
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The presence of 5-hydroxymethyl cytosine in DNA has been previously associated with ageing. Using in silico analysis of normal liver samples we presently observed that in 5-hydroxymethyl cytosine sequences, DNA methylation is dependent on the co-presence of G-quadruplexes and palindromes. This association exhibits discrete patterns depending on G-quadruplex and palindrome densities. DNase-Seq data show that 5-hydroxymethyl cytosine sequences are common among liver nucleosomes (p < 2.2x10−16) and threefold more frequent than nucleosome sequences. Nucleosomes lacking palindromes and potential G-quadruplexes are rare in vivo (1%) and nucleosome occupancy potential decreases with increasing G-quadruplexes. Palindrome distribution is similar to that previously reported in nucleosomes. In low and mixed complexity sequences 5-hydroxymethyl cytosine is frequently located next to three elements: G-quadruplexes or imperfect G-quadruplexes with CpGs, or unstable hairpin loops (TCCCAY6TGGGA) mostly located in antisense strands or finally A-/T-rich segments near these motifs. The high frequencies and selective distribution of pentamer sequences (including TCCCA, TGGGA) probably indicate the positive contribution of 5-hydroxymethyl cytosine to stabilize the formation of structures unstable in the absence of this cytosine modification. Common motifs identified in all total 5-hydroxymethyl cytosine-containing sequences exhibit high homology to recognition sites of several transcription factor families: homeobox, factors involved in growth, mortality/ageing, cancer, neuronal function, vision, and reproduction. We conclude that cytosine hydroxymethylation could play a role in the recognition of sequences with G-quadruplexes/palindromes by forming epigenetically regulated DNA ‘springs’ and governing expansions or compressions recognized by different transcription factors or stabilizing nucleosomes. The balance of these epigenetic elements is lost in hepatocellular carcinoma.
Introduction
DNA cytosine methylation is an enzymatic process that plays a critical role in regulating the differentiation of gene expression [Citation1], splicing and alternative splicing [Citation2,Citation3], as well as the expression of transcript variants resulting from different exonic CpG islands [Citation4]. DNA methyl-binding proteins play a key role in the recognition of methyl-cytosine (5mC) in normal cells, neurological disorders, and cancer [Citation5]. The presence of 5mC depends principally on the enzymatic activity of two different enzymes, DNA methyltransferase 1 (DNMT1) and DNA methyltransferase 3 (DNMT3) [Citation6]. Oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) occurs via a multistep process in which DNA methyltransferases and ten-eleven-translocation proteins (TET) are involved (reviewed by [Citation7]). 5hmC is a stable product processed by formylation and carboxylation [Citation7]. 5mC oxidation products affect replication [Citation8] and are probably significant regulators in the differentiation process [Citation9]. 5hmC has also been shown to be associated with several neurological disorders [Citation10], cancer [Citation11] and ageing [Citation12]. Cytosine modifications are tissue-specific [Citation13] and involved in differentiation [Citation14]. The biological and environmental factors which are involved in this oxidative process remain unknown. TETs promiscuity for dsDNA [Citation15] might indicate that sequence elements, which can potentiate changes in the DNA conformation by modifying its helical integrity, could also modify the rates and efficiency of these enzymes. Ngo et al. have shown that 5mC modifies helical flexibility [Citation16]. To this effect, other elements such as G-quadruplexes (G4s), which can modify the helical DNA configuration, have also been shown to modify DNA expression dynamics in the case of mitochondrial DNA [Citation17].
G4s are parallel or antiparallel helical structures that can be formed by single-stranded DNA or RNA. These structures are characterized by the number of G-tetrads that they can form and vary depending on the orientation of the strands and their inter- or intramolecular folding [Citation18]. G4s can be considered as landmarks of the nucleic acid structure, which are associated with DNA methylation [Citation19]. Recently, Cheng et al. [Citation20] showed that a principle parameter associated with the stability of these structures is loop permutation, or else their ability to form polymorphic loops of different lengths and stability, depending on their primary sequence. However, there is no information on the association of the tentative loop swapping with the presence of the other DNA elements which contribute to its expression dynamics, such as the presence of DNA methylation and other intermediate oxidation states such as 5hmC. The transient formation of G4s [Citation21], their permutation [Citation20] and their complex interactions in-vivo [Citation22] introduce challenges in relevant research. Potential G4 formation has been shown to be involved in nucleosome occupancy [Citation23] similarly to the methylation density [Citation24].
The loop length and sequence of G4s determine their interaction with small molecules and proteins [Citation25]. Nucleosome remodelling in association with epigenetic modifications is probably involved in negative feedback loops in ageing and cancer [Citation26]. Information regarding the association of epigenetic characteristics such as cytosine methylation and 5hmC, with the potential formation of distinct structures such as G4s and hairpins, would facilitate studies on relevant chemical interventions and biological applications [Citation18].
Although the impact of methylation changes varies, it is likely that such changes in sequences with a potential to form G4s could lead to deregulation of transcription and genome instability [Citation27]. In addition, they could modify the activity of TETs due to the asymmetric double-/single-strand equilibrium [Citation28] and the changes in oxidation dynamics [Citation15] associated with G4 formation.
Palindromes (Pals) or inverted repeats and G4s are additional elements which have recently acquired extensive interest as tentative regulators of the nucleosome, nucleosome positioning, rearrangements, and cellular reprogramming [Citation29]. Furthermore, specific sequence motifs have been proposed as nucleosome positioning determinants [Citation30].
We presently investigated the distribution and sequence characteristics of G4s and Pals in sequences of known 5mC and 5hmC levels. Moreover, we identified their association with nucleosome-related parameters and identified over-represented motifs relative to their frequency in the genome. Our study was based on whole-genome analysis data from normal liver, which has been previously shown to exhibit distinct differences from its corresponding cancerous tissue [Citation31]. The present analysis revealed a strong association between G4s and Pals with the sites of epigenetic modifications particularly in low complexity sequences, within sequence limits which could define a nucleosome entity and association of the sequences involved with specific growth regulatory signals. These findings provide new perspectives to the understanding of epigenetically regulated processes, such as genomic rearrangement, transcription, and alternative gene expression, as well as cellular differentiation, cancer, genome repair, toxicity, and mutagenesis.
