
ABSTRACT
Acute myeloid leukaemia (AML) is a heterogeneous myeloid malignancy characterized by recurrent clonal events, including mutations in epigenetically relevant genes such as DNMT3A, ASXL1, IDH1/2, and TET2. Next-generation sequencing analysis of a mother and son pair who both developed adult-onset diploid AML identified a novel germline missense mutation DNMT3A p.P709S. The p.P709S protein-altering variant resides in the highly conserved catalytic DNMT3A methyltransferase domain. Functional studies demonstrate that the p.P709S variant confers dominant negative effects when interacting with wildtype DNMT3A. LINE-1 pyrosequencing and reduced representation bisulphite sequencing (RBBS) analysis demonstrated global DNA hypomethylation in germline samples, not present in the leukaemic samples. Somatic acquisition of IDH2 p.R172K mutations, in concert with additional acquired clonal DNMT3A events in both patients at the time of AML diagnosis, confirms the important pathogenic interaction of epigenetically active genes, and implies a strong selection and regulation of methylation in leukaemogenesis. Improved characterization of germline mutations may enable us to better predict malignant clonal evolution, improving our ability to provide customized treatment or future preventative strategies.
Introduction
Acute myeloid leukaemia (AML) is a haematopoietic malignancy characterized by arrested maturation and uncontrolled proliferation of immature clonal myeloid cells. Cooperating genomic events within various functional gene categories such as epigenetic regulators, master haematopoietic transcription factors, key tumour suppressors, and cell signalling and proliferation pathways have been recognized as underlying causes of the notable clinical and biological heterogeneity of AML. Concurrent with improved molecular annotation of AML, the past decade of next-generation sequencing has led to an increased recognition of inherited leukaemia predispositions, with evidence suggesting that 5–10% of individuals with a haematologic malignancy may have an underlying germline predisposing mutation[Citation1]. Nearly a dozen adult-onset inherited leukaemia predisposition syndromes are now defined, including the myeloid-predominant leukaemia syndromes related to RUNX1, GATA2, CEBPα, ETV6, ANKRD26 and DDX41[Citation2]. Germline mutations in these genes are associated with >30% of the familial MDS/AML pedigrees, however additional characterizations of these mutations have been lacking[Citation3].
DNMT3A is a highly conserved 130 kDa protein encoded by 23 exons[Citation4]. Somatic mutations in DNMT3A occur in 12–35% of MDS and AML, with a hotspot mutation located at arginine 882 (R882) in the catalytic domain and additional pathogenic variants distributed throughout the gene[Citation5]. DNMT3A mutations are typically identified at higher variant allele frequency (VAF) than other accompanying mutations in myeloid malignancies, suggestive of a founding mutational event [Citation6,Citation7]. Somatic mutations in DNMT3A provide a modest haematopoietic stem cell (HSC) fitness advantage, contributing over time to clonal haematopoiesis which can be identified in >10% of hematologically-normal individuals ≥80 years of age [Citation8,Citation9]. DNMT3A mutations alone, however, do not appear sufficient for the development of bona fide haematologic malignancy.
DNMT3A belongs to the DNA methyltransferase family (including DNMT1 and DNMT3B) with a specific role of replication independent, de novo addition of methyl groups to cytosine residues within CpG dinucleotides. Murine models of conditional deletions of Dnmt3a in haematopoietic stem cells (HSC) favour HSC self-renewal over differentiation and out-compete wild-type DNMT3A stem cells, with expansion of the HSC population within the bone marrow. Notably the animals were not reported to develop leukaemia[Citation10]. DNMT3A-null HSCs promote DNA hypomethylation, particularly at CpG islands and canyon regions enriched for genes associated with HSC self-renewal that are overexpressed in cancer (i.e. Hoxa9, Meis1 and Mecom) [Citation11–13].
