the specific experimental conditions in which SIRT1 modulation of proinflammatory cytokines is assessed. In agreement with this, in the liver tissue analysed in our study, GCs may impact differentially SIRT1 in several cell types, e.g., hepatocytes, fibroblasts, Kupffer cells. Indeed, the level of cytokine production observed in the liver could result from distinct SIRT1 responses to GCs in these liver cells. In agreement with this hypothesis, the impact of GC on isolated cells cultivated in vitro is different from the impact of GC on tissues isolated from treated animals. Intravenous infusion of LPS into the isolated perfused rat liver (IPRL) leads to dose-dependent increases in TNF and IL-6 concentrations in the effluent [Citation56]. When corticosterone is added to LPS-treated IPRL, the lower dose of GCs enhanced the release of cytokines, whereas the higher dose of GCs suppressed the release of cytokines, and this effect was mediated via Kupffer cells [Citation56]. We observed a similar pattern in our liver samples, where treatment with low doses of prednisolone resulted in higher expression of TNF and IL-1 beta (although non-significant), in contrast to high doses of prednisolone that decreased expression of both proinflammatory cytokines (significant for IL-1 beta only) ()), pointing towards a potential role for Kupffer cells in the observed results. Thus, although GCs affect the immune cells usually by inhibiting the production of proinflammatory cytokines in vitro, GCs can in some cases enhance the production of proinflammatory cytokines in tissues. To further add complexity to the cytokine gene expression, besides SIRT1, several histone deacetylases (HDACs) including HDAC1 and HDAC2 are involved in the regulation of gene expression of proinflammatory cytokines [Citation56]. This is demonstrated by the fact that multiple HDAC inhibitors reduced the production of circulating proinflammatory cytokines released from PBMCs, for instance TNF, IL-1 beta, IL-6, and IFN-γ in vivo and in vitro [Citation57–59].
In conclusion, we show here that both SIRT1 expression and activity are increased in PBMCs isolated from AIA rats compared to healthy rats and subsequently decreased under GC treatment. Similarly, increased SIRT1 activity was observed in liver isolated from AIA rats compared to healthy rats and eventually reduced under GC treatment. Our results also indicate that the trends of SIRT1 activity and TNF and IL1 beta production are similar in a specified tissue (e.g., liver), indicating that SIRT1 activity and proinflammatory cytokine expression might be regulated by a common cellular pathway (e.g., NF-kB pathway) which could be controlled therapeutically by GCs. Future studies will be undertaken to determine the exact role of SIRT1 and GCs and the underlying molecular mechanisms involved in the release of tissue proinflammatory cytokines with the ultimate goal of developing new therapeutic approaches to cure RA.
Supplemental Material
Download MS Word (69.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 2013;9:141–153.
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442.
- McInnes IB, Liew FY. Cytokine networks–towards new therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:31–39.
- Vasanthi P, Nalini G, Rajasekhar G. Role of tumor necrosis factor-alpha in rheumatoid arthritis: a review. APLAR J Rheumatol. 2007;10:270–274.
- Kay J, Calabrese L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2004;43(Suppl 3):iii2–9.
- Nalbant S, Birlik AM. Cytokines in Rheumatoid Arthritis (RA). New developments in the pathogenesis of rheumatoid arthritis. 2017. DOI:10.5772/65893.
- Selmi C, De Santis M, Gershwin ME. Liver involvement in subjects with rheumatic disease. Arthritis Res Ther. 2011;13:226.
- Brzustewicz E, Bryl E. The role of cytokines in the pathogenesis of rheumatoid arthritis–Practical and potential application of cytokines as biomarkers and targets of personalized therapy. Cytokine. 2015;76:527–536.
- Herbein G, Wendling D. Histone deacetylases in viral infections. Clin Epigenetics. 2010;1:13–24.
- Holroyd C, Harvey N, Dennison E, et al. Epigenetic influences in the developmental origins of osteoporosis. Osteoporos Int. 2012;23:401–410.
- Grabiec AM, Tak PP, Reedquist KA. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther. 2008;10:226.
- Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23:2369–2380.
- Niederer F, Ospelt C, Brentano F, et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann Rheum Dis. 2011;70:1866–1873.
- Kotas ME, Gorecki MC, Gillum MP. Sirtuin-1 is a nutrient-dependent modulator of inflammation. Adipocyte. 2013;2:113–118.
- Wang W, Yan C, Zhang J, et al. SIRT1 inhibits TNF-α-induced apoptosis of vascular adventitial fibroblasts partly through the deacetylation of FoxO1. Apoptosis. 2013;18:689–701.
- Tanno M, Sakamoto J, Miura T, et al. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–6832.
- Correia M, Perestrelo T, Rodrigues AS, et al. Sirtuins in metabolism, stemness and differentiation. Biochim Biophys Acta. 2017;1861:3444–3455.
- Tseng P-C, Hou S-M, Chen R-J, et al. Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J Bone Miner Res. 2011;26:2552–2563.
- C-M B, Li Y, Lindgren U, et al. Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. J Bone Miner Res. 2006;21:993–1002.
- Taylor DM, Maxwell MM, Luthi-Carter R, et al. Biological and potential therapeutic roles of sirtuin deacetylases. Cell Mol Life Sci. 2008;65:4000–4018.
- Zeng L, Chen R, Liang F, et al. Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr Gerontol Int. 2009;9:7–15.
- He X, Andersson G, Lindgren U, et al. Resveratrol prevents RANKL-induced osteoclast differentiation of murine osteoclast progenitor RAW 264.7 cells through inhibition of ROS production. Biochem Biophys Res Commun. 2010;401:356–362.
- Shakibaei M, Buhrmann C, Mobasheri A. Resveratrol-mediated SIRT-1 interactions with p300 modulate receptor activator of NF-kappaB ligand (RANKL) activation of NF-kappaB signaling and inhibit osteoclastogenesis in bone-derived cells. J Biol Chem. 2011;286:11492–11505.
- Toussirot E, Abbas W, Khan KA, et al. Imbalance between HAT and HDAC activities in the PBMCs of patients with ankylosing spondylitis or rheumatoid arthritis and influence of HDAC inhibitors on TNF alpha production. PLoS ONE. 2013;8:e70939.
- Yang S-R, Wright J, Bauter M, et al. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–576.
- Yang H, Zhang W, Pan H, et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS ONE. 2012;7:e46364.
- Knobloch J, Wahl C, Feldmann M, et al. Resveratrol attenuates the release of inflammatory cytokines from human bronchial smooth muscle cells exposed to lipoteichoic acid in chronic obstructive pulmonary disease. Basic Clin Pharmacol Toxicol. 2014;114:202–209.
- Rajendrasozhan S, Yang S-R, Kinnula VL, et al. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870.
- Kim HY, Park SY, Lee SW, et al. Inhibition of HMGB1-induced angiogenesis by cilostazol via SIRT1 activation in synovial fibroblasts from rheumatoid arthritis. PLoS ONE. 2014;9:e104743.
- Katto J, Engel N, Abbas W, et al. Transcription factor NFκB regulates the expression of the histone deacetylase SIRT1. Clin Epigenetics. 2013;5:11.
- Dvir-Ginzberg M, Gagarina V, Lee EJ, et al. Tumor necrosis factor α-mediated cleavage and inactivation of SirT1 in human osteoarthritic chondrocytes. Arthritis Rheum. 2011;63:2363–2373.
- Fernandes CA, Fievez L, Neyrinck AM, et al. Sirtuin inhibition attenuates the production of inflammatory cytokines in lipopolysaccharide-stimulated macrophages. Biochem Biophys Res Commun. 2012;420:857–861.
- Zhang T, Kraus WL. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta. 2010;1804:1666–1675.
- Chen J, Xavier S, Moskowitz-Kassai E, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol. 2012;180:973–983.
- Fusco S, Maulucci G, Pani G. Sirt1: def-eating senescence? Cell Cycle. 2012;11:4135–4146.