Material and methods
Putative structural DNA elements were computationally identified on different DNA methylation data used in cancer and ageing studies and from both array-based and next-generation sequencing platforms. Whole-genome bisulphite sequencing data of three liver normal/tumour sample pairs were downloaded from Gene Expression Omnibus (GEO, Data Series: GSE70090) [Citation31]. The level of DNA methylation in these samples was estimated at single cytosine resolution by the fraction of cytosine-reporting reads vs the total number of mapped reads. In addition, the 5hmC-containing landscape of the same samples was downloaded and associated with the abundance of the incorporated structural DNA features. For each hydroxymethylated region, the mean DNAm level was calculated using bedtools v2.29.2 [Citation32] after discarding poorly covered CpGs (less than five sequencing reads).
G4s and Pals were computationally identified in genomic regions overlapping DNA methylation sites and correlated with the corresponding methylation levels. Pqsfinder was used to identify G4s in 200nt sequences centred at the methylation sites using the default parameters [Citation33]. To detect palindromic sequences neighbouring DNA methylation sites, we used the Biostrings R package [Citation34]. A palindromic sequence is defined in 20nt regions with 5nt minimum arm and maximum loop length, allowing one mismatch between the two arms. To perform batch G4 and Pals analysis of the DNA methylation datasets we used MeinteR [Citation35]. In addition, we calculated the density of G4s (dG4) and Pals (dPals) in the 5hmC-containing regions identified by Li et al. in normal/tumour liver samples [Citation31].
dG4 and dPals are defined by the number of G4 and Pals in 100nt regions, respectively. 5hmC-containing regions in normal/tumour liver samples were further analysed with respect to the nucleosome occupancy. To detect the nucleosome density in DNA sequences we applied the nucleosome-DNA interaction model proposed by Kaplan et al. [Citation36]. 5hmC-containing regions of length 50–300nt were selected and expanded to 300nt to avoid misleading comparisons. The presence of nucleosomes was correlated with the abundance of G4 and Pals in normal liver and hepatoma cells using in-house R scripts. In addition, we mapped nucleosome positioning loci from the liver (right lobe) of a 53-year-old female. The DNase-Seq data were downloaded from the ENCODE Portal (Phase 4, experiment id: ENCSR909HFI) [Citation37]. Peaks corresponding to the accessibility of the DNA minor and major groove along the nucleosome were mapped to the hydroxymethylation regions of the normal liver, following a lift over step to map genome assemblies. Of the 96,869 peaks (99.6% of all hg38 peaks) mapped to hg19 21,938 overlapped (at least one nucleotide) with hydroxymethylated regions of normal liver.
To identify over-represented motifs in 5hmC-containing regions of normal liver we fetched DNA sequences using UCSC Genome Browser and built two subsets of sequences containing up to six Pals that include either zero or one G4. Each subset was split into three subsets (high, mixed, low) depending on the sequence complexity level (as identified by RepeatMasker [Citation38] and Tandem Repeats Finder [Citation39] with a period of 12 or less). The MEME algorithm was used to identify over-represented motifs in each subset using the default parameters [Citation40]. The whole set of 5hmC-containing sequences in normal liver was scanned for the best scoring motifs with a p-value less than 0.0001, using the FIMO algorithm (v. 5.0.5) [Citation41]. To identify putative transcription factor binding sites in the most frequent sequences among those detected by FIMO we applied JASPAR scanning method against human core collection [Citation42], using the default parameters. Custom R scripts were developed to analyse and visualize the results of the analysis [Citation43].
Results
Association of Pals, G4s and DNA methylation in 5hmC-containing sequences obtained from normal liver
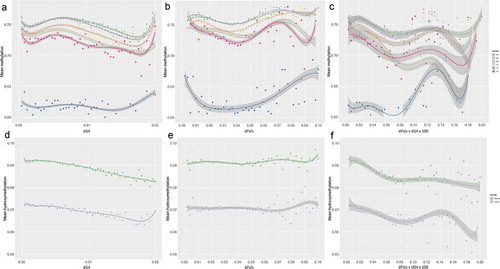
The association between G4s and Pals with nucleosome positioning was first investigated in 5hmC-containing sequences of normal liver (Suppl. File 1) by evaluating the dependence of average DNAm on the density of Pals and G4s (dPals and dG4s, respectively). ) shows that for dPals ranging between 0.01 and 0.05 the mean DNAm exhibits very small spread out from the average value, i.e.,, variance and follows similar DNAm pattern in all samples. Variance increases for . According to previous studies [Citation44], the minimum average DNAm expected for a nucleosome would correspond to a density of ~0.048 (7 Pals/complete nucleosome core of 145nt), while maximum DNAm values are observed at the nucleosome core centre [Citation45]. Thus, the observed low variation of mean methylation values for
could reflect the association of the 5hmC-containing sequences with the nucleosome structural characteristics.
Figure 1. Mean DNAm (a,b) and hydroxymethylation (d,e) with respect to the tentative palindrome (dPals) and G-quadruplex (G4) densities in three normal liver samples. (c) and (f) show the mean DNAm and hydroxymethylation relative to the co-presence of tentative Pals and G4 densities (dPals x dG4), respectively. Regression lines of the smoothed conditional means and shaded standard errors are shown in each plot using a linear model as smoothing function
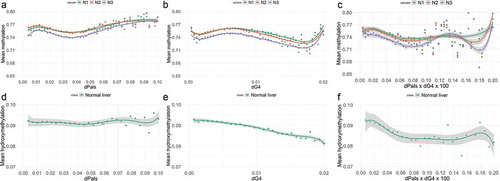
A similar analysis shows again a strict association between mean DNAm and dG4 ()). Maximum mean DNAm is observed at approximately corresponding to one G4 per nucleosome core (~145nt). For
the mean DNAm decreases and for
DNAm exhibits high variance levels. The above data again support a critical role of G4s in DNAm and most probably in nucleosome positioning.
In order to determine whether DNAm depends on the co-presence of G4s and Pals, we further investigated the association between DNAm and . Low variance of the
could result if these parameters were interdependent in forming a common single frame which determines DNA methylation. ) reveals that the mean DNAm exhibits very low variance in
and is significantly increased for higher
values. It should be noted that the increased DNAm variance cannot be attributed to a limited number of sequences.
Using the same approach, we then investigated the association of dPals and dG4 with the 5hmC levels. ) shows that hydroxymethylation exhibits very small variance across all samples for and the mean hydroxymethylation decreases for
. It is also evident that 5hmC levels exhibit an overall decrease with low fluctuation with respect to
. These observations could be interpreted by assuming that within certain dPals and dG4 limits
both tentative structural elements are involved in nucleosome formation. Moreover, these results reveal that in normal liver, the probability of a sequence of specific G4s and Pals densities to form (or occupy) a nucleosome is related to DNAm and its oxidation to 5hmC.