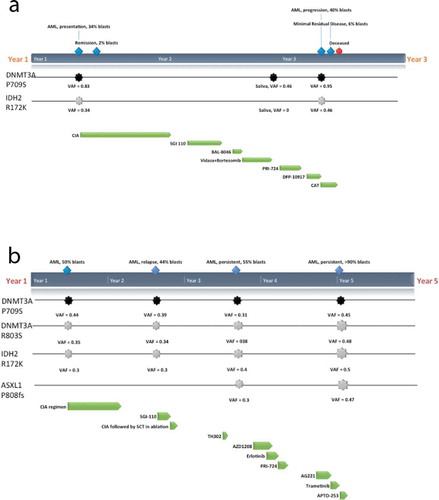
We studied a mother (I-2) and son (II-1) of European ancestry treated at our institution for cytogenetically normal (CN) AML with FLT3 internal tandem duplication (ITD) and NPM1 wild-type status, presenting at the ages of 50 and 30, respectively. Both patients were treated with cytarabine-based intensive induction/consolidation chemotherapy with attainment of an initial complete remission followed by AML relapse (complete remission duration of 8 months for I-2, and 12 months for II-2) (). Whole exome sequencing of samples at AML diagnosis, remission, and relapse in I-2 and II-1, as well as germline samples including cultured skin fibroblasts and saliva in II-1, and saliva in I-2, were performed.
Figure 1. Clinical course and mutations detected in I-2(a) and II-1(b). Germline mutation in DNMT3A P709S (black) and predominant somatic mutations in IDH2 R172K and ASXL1 P808fs (grey) that were detected within bone marrow samples at various timepoints (blue) along with the regimens and protocols given (green): CIA (induction of clofarabine, idarubicin and cytarabine), SGI-110 (DNMT inhibitor), BL-8040 (CXCR4 inhibitor), TH302 (Evofosfamide, replication inhibitor), AZD1208 (Pim-kinase inhibitor), Erlotinib (epidermal growth factor receptor (EGFR) inhibitor), PRI-724 (Wnt inhibitor), J0894 + J9100 (Decitabine + Cytarabine), AG221 x 2 cycles (IDH2 inhibitor), Trametinib (MEK inhibitor), APTO-253 (MTF-1 inhibitor) are shown
Results
Somatic identification of missense DNMT3A p.P709S
Two protein-altering rare variants (population MAF<0.01) in the germline and remission samples of both I-2 and II-1 were identified: DNMT3A p.P709S and MSH2 p.A689T. No damaging or potentially damaging germline mutations in known hereditary cancer predisposition and/or bone marrow failure genes were identified. Immunohistochemistry demonstrated that MSH2 protein levels were retained within the germline samples and leukaemic blasts, and no allelic shifts were identified on microsatellite instability (MSI) testing consistent with MSI-stable phenotype (data not shown). Since there was no demonstrable functional impact of the A689T MSH2 variant, we turned our attention to the DNMT3A p.P709S candidate variant. This missense variant is absent from public databases including dbSNP129 (also not found in >1% dbSNP146)[Citation14], the 1000 Genomes Project [Citation15] and the Exome Variant Server[Citation16]. Additionally this variant has not been identified as a somatic variant in >4500 AML samples within our institution and >2800 European cases (E. Papaemmanuil and P. Campbell, personal communication).
Deficient methylation activity in DNMT3A p.P709S
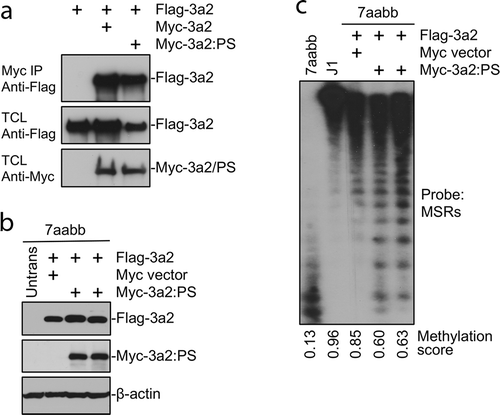
The detected germline p.P709S alteration in both I-2 and II-1 resides in the DNMT3A SAM-dependent methyltransferase C5-type catalytic domain (amino acids 634–912) at the PC motif (P709/C710) (). We performed functional studies of the p.P709S mutation using the corresponding to murine Dnmt3a2:P705S mutant. WT murine Dnmt3a and Dnmt3a:P705S were stably expressed in Dnmt3a/3b double knockout (7aabb) embryonic stem (ES) cells. Methylation analysis by Southern blot with a probe specific for major satellite repeats (MSRs) () demonstrated that WT Dnmt3a was largely able to restore DNA methylation, whereas the Dnmt3a:P705S mutant failed to do so. In addition to near complete inactivity of the P705S protein, co-transfection of P705S and WT proteins resulted in less efficient rescue of DNA methylation compared to expression of WT Dnmt3a alone (). Therefore, Dnmt3a:P705S inhibits wild-type DNMT3A, attenuating normal levels of DNA methylation in a dominant-negative manner[Citation17].