- Wendling D, Vidon C, Abbas W, et al. Sirt1 activity in peripheral blood mononuclear cells from patients with rheumatoid arthritis. Joint Bone Spine. 2014;81:462–463.
- Wendling D, Abbas W, Godfrin-Valnet M, et al. Dysregulated serum IL-23 and SIRT1 activity in peripheral blood mononuclear cells of patients with rheumatoid arthritis. PLoS ONE. 2015;10:e0119981.
- Burmester GR, Feist E, Dörner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10:77–88.
- Listing J, Kekow J, Manger B, et al. Mortality in rheumatoid arthritis: the impact of disease activity, treatment with glucocorticoids, TNFα inhibitors and rituximab. Ann Rheum Dis. 2015;74:415–421.
- Verhoeven F, Prati C, Maguin-Gaté K, et al. Glucocorticoids and endothelial function in inflammatory diseases: focus on rheumatoid arthritis. Arthritis Res Ther. 2016;18:258.
- Verhoeven F, Totoson P, Maguin-Gaté K, et al. Glucocorticoids improve endothelial function in rheumatoid arthritis: a study in rats with adjuvant-induced arthritis. Clin Exp Immunol. 2017;188:208–218.
- Sakaguchi N, Takahashi T, Hata H, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460.
- Abdin AA, Abd El-Halim MS, Hedeya SE, et al. Effect of atorvastatin with or without prednisolone on Freund’s adjuvant induced-arthritis in rats. Eur J Pharmacol. 2012;676:34–40.
- Engler A, Tange C, Frank-Bertoncelj M, et al. Regulation and function of SIRT1 in rheumatoid arthritis synovial fibroblasts. J Mol Med. 2016;94:173–182.
- Paolino S, Cutolo M, Pizzorni C. Glucocorticoid management in rheumatoid arthritis: morning or night low dose? Reumatologia. 2017;55:189–197.
- Cutolo M. Glucocorticoids and chronotherapy in rheumatoid arthritis. RMD Open. 2016;2. DOI:10.1136/rmdopen-2015-000203
- Poulsen RC, Watts AC, Murphy RJ, et al. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann Rheum Dis. 2014;73:1405–1413.
- Suzuki S, Iben JR, Coon SL, et al. SIRT1 is a transcriptional enhancer of the glucocorticoid receptor acting independently to its deacetylase activity. Mol Cell Endocrinol. 2018;461:178–187.
- Moon M-H, Jeong J-K, Lee Y-J, et al. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthr Cartil. 2013;21:470–480.
- Liu TF, McCall CE. Deacetylation by SIRT1 reprograms inflammation and cancer. Genes Cancer. 2013;4:135–147.
- Park SY, Lee SW, Lee SY, et al. SIRT1/adenosine monophosphate-activated protein kinase α signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front Immunol. 2017;8:1135.
- Liu T, Zhang L, Joo D, et al. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2. DOI:10.1038/sigtrans.2017.23
- Volonte D, Zou H, Bartholomew JN, et al. Oxidative stress-induced inhibition of Sirt1 by Caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6). J Biol Chem. 2015;290:4202–4214.
- Hah Y-S, Cheon Y-H, Lim HS, et al. Myeloid deletion of SIRT1 aggravates serum transfer arthritis in mice via nuclear factor-κB activation. Plos One. 2014;9:e87733.
- Zhu X, Liu Q, Wang M, et al. Activation of Sirt1 by resveratrol inhibits TNF-α induced inflammation in fibroblasts. PLoS ONE. 2011;6:e27081.
- Liao J, Keiser JA, Scales WE, et al. Role of epinephrine in TNF and IL-6 production from isolated perfused rat liver. Am J Physiol. 1995;268:R896–901.
- Villalba JM, Alcaín FJ. Sirtuin activators and inhibitors. Biofactors. 2012;38:349–359.
- Leoni F, Zaliani A, Bertolini G, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA. 2002;99:2995–3000.
- Choo QY, Ho PC, Lin HS. Histone deacetylase inhibitors: new hope for rheumatoid arthritis? Curr Pharm Des. 2008;14:803–820.