5hmC-containing sequences without G4s and Pals, are significantly more frequent in chromosome X, compared to normal liver autosomes and methylation is less dependent on the G4 density (Suppl. File 2, Table). However, the presence of 5hmC is again associated with the co-presence of G4s and Pals , particularly at low dG4 and dPals (Suppl. File 2, Figure).
In silico evaluation of nucleosome occupancy in normal liver relative to 5hmC, G4s and Pals
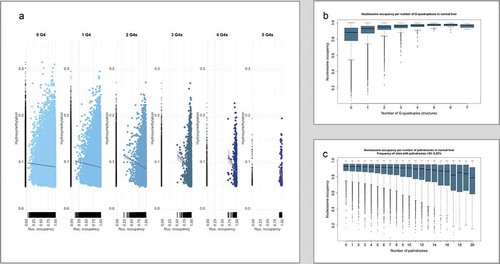
Nucleosome occupancy was investigated in silico for small length 5hmC-containing sequences extracted from Suppl. File 1. In order to avoid biased estimation of nucleosome abundance due to the variable 5hmC-containing sequence length, we limited the analysis to sequences of 50–300nt that are expected to carry up to one nucleosome core and one complete linker region. In addition, we identified the G4s and Pals in the above sequences after expansion to 300nt in which the hydroxymethylated CpGs were centred. The results are included in Suppl. File 3.
shows that when more than two G4s are present in a 5hmC-containing sequence, the nucleosome occupancy increases (>0.5). On the contrary, the increase of Pals leads to a small nucleosome occupancy decrease, indicating that sequences with an increasing number of Pals are probably less likely to form nucleosomes. This finding is particularly evident when Pals>6 are present in a 300nt sequence ()).
Figure 2. (a) Hydroxymethylation levels and their linear regression lines in normal liver with respect to nucleosome occupancy in 300nt sequences centred at each single CpG, relative to their G4 content (0 to 5 G4s). (b) Boxplots of the nucleosome density relative to the number of G4s per sequence and (c) Boxplots of the nucleosome density relative to the number of palindromes per sequence
Quantitation of G4s and Pals in variable length 5hmC-containing sequences. Correlation with 5hmC and 5mC levels
In a previous study on nucleosome reconstitution [Citation46], it was observed that cytosine oxidation to 5hmC occurs at the linker region. Based on this finding it is probable that a significant percentage of sequences would include at least a part of the linker region, as well as the nucleosome core. We therefore investigated the presence of G4s and Pals, which appears to be elements defining nucleosome resting (and nucleosome occupancy, ) in 5hmC-containing sequences of increasing lengths
.
The results in ) (left) reveal that one-third of the sequences with contains a single G4 and only a small number (<20%) of sequences
, that can accommodate up to one complete nucleosome core ) (right), contain two G4s.
Figure 3. Frequency of G4s (a) and Pals (b) in sequences of incrementally increasing lengths. (c) 5hmC and 5mC in sequences of incrementally increasing lengths. (d) Hydroxymethylation relative to different content in sequences with average methylation
and limited length (60–80nt)
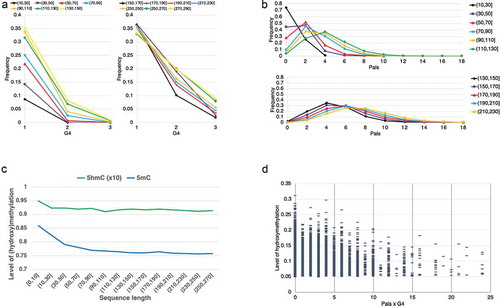
The frequency of Pals exhibits a steady increase with sequence length up to 170nt, while there is less Pals variation in longer sequences ()). These data are compatible with the assumption that in a 5hmC-containing sequence Pals are mostly contained within 170nt. Longer sequences (170–230nt, ), right) that can accommodate an average nucleosome and a linker contain an average number of 6–8 Pals and up to 14 Pals.
A more detailed analysis of methylation and hydroxymethylation in ) reveals that the presence of 5hmC and 5mC levels remains constant for sequences >90nt but can be higher among smaller sequences ()).
Finally, in order to further investigate the balance between hydroxymethylation, G4, and Pals, we examined these elements in an ensemble of small representative sequences with average epigenetic modification similar to the observed in ), (0.75β +10%) and sequence length 70–135nt that might correspond to the ‘tail’ of the core if the analysed segments would correspond to the nucleosome core ()). In these sequences, the presence of 5hmC decreases with increasing numbers of G4s and Pals. Most of them contain approximately six Pals and one G4, but few contain more Pals (<11). This strictly defined sample corresponds to approximately 30% of all G4- and Pals-containing sequences of this length (6,622/20,867) and complies with the assumption that in G4-containing sequences, G4 and Pals are jointly responsible for 5hmC modification of the helix.
Nucleosome localization in 5hmC-containing sequences
In order to evaluate the actual association of 5hmC with the presence of structural elements (Pals, G4s) and actual nucleosome occupancy, we also used DNase-Seq data obtained from the liver of a female adult (ENCODE project, ENCSR909HFI). The analysis of the experimental data revealed that a very significant percentage of the total nucleosome sequences (22.64%, p < 2.2x10−16) overlaps with 5hmC-containing sequences (Suppl. File 4). However, the percentage of the 5hmC sequences occupied by single nucleosomes is only 6.32%, indicating that the majority of 5hmC sequences are probably responsible for regulating additional processes. Only 1.03% of 5hmC sequences contains more than one nucleosome.
In order to further investigate the association of G4s and Pals with nucleosome occupancy, we compared the frequencies of 5hmC sequences with up to two G4s, and up to six Pals that contain nucleosomes (NC+) to those that lack nucleosomes (NC-) (). NC+ sequences with these characteristics are limited compared to NC+ sequences that have more G4s and Pals (data not shown). NC+ sequences without G4s and Pals are rare (NC+/G4(0)/Pals(0) vs NC-/G4(0)/Pals(0) is 1 × 10−2). NC+ sequences with limited G4s (1–2) and Pals(1–6) are also infrequent (NC+/G4(1-2)/Pals(1-6) vs NC-/G4(1-2)/Pals(1-6) is 2.12 × 10−2) but less infrequent compared to NC+ sequences with more G4s and Pals (6,32%). The average size of NC+ and NC- sequences lacking G4s and Pals is very similar (39.2nt and 39.5nt). On the contrary, the average size of the corresponding 5hmC sequences exceeds that of the nucleosome core (197.7 > 150nts), probably indicating their presence close to the linker and in adjacent sequences. These data show that 5hmC is frequent in nucleosomes, and its presence probably depends on the structural characteristics of the sequence. These findings are in agreement with those from in silico analysis, with regards to the nucleosome increase among 5hmC-containing sequences when G4 > 2.