Figure 2. Position of the germline DNMT3A P709 within (a) the SAM-dependent methyltransferase (arrow) (cBioPortal) [Citation45,Citation46] and (b) the substrate-binding pocket of the DNA methyltransferase in the crystal structure (CN3D) [Citation47] and its positional relationship to the R882 canonical mutation
![Figure 2. Position of the germline DNMT3A P709 within (a) the SAM-dependent methyltransferase (arrow) (cBioPortal) [Citation45,Citation46] and (b) the substrate-binding pocket of the DNA methyltransferase in the crystal structure (CN3D) [Citation47] and its positional relationship to the R882 canonical mutation](/cms/asset/f3326a6b-f32c-41f0-ad03-3ac6483999c7/kepi_a_1809871_f0002_c.jpg)
Figure 3. The P705S mutation severely impairs the ability of Dnmt3a to methylate DNA
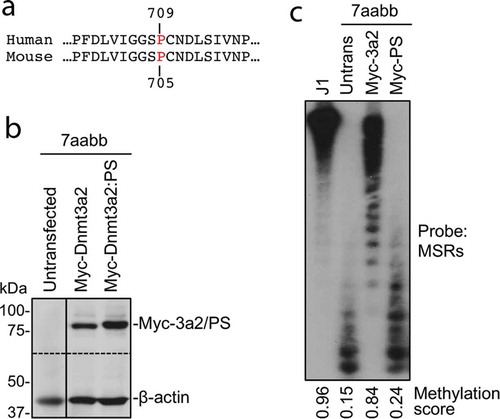
Figure 4. The Dnmt3a2:P705S mutant protein interacts with and antagonizes wild-type Dnmt3a2
Global hypomethylation in mother and son with DNMT3A p.P709S
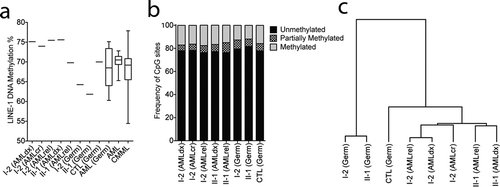
Given the inhibitory nature of DNMT3A p.P709S, we next assessed DNA methylation in the germline and AML samples of both individuals. LINE-1 methylation was evaluated as a surrogate for global DNA methylation levels. Compared to oral gDNA from a healthy donor, lower germline methylation of both I-2 and II-1 was observed, as would be expected from a loss-of-function mutation (). Next, we evaluated genome-wide DNA methylation by Reduced Representation Bisulphite Sequencing (RRBS). This analysis identified I-2 and II-1 germline samples to be the least methylated, with respectively 79.5% and 81.7% of all evaluated CpG sites classified as unmethylated (defined as < 25% methylation), compared to 77.9% in the germline control and 76.4% to 78% in AML (). Concordantly, average DNA methylation per major genomic compartments (upstream, promoter, gene body, intergenic, non-CpG) identified significant and relative hypomethylation of patients I-2 and II-1 germline DNA (Supplementary Figure 1). Hierarchical clustering was performed on the top 1% most variably methylated CpG sites; I-2 and II-1 germline samples were most similar to one another. AML samples clustered apart from germline and were sub-grouped per individual (). Consistent with loss of function activity, the DNMT3A p.P709S variant is associated with decreased DNA methylation in germline samples.
Figure 5. DNMT3A p.P709S is associated with decreased DNA methylation in germline samples
Similar somatic evolution of AML in mother and son
Additional sequencing of I-2 and II-1 samples was performed to identify acquired genomic events and evaluate for clonal evolution during therapy. Of interest, a somatic IDH2 p.R172K mutation at AML diagnosis was identified in both diagnostic samples, present at a VAF of 36% in I-2 and 32% in II-1 (). Also of note, the diagnostic sample of I-2 demonstrated reduction to homozygosity of the mutant p.P709S allele (VAF 84%, copy neutral loss of heterozygosity), whilst in II-1 the acquisition of an additional, somatic p.R803S mutation in DNMT3A was identified (VAF 31%) in addition to the p. P709S mutation (VAF 41%) by 300-gene panel sequencing. Additional somatic variants in commonly-mutated genes were detected, several at ultra-low allele frequencies by 28-gene panel sequencing (Supplementary Table 1).