Table 1. Summary statistics of the nucleosomes overlapping 5hmC sequences
Common motifs in small 5hmC-containing sequences of high, mixed and low complexity
In order to identify the presence and order of common motifs among 5hmC-containing sequences we first examined sequences of limited length (60–80nt), that might correspond the methylation-rich centre of the nucleosome core (see Materials and Methods, Suppl. File 5A). Sequences were classified in three categories, according to their complexity: high complexity (HC) sequences that potentially code for expressed regions, repetitive or low complexity (LC) sequences that are probably non-coding and sequences with mixed (MC) complexity, possessing both high and low complexity segments. Each category was also subdivided into G4-containing and G4-free sequences for sequences that include and do not include G4s, respective. The most significant motifs in the smaller sequence array, their frequencies and probabilities are included in ). Their common presence in a sequence identified by MEME is shown in ).
Figure 4. (a) Similarities among overrepresented motifs in 5hmC-containing sequences of limited length (60–80nt). Sequences are classified relative to their complexity to HC, MC and LC corresponding to high, mixed and low complexities respectively. S: motifs observed in sense strand; A: motifs observed in antisense strand; G4 indicates the tentative presence of G-quadruplex(es) in the sequence. Co-existing motifs are infrequent in high complexity sequences (HC) and are not included. E-value: Statistical significance of the motif presence based on its log-likelihood ratio, width, sites the background letter frequencies and the length of the training set. (b) Co-presence of different motifs in MC and LC sequences. S/S: both motifs are present in sense strand. S/A: motifs present in antisense strands
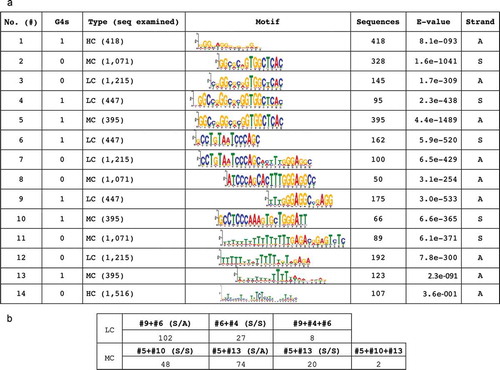
The results, included in , reveal that characteristic motifs are more frequent in MC and LC sequences compared to HC sequences. Four of the prevalent motifs include multiple CpGs even at low frequencies (#3-#5 in ). Furthermore, the motifs can be classified in three categories: A: those containing either a single G4 sequence (#1, #4 and #5) or, frequently, a shorter part of the same G4-containing motif (#4 and #5), with only two or three GG pairs (motifs #2 and #3) and sites of tentative epigenetic modification (e.g., CpGs). B: TCCCAY6TGGGA (#7, #8 and #10) sequences or sequences containing segments of the above motifs (#6 and #9 in the same strand or #9 and #4 in antisense strands, see inserted Table); the former can give rise to an unstable hairpin structure. C: A-rich or T-rich sequences (#11-#14; ); A-rich sequences have been previously identified as tentative H1-histone binding sites [Citation47]. Notably, 5hmC is present in sequences which include imperfect G4s and in antisense sequences corresponding to i-motifs formed opposite G4s [Citation48]. In 59% of HC the sequences lacking G4s, the tentative 5mC/5hmC sites are located next to the only common motif (T-rich, #14 in ).
Analysis of the proximity of these motifs on the basis of their common presence in these short sequences (60–80nt, )) reveals the following: A: some complementary sequences containing the TGGGA and TCCCA motifs are always present in antisense directions (#9 and #6), B: other motifs, which include a G4 and a potential hairpin structure related to the presence of TGGGA and TCCCA, are always present on the same strand (#5 and #10) and in close proximity, C: few sequences of mostly low complexity contain more than two of the above motifs.
Prevalence of motifs in all 5hmC-containing sequences
The frequent presence of the motifs in was also verified in the total 5hmC-containing sequence array (297,717 sequences) using analysis by FIMO (Suppl. File 6). Seven different motifs were identified using this approach (); one of these motifs was common between mixed and low complexity sequences containing G4 (#6). The principle common characteristics identified are:
Table 2. Prevalence of motifs in the 5hmC-containing sequences. Repeated or repeated/complementary sequences are shown in bold; complementary/antisense sequences are shown with arrows. C: complexity; LC, HC MC: low, high, mixed complexity sequences, respectively. S/A: antisense repeated sequences
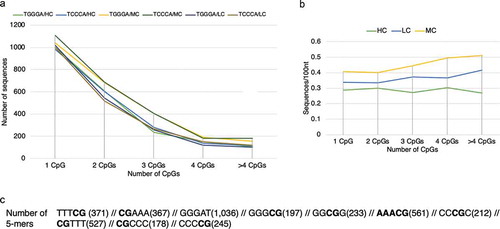
a) very frequent presence of common motifs in all 5hmC-containing sequences (1,420,618), b) very high frequency of uninterrupted A- or T-rich motifs in MC and LC sequences and (A)nG-repeats (#1) in HC sequences (total 650,775), c) common presence of palindromes in antisense directions (#2 and #5) within a single sequence motif in proximity with CpGs (total 426,085), or in different motifs (#4, #6, #7 and #8, , total 565,996), and d) very high frequency of two specific 5-mers TCCCA and TGGGA (629,260). These results reveal that the proximity of elements tentatively introducing steric modifications, e.g., deviations from the double helix such as G4s or different types of palindromes, is a common characteristic of 5hmC-containing sequences.
In order to reinforce the hypothesis that TGGGA/TCCCA motifs are inter-related and related to the CpG presence, we finally investigated their presence in sequences of moderate length (60–300nt) that can accommodate up to a single nucleosome and adjacent linkers (). The densities of these motifs, shown in ), are almost identical for sequences with similar CpG densities, and decrease with increasing CpG density ()). Moreover, the CpG density increases with increasing overall sequence complexity (HC, MC, or LC, )). Most common 5mers include nucleotide repeats in addition to CpGs (TTTs, AAAs, GGGs, or CCCs, )). Although TGGGAs and TCCCAs do not form stable dimers in unmodified DNA sequences, their identical frequencies most probably reveal that they participate in common stable formations under specific conditions. These data corroborate those in ) with regards to the association of 5hmC with stable palindromes.
Figure 5. (a) Number of TGGGA and TCCCA oligonucleotides in high (HC), mixed (MC) and low (LC) complexity hydroxymethylated sequences of specific length limits (60–300nt) relative to the number of CpGs (total sequences: 10,338). (b) Frequency of TGGGA and TCCCA oligonucleotides in hydroxymethylated sequences of normal liver relative to the CpG density. (c) Prevalence of various 5mers neighbouring CpGs among the above sequences
Transcription factors recognizing the frequent motifs in 5hmC-containing sequences
Finally, we identified transcription factors that recognize the sequence motifs in the complete 5hmC-containing sequence ensemble (Suppl. File 6). Transcription factors binding sites which exhibit high homology (>85%) are included among the sequences in . These motifs exhibit high homology for transcription factors involved in mortality/ageing and apoptosis, as well as cancer, behavioural and neurological disorders, vision, and reproduction.
Table 3. Transcription factors that bind HC (uppercase) and LC (lowercase) sequences (motif similarity >0.85%)
Both TGGGA and TCCCA 5mers are found in very high and similar frequencies, in neighbouring positions of the same strand separated by six nucleotides (: motif #8 in LC and motif #10 in MC sequences; : motif 2 in LC sequences) or in antisense strands. Although these motifs do not include CpGs, 5hmC is probably located close to these repeats (present in 20–26nt long motifs of short, 50–70nt sequences, ). A single TCCCA motif is also frequently present next to a CpG (#7 in ) in LC sequences. Motifs #7 and #9 in are similar to #8 but less preserved or smaller. 5hmC is found either in the sense or antisense strand.
Disruption of the 5hmC/5mC and palindrome/G4 balance in hepatocellular carcinoma
The previously described 5mC and 5hmC balance with palindromes and G4s in normal cells () is disrupted in hepatocellular carcinoma. As shown in the association of dG4s, dPals and dG4*dPals with methylation in 5hmC-containing sequences collapses (), Suppl. File 3). Similarly, we observed changes in the average 5hmC dependence on G4s, Pals, and dG4s*Pals ()). This fact reveals major changes in the epigenetic characteristics of sequences adjacent to G4s and Pals and in transcription factor binding sites regulating cell-growth and death in cancer and reflects the deregulation of the previously described epigenetic balance.
Figure 6. Mean DNAm (a,b) and hydroxymethylation (d,e) with respect to the tentative palindrome (dPals) and G-quadruplex (G4) densities in three normal liver samples. (c) and (f) show the mean DNAm and hydroxymethylation relative to the co-presence of tentative Pals and G4 densities (dPals x dG4), respectively. Regression lines of the smoothed conditional means and shaded standard errors are shown in each plot using a linear model as smoothing function
Discussion
Alternative epigenetic modifications are critical for studying cell differentiation but poorly understood. Similarly, the impact of non-canonical DNA structures and their association with dynamic nucleosome changes remains unclear. Investigation of these structural elements with respect to the functional role in sequences where they commonly reside can promote our understanding of the development and its deregulation in complex diseases, such as cancer and neurological disorders, as well as mutagenesis.
In this study, we show that diverse epigenetic modifications (5hmC and/or 5mC) are frequent in sequences which can form G4s and Pals. Moreover, we report that these non-canonical secondary DNA structures are observed in, or near, transcription factor binding sites that are involved in cell differentiation and death, or cancer. The distribution and frequency of these elements and their contiguity are related to nucleosome occupancy. Their presence next to potential histone H1 binding sites, (A)n or (T)n sequences, is most probably critical for nucleosome positioning.
The frequent TGGGA sequence has been previously identified as the binding site for the Cys2His2 type zinc fingers, similar to those observed in TFIIIA transcription factor in Xenopus [Citation49]. This type of zinc fingers, which are frequent in many organisms including humans, bind to the DNA 5mer [Citation50] inducing, in the case of TFIIIA, compaction of the promoter into a precise three-dimensional hairpin-shaped structure [Citation51]. Usually, there are several such zinc fingers present in a DNA-binding protein. In these cases, only one zinc finger binds to the 5mers, while the second zinc finger is restricted from binding. Arrangement of zinc fingers in this fashion is considered critical for differentiation procedures [Citation49]. The presence of frequent, regularly arranged TGGGA and TCCCA in a single, or in complimentary 5hmC-containing sequences, probably facilitates the formation of such hairpin-shaped structures [Citation50,Citation51] similar to those in , possibly stabilized by the zinc ion [Citation52]. In these cases, 5hmC probably relaxes the helical distortion associated with zinc finger binding [Citation51]. In short, the observed motifs are probably compatible with an elegant bimodal (or multimodal depending on the repeat number and arrangement) system related to the regulation of transcription by involving zinc finger proteins, which is facilitated by the 5hmC presence. G4 formation, frequent in high complexity sequences, is also probably facilitated by the 5hmC modification.
Figure 7. (a–b) Conceptual model for the formation of TGGGA/TCCCA duplexes and neighbouring-complementary strand exposure associated with: hairpin formation (a) or strand slippage (b). Hairpin formation could be facilitated by the 5mC → 5hmC modification. (c–e) Conceptual model for the distribution of G4s and palindromes in 5hmC-containing sequences identified in silico relative to histone H1 binding sites and to different nucleosome rearrangements. (c) Transcriptional retardation due to the presence of 5hmC/5mC in sequences including G4s/imperfect G4 structures and Pals (possibly associated with transcript processing). (d) Potential transcriptional arrest could result to H1 binding interference by MeCP2. (e) Densely arranged Pals and formation of single-stranded sequences complementary and tentative exposure to transcription factors for transcriptional activation/deactivation. Two-six palindromes and a single G4 or imperfect G4 are shown (upper tentative limits: 3 G4s and 14 Pals, respectively)
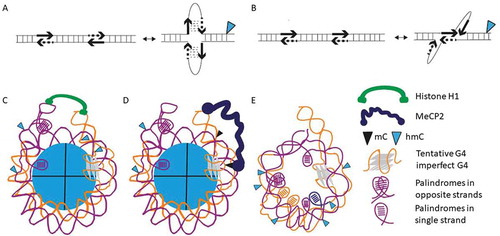
These findings are summarized in . The almost identical frequency of TGGGA and TCCCAs is in agreement with the formation of a 5bp double-stranded regions in the presence of 5hmC [Citation53], and occasional strand slippage, exposing variable single-stranded regions depending on the intervening sequences. The plasticity is enhanced by increasing the 5hmC/CpG ratio near TGGGA/TCCCAs, particularly among LC sequences ()). Nucleosome occupancy is low in less complex sequences (low G4 and Pals), while in more complex sequences DNA accessibility and recognition by transcription factor depends on the 5mC/5hmC ratio ()).