Discussion
More than half of DNMT3A mutations in myeloid malignancies occur at residue p.R882 [Citation18,Citation19]. The p.R882 mutation, among other recurrent DNMT3A mutations in AML, leads to reduced de novo methyltransferase activity, with focal hypomethylation at clustered CpGs in CpG islands due to the inability to form active homo-tetramers[Citation20]. Despite being a heterozygous somatic mutation, the p.R882 exerts a disproportionate impact due to dominant-negative activity, leading to a retained DNMT3A wild-type function of only ~20% [Citation13,Citation17]. The germline mutation p.P709S identified here has closer contact with DNA substrate than the p.R882 variant and also exerts dominant negative effects that were associated with global hypomethylation.
Mutations in genes encoding essential enzymes in 5-hydroxymethylation (TET2, IDH1, IDH2) frequently co-occur with DNMT3A mutations in AML [Citation21,Citation22]. Co-occurrence with IDH2 mutations are especially common and are associated with a unique gene-expression and DNA methylation profile [Citation23,Citation24]. IDH2 mutations are common somatic mutations in AML, occurring in ~12% of AML, with increasing frequency with increasing age and in patients with normal karyotype [Citation5,Citation25,Citation26]. IDH2 mutations occur at ‘hot spot’ arginine residues within the conserved enzymatic active site, namely at p.R140 or p.R172, and cause the aberrant production of the onco-metabolite 2-hydroxyglutarate[Citation27]. Via competitive inhibition of aKG, 2HG accumulation blocks TET2-dependent hydroxymethylation and histone demethylation, and leads to a distinct and globally ‘hypermethylated DNA signature’ [Citation21,Citation28]. Of interest, while common in certain T-cell lymphomas, the specific IDH2-R172 mutation is rare in AML, representing only 1% of AML overall[Citation23]. The presence of IDH2-R172 and DNMT3A mutations are strongly correlated in both AML and lymphomas, and our finding of an IDH2 p.R172K mutation in both I-2 and II-1 at the time of AML diagnosis in the context of germline DNMT3A p.P709S is notable and supports the belief that these specific epigenetic and metabolically-active mutations may interact to contribute to malignant progression in patients with pre-leukaemic stem cells.
An overgrowth syndrome now termed Tatton-Brown-Rahman overgrowth syndrome (TBRS, OMIM:615,879) [Citation29] has been described in individuals with key features including heavy horizontal eyebrows, tall stature and intellectual disability [Citation30–32]. In these patients, constitutional DNMT3A mutations in functional domains have been identified that parallel those found in haematologic malignancies[Citation33]. In our series, I-2 was of average height (169 cm, 5ʹ6”) and II-1 was tall (191 cm, 6ʹ3”). Neither patient had evidence or history of developmental delay, cognitive impairment, or dysmorphology. Therefore, the development of TBRS may not be solely dependent on germline DNMT3A mutations, or alternatively this family may represent a phenotypically silent version of TBRS. To date, three patients with TBRS have been reported to develop acute myeloid leukaemia, two with the p.R882S missense and one with p.Y735S [Citation30,Citation34,Citation35]. Future studies are needed to more confidently ascertain the risk of developing haematologic disorders such as AML in patients with constitutional DNMT3A mutations.
In summary, we identified an inherited germline missense mutation in DNMT3A p.P709S driving a global decrease in DNA methylation and predisposing to familial AML. Functional studies confirm the dominant inhibitory activity of this mutation and related global hypomethylation of the constitutional samples. We hypothesize that the requirement for methylation reprogramming during haematopoiesis may result in selection for cells that have acquired somatic alterations to sufficiently increase methylation levels to compensate for this germline deficiency. The nature of these secondary mutations, which in this family was an oncogenic IDH2 p.R172K, could result in haematologic malignancy. These data suggest that there is strong selection for the degree of methylation in leukaemogenesis resulting in either gain of loss-of-function DNMT3A p.P709S or the acquisition of DNMT3A p.R803S. This suggests that future characterization of germline mutations may enable us to better predict malignant clonal evolution and improve our ability to provide customized treatment strategies.