A conceptual depiction of a G4-containing nucleosome in agreement with the previously analysed epigenetic and structural element balance is shown in ). It is noted that less than half of the 5hmC-containing sequences include a potential G4 motif. An average number of six palindromes is included per nucleosome. Sites of 5hmC are deduced from the CpG sites in small length sequences: One or three clustered CpGs, or CpGs adjacent to motifs with palindromes. The A-rich or T-rich motif (tentative H1 histone-binding site [Citation47]), and the single G4 are in proximity. Sites complementary to G4 lack secondary structure (i-motif configuration) and are also available for protein recognition.
Binding of MeCP2, or possibly another methyl-binding protein, to the epigenetically modified sites interferes with H1 binding and transcription is possibly retarded ()). In addition, ) shows that MeCP2 binding is probably associated with 5hmC decrease [Citation54]. This condition is met in active promoters which are deployed of nucleosomes [Citation55]. Motifs containing these sequences (e.g., in HOX genes) are recognized by Homeobox proteins as well as Jun and Fos-regulated, cell-specific and often associated with ageing.
The proposed model is in compliance with the observations of Collings and Anderson [Citation24] regarding the presence of a methylation core in nucleosomes and predicts a palindrome density similar to that previously proposed by [Citation44]. Palindromic motifs in 5hmC-containing sequences are non-random, contrary to those previously described as AT-rich regions [Citation44]. In agreement with previous findings, 5hmC is shown close to the H1-histone binding site [Citation46].
In ), MeCP2 is proposed as a factor that could be associated with 5hmC presence in nucleosomes. MeCP2 regulates the conversion of 5mC to 5hmC [Citation56] and mediates nucleosome transformations through its cooperative binding to epigenetically modified DNA acting as an histone H1 competitor [Citation57]. Its binding sites exhibit different affinities for the two epigenetically modified cytosines, and one of the previously reported binding motifs, the AT repeat (hook1 motif) [Citation58]. Furthermore, MeCP2 traps the nucleosome in a more compact, mononucleosome structure [Citation59]. These requirements are met in the conceptual model in and are in agreement with the presence of the A- and T-rich motifs. The presence of a G4 in contiguity with the H1 histone binding site could account for the reported MeCP2-binding cooperativity to the nucleosome, in the presence of metals [Citation57]. Regulation by MeCP2 is cell-specific [Citation60] similarly to 5hmC presence [Citation14]. These characteristics of MeCP2 support its tentative role in 5mC/5hmC discrimination [Citation61].
However, the biological role of MeCP2 is complicated by its presence in two different isoforms [Citation62], with varying biological roles. In addition, there are three different TET enzymes which are actually activated at specific instances during embryogenesis [Citation63]. Furthermore, epigenetically regulated mobilization of histone deacetylases and corepressors are probably involved in this process [Citation64]. Thus, the parameters involved in the regulation of demethylation are multiple and complex.
G4 and Pals structures probably expose part of the complementary sequence to binding of different factors, including transcriptional regulators, positive or negative (). This is in agreement with previous studies [Citation27,Citation65,Citation66]. Moreover, both G4s and Pals could also ‘translate’ the presence of environmental factors to immediate conformational changes [Citation67], and adjust the DNA expression to environmental needs under normal conditions [Citation66]. This would account for the distribution of repeated complementary motifs in antiparallel strands (). The data presented above indicate that the previously reported, cancer-related changes of 5hmC [Citation26] could be associated with nucleosome deregulation.
The distinct roles of 5mC and 5hmC in nucleosome formation, together with G4s and Pals can be ascribed to the fact that 5mC, 5hmC, and 5-formylcytosine exert different effects on DNA flexibility [Citation16]. These epigenetic modifications could also contribute to the interplay of different structures, which as a result of the 5mC:5hmC balance between the Pals and G4s [Citation68], could be critical for the binding of modifiers and contribute to histone modifications. On the other hand, disruption of this balance could account for compromising of epigenetic memory as a result of repetitive DNA replication cycles when associated with the formation of stable G4 structures and methylation [Citation69] and for the frequently observed silent mutations associated with the formation/loss of palindromes in the coding region of cancer-related genes [Citation70]. Finally, it could account for the common phenomenon of deregulation in gene bodies and enhancers where 5hmC is observed [Citation71].
Multiple transient helical formations such as G4s and Pals, which are evidently related to the balance of epigenetic modifications, could be viewed as chromatin ‘compressions’ and ‘expansions’ [Citation72] involved in cellular reprogramming [Citation29]. Alternating 5mC and 5hmC epigenetic signals would probably affect the proposed model for nucleosome occupancy changes [Citation69,Citation73] and transcription factor binding. The non-duplex structures complementary to G4s could emerge as mediators of epigenetic modifications particularly in gene bodies and in enhancers where 5hmC is detected [Citation74]. In addition, the non-duplex regions could serve as binding sites for RNA repressors such as polycomb repressive complex 2 (PRC2), that acts as a histone methyltransferase during transcription, recognizing interspersed regions of higher and lower flexibility [Citation75], depending on the presence of epigenetically modified DNA sequences. Finally, variations of 5hmC and 5mC near the linker might contribute to the binding of the different H1 variants, a process which is also sensitive to chromatin expansions and contractions [Citation76].
The different frequency of G4 in X chromosome has been previously reported by Lin et al. [Citation77] for the whole genome. In addition, it has been previously shown that sex chromosomes are mostly depleted of 5hmC [Citation78,Citation79]. However, according to our findings, the nucleosomes formed in X chromosome exhibit the same G4/Pals balance observed in autosomes.
In conclusion, the universal structural characteristics presently analysed in association with epigenetic cytosine modifications, reveal that the epigenetic 5mC/5hmC balance relative to the presence of G4s and Pals can act as epigenetic molecular springs. Such structures could play a catalytic role in changing the accessibility of the helix during replication, transcription, and probably, co-transcriptional splicing. These data contribute to our understanding of the chromatin plasticity and constitutes a useful background for understanding expression mechanisms, the impact of external elements, cancer, and ageing and could contribute to the design of new compounds of pharmaceutical interest.