Methods
Sample collection
Patient samples were collected under informed consent through a protocol approved by the University of Texas MD Anderson Cancer Centre Institutional Review Board. From I-2, we collected unsorted bone marrow samples at presentation, remission, and progression, saliva at remission, and a skin punch at progression. From II-1, we collected bone marrows during presentation of AML, at multiple relapse time points, and saliva at progression.
Next generation sequencing
Isolation of genomic DNA (gDNA) from whole blood, bone marrow or saliva collected in Oragene® Discover devices was performed using the automated ReliaPrep™ 96 gDNA Miniprep HT System (Promega Corporation, Madison, WI). Proteinase K and a specially formulated Cell Lysis Buffer is added to the blood/bone marrow specimens and subjected to heating and shaking cycles to liberate sample DNA. Released DNA is bound to the ReliaPrep™ particles in the presence of the Binding Buffer. The gDNA bound to Resin is captured by a magnet and contaminants are removed by washing with the Wash Buffer and ethanol. The gDNA is then eluted from the paramagnetic particles with 25 mM Tris-HCl. Fibroblasts were grown from the skin punch biopsy and were cultured following procedures suggested by the EuroBioBank (Besta Institute, Italy). gDNA from cultured fibroblasts were isolated using QIAamp DNA Mini kit (Qiagen Inc., Germantown, MD). For all gDNA samples, library preparation was done using SureSelect Human All Exon V4 library preparation (Agilent Technologies, Inc., Santa Clara, CA) with 76 bp short-read sequencing on Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA) to a target coverage of 200X for bone marrow samples and 100X for germline saliva and fibroblast samples. After mapping of reads (hg19 reference genome) (BWA-MEM) [Citation36], variant calling was done using Platypus[Citation37]. To determine high quality and rare variants, exonic variants with a minimum read depth of 15 and minor allele frequencies (MAF) <0.01 in ESP6500 [Citation16] and 1000Genomes [Citation15] were examined. For copy number calls, segments were generated using Circular Binary Segmentation (CBS) [Citation38] and log2 ratio scores derived with a modified version of HMMcopy[Citation39]. Gains (log2 > 0.5) and losses (log2 < −0.5) were scored. Clinical testing was done on I-2 bone marrow using a 28-gene panel (CMS28) at the Medical Diagnostics Laboratories. Panel sequencing on 295 genes related to haematologic malignancies was reported previously[Citation40].
Functional studies of murine DNMT3A P705S mutant
The effects of the murine DNMT3A p.P705S (equivalent to human P709S) mutation were evaluated using DNMT3A/DNMT3B double KO ES cells, as described previously[Citation17].
Patient DNA methylation analyses
LINE-1 methylation was evaluated using pyrosequencing methylation analysis as previously described[Citation41]. Reduced Representation Bisulphite Sequencing (RRBS) library preparation and sequencing were done at the UT MD Anderson Cancer Centre’s DNA Methylation Analysis Core and Sequencing and Microarray Facility, according to published protocols [Citation42,Citation43]. Sequencing reads were mapped to the human reference genome hg19 using Bismark[Citation44]. Definition of gene promoter regions were based on hg19 RefSeq gene annotation, with regions starting 1 kb upstream of the annotated transcription start site (TSS) and extending 500 base pairs downstream of TSS. Exons, introns, and CpG islands coordinates were collected from the USCS Genome Browser hg19 human reference genome. Hierarchical clustering analysis was performed for the top 1% most variable CpG sites across the 8 samples evaluated by RRBS, using Euclidean distance and complete linkage parameters.
Authorship contributions
CDD, HCB, ME, SAB, and PAF conceived of the study and validation strategies. CG conducted the next generation sequencing studies through the Cancer Genomics Laboratories. ME, TG, and KK conducted the patient methylation studies. SH and TC conducted the murine functional assays. All authors were involved in data analysis and interpretation. CDD, HCB, ME, TC, SAB, and PAF drafted the article. All authors reviews and gave final approval of the version to be published.
Data availability
Whole exomes are deposited under accession number EGAS00001002940 at the European Genome-phenome Archive (EGA), hosted at the EBI and the CRG.
Supplemental Material
Download Zip (728.3 KB)Acknowledgments
This work was supported in part by the MD Anderson Cancer Center Support Grant P30 CA016672 and by generous philanthropic contributions to MD Anderson’s MDS/AML Moon Shot Program. CDD is also supported by the Jeanne F. Shelby Scholarship Fund which has supported her