Supplemental Material
Download Zip (28.7 MB)Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: in the right place at the right time. Science. 2018;361:1336–1340.
- Anastasiadou C, Malousi A, Maglaveras N, et al. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30:267–275.
- Lev Maor G, Yearim A, Ast G. The alternative role of DNA Methylation in splicing regulation. Trends Genet. 2015;31:274–280.
- Salami F, Qiao S, Homayouni R. Expression of mouse Dab2ip transcript variants and gene methylation during brain development. Gene. 2015;568:19–24.
- Parry L, Clarke AR. The roles of the methyl-CpG binding proteins in cancer. Genes Cancer. 2011;2:618–630.
- Malygin EG, Hattman S. DNA methyltransferases: mechanistic models derived from kinetic analysis. Crit Rev Biochem Mol Biol. 2012;47:97–193.
- Klungland A, Robertson AB. Oxidized C5-methyl cytosine bases in DNA: 5-hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radic Biol Med. 2017;107:62–68.
- Ji D, You C, Wang P, et al. Effects of tet-induced oxidation products of 5-methylcytosine on DNA replication in mammalian cells. Chem Res Toxicol. 2014;27:1304–1309.
- Melamed P, Yosefzon Y, David C, et al. Tet enzymes, variants, and differential effects on function. Front Cell Dev Biol. 2018;6:22.
- Shukla A, Sehgal M, Singh TR. Hydroxymethylation and its potential implication in DNA repair system: a review and future perspectives. Gene. 2015;564:109–118.
- Li W, Zhang X, Lu X, et al. 5-hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017;27:1243–1257.
- Tammen SA, Dolnikowski GG, Ausman LM, et al. Aging alters hepatic DNA hydroxymethylation, as measured by liquid chromatography/mass spectrometry. J Cancer Prev. 2014;19:301–308.
- Ponnaluri VKC, Ehrlich KC, Zhang G, et al. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics. 2017;12:123–138.
- Gackowski D, Zarakowska E, Starczak M, et al. Tissue-specific differences in DNA modifications (5-hydroxymethylcytosine, 5-formylcytosine, 5-carboxylcytosine and 5-hydroxymethyluracil) and their interrelationships. PloS One. 2015;10:e0144859.
- DeNizio JE, Liu MY, Leddin EM, et al. Selectivity and promiscuity in TET-mediated oxidation of 5-methylcytosine in DNA and RNA. Biochemistry. 2019;58:411–421.
- Ngo TTM, Yoo J, Dai Q, et al. Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat Commun. 2016;7:10813.
- Falabella M, Kolesar JE, Wallace C, et al. G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci Rep. 2019;9:5605.
- Kwok CK, Merrick CJ. G-quadruplexes: prediction, characterization, and biological application. Trends Biotechnol. 2017;35:997–1013.
- Mao S-Q, Ghanbarian AT, Spiegel J, et al. DNA G-quadruplex structures mold the DNA methylome. Nat Struct Mol Biol. 2018;25:951–957.
- Cheng M, Cheng Y, Hao J, et al., Loop permutation affects the topology and stability of G-quadruplexes. Nucleic Acids Res. 2018;46:9264–9275.
- Guo S, Lu H. Conjunction of potential G-quadruplex and adjacent cis-elements in the 5ʹ UTR of hepatocyte nuclear factor 4-alpha strongly inhibit protein expression. Sci Rep. 2017;7:17444.
- Hegyi H. Enhancer-promoter interaction facilitated by transiently forming G-quadruplexes. Sci Rep. 2015;5:9165.
- Halder K, Halder R, Chowdhury S. Genome-wide analysis predicts DNA structural motifs as nucleosome exclusion signals. Mol Biosyst. 2009;5:1703–1712.
- Collings CK, Anderson JN. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenetics Chromatin. 2017;10:18.
- Tippana R, Xiao W, Myong S. G-quadruplex conformation and dynamics are determined by loop length and sequence. Nucleic Acids Res. 2014;42:8106–8114.
- Watanabe R, Kanno S-I, Mohammadi Roushandeh A, et al. Nucleosome remodelling, DNA repair and transcriptional regulation build negative feedback loops in cancer and cellular ageing. Philos Trans R Soc Lond B Biol Sci. 2017;372:20160473.
- Hänsel-Hertsch R, Beraldi D, Lensing SV, et al. G-quadruplex structures mark human regulatory chromatin. Nat Genet. 2016;48:1267–1272.
- Morgan RK, Molnar MM, Batra H, et al. Effects of 5-hydroxymethylcytosine epigenetic modification on the stability and molecular recognition of VEGF I-Motif and G-Quadruplex structures. J Nucleic Acids. 2018;2018:9281286.
- Varizhuk A, Isaakova E, Pozmogova G. DNA G-quadruplexes (G4s) modulate epigenetic (Re)programming and chromatin remodeling: transient genomic g4s assist in the establishment and maintenance of epigenetic marks, while persistent G4s may erase epigenetic marks. BioEssays. 2019;41:e1900091.
- Liu M-J, Seddon AE, Tsai ZT-Y, et al. Determinants of nucleosome positioning and their influence on plant gene expression. Genome Res. 2015;25:1182–1195.
- Li X, Liu Y, Salz T, et al. Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver. Genome Res. 2016;26:1730–1741.
- Quinlan AR. BEDTools: the swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:11.12.1–34.
- Hon J, Martínek T, Zendulka J, et al. Pqsfinder: an exhaustive and imperfection-tolerant search tool for potential quadruplex-forming sequences in R. Bioinformatics. 2017;33:3373–3379.
- Pages H, Aboyoun P, Gentleman R, et al., Biostrings: efficient manipulation of biological strings. 2019.
- Malousi A, Kouidou S, Tsagiopoulou M, et al. MeinteR: A framework to prioritize DNA methylation aberrations based on conformational and cis-regulatory element enrichment. Sci Rep. 2019;9:19148.
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366.
- Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
- Smit AFA, Hubley R. RepeatModeler Open-1.0. 2008–2015; software Available from: http://www.repeatmasker.org.
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580.
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018.
- Chiu T-P, Xin B, Markarian N, et al., TFBSshape: an expanded motif database for DNA shape features of transcription factor binding sites preprint. 2019.
- R Core Team. a language and environment for statistical computing.: R foundation for statistical computing. 2018; software Available from: https://www.R-project.org.
- Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20:267–273.
- Ma Q, Xu Z, Lu H, et al. Distal regulatory elements identified by methylation and hydroxymethylation haplotype blocks from mouse brain. Epigenetics Chromatin. 2018;11:75.
- Kizaki S, Zou T, Li Y, et al. Preferential 5-methylcytosine oxidation in the linker region of reconstituted positioned nucleosomes by tet1 protein. Chemistry. 2016;22:16598–16601.
- Roque A, Orrego M, Ponte I, et al. The preferential binding of Histone H1 to DNA scaffold-associated regions is determined by its c-terminal domain. Nucleic Acids Res. 2004;32:6111–6119.
- Sedghi Masoud S, Nagasawa K. I-motif-binding ligands and their effects on the structure and biological functions of I-motif. Chem Pharm Bull (Tokyo). 2018;66:1091–1103.
- Klug A. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation, Quart. Rev Biophys. 2010;43: 1–21.
- Martiénez-Balbás MA, Jiménez-Garciéa E, Azorin F. Zinc(II) ions selectively interact with DNA sequences present at the TFIIIA binding site of the Xenopus 5S-RNA gene. Nucl Acids Res. 1995;23:2464–2471.
- Brown ML, Schroth GP, Gottesfeld JM, et al. Protein and DNA requirements for the transcription factor IIIA-induced distortion of the 5 S rRNA gene promoter. J Mol Biol. 1996;262:600–614.
- Brukner I, Susic S, Dlakic M, et al. Physiological concentration of magnesium ions induces a strong macroscopic curvature in GGGCCC-containing DNA. J Mol Biol. 1994;236:26–32.
- Yu D, Sawitzke JA, Ellis H, et al. Recombineering with overlapping single-stranded DNA oligonucleotides: testing a recombination intermediate. Proc Natl Acad Sci U S A. 2003;100:7207–7212.
- Ludwig AK, Zhang P, Hastert FD, et al. Binding of MBD proteins to DNA Blocks Tet1 function thereby modulating transcriptional noise. Nucleic Acids Res. 2017;45:2438–2457.
- Owen-Hughes T, Gkikopoulos T. Making sense of transcribing chromatin. Curr Opin Cell Biol. 2012;24:296–304.
- Zheng Z, Ambigapathy G, Keifer J. MeCP2 regulates Tet1-catalyzed demethylation, CTCF binding, and learning-dependent alternative splicing of the BDNF gene in turtle. eLife. 2017;6. DOI:10.7554/eLife.25384
- Khrapunov S, Tao Y, Cheng H, et al. MeCP2 binding cooperativity inhibits DNA modification-specific recognition. Biochemistry. 2016;55:4275–4285.
- Lei M, Tempel W, Chen S, et al. Plasticity at the DNA recognition site of the MeCP2 MCG-binding domain, biochimica et biophysica acta. Gene Regul Mech. 2019;1862:194409.
- Riedmann C, Fondufe-Mittendorf YN. Comparative analysis of linker Histone H1, MeCP2, and HMGD1 on nucleosome stability and target site accessibility. Sci Rep. 2016;6:33186.
- Mellén M, Ayata P, Dewell S, et al. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430.
- Yang Y, Kucukkal TG, Li J, et al. Binding analysis of methyl-CpG binding domain of MeCP2 and rett syndrome mutations. ACS Chem Biol. 2016;11:2706–2715.
- Schmidt A, Zhang H, Cardoso MC. MeCP2 and Chromatin Compartmentalization. Cells. 2020;9:878.
- Khoueiry R, Sohni A, Thienpont B, et al. Lineage-specific functions of TET1 in the postimplantation mouse embryo. Nat Genet. 2017;49:1061–1072.
- Della Ragione F, Filosa S, Scalabrì F, et al. MeCP2 as a genome-wide modulator: the renewal of an old story. Front Genet. 2012;3:181.
- Campilongo R, Fung RKY, Little RH, et al. One ligand, two regulators and three binding sites: how KDPG controls primary carbon metabolism in pseudomonas. PLoS Genet. 2017;13:e1006839.
- Joseph SR, Pálfy M, Hilbert L, et al. Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. eLife. 2017;6. DOI:10.7554/eLife.23326
- Perriaud L, Marcel V, Sagne C, et al. Impact of G-quadruplex structures and intronic polymorphisms Rs17878362 and Rs1642785 on basal and ionizing radiation-induced expression of alternative P53 transcripts. Carcinogenesis. 2014;35:2706–2715.
- Czech A, Konarev PV, Goebel I, et al. Octa-repeat domain of the mammalian prion protein mRNA forms stable a-helical hairpin structure rather than G-quadruplexes. Sci Rep. 2019;9:2465.
- Mukherjee AK, Sharma S, Chowdhury S. Non-Duplex G-Quadruplex structures emerge as mediators of epigenetic modifications. Trends Genet. 2019;35:129–144.
- Kouidou S, Malousi A, Maglaveras N. Methylation and repeats in silent and nonsense mutations of P53. Mutat Res. 2006;599:167–177.
- Stroud H, Feng S, Morey Kinney S, et al. 5-hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54.
- Farnung L, Vos SM, Wigge C, et al. Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature. 2017;550:539–542.
- McGinty RK, Tan S. Nucleosome structure and function. Chem Rev. 2015;115:2255–2273.
- Williams K, Christensen J, Pedersen MT, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348.
- Wang X, Goodrich KJ, Gooding AR, et al. Targeting of polycomb repressive complex 2 to RNA by short repeats of consecutive guanines. Mol Cell. 2017;65:1056–1067.e5.
- Öberg C, Izzo A, Schneider R, et al. Linker histone subtypes differ in their effect on nucleosomal spacing in vivo. J Mol Biol. 2012;419:183–197.
- Eddy J, Vallur AC, Varma S, et al. G4 motifs correlate with promoter-proximal transcriptional pausing in human genes. Nucleic Acids Res. 2011;39:4975–4983.
- Lin I-H, Chen Y-F, Hsu M-T. Correlated 5-hydroxymethylcytosine (5hmC) and gene expression profiles underpin gene and organ-specific epigenetic regulation in adult mouse brain and liver. PloS One. 2017;12:e0170779.
- Yu B, Russanova VR, Gravina S, et al. DNA methylome and transcriptome sequencing in human ovarian granulosa cells links age-related changes in gene expression to gene body methylation and 3ʹ-End GC Density. Oncotarget. 2015;6:3627–3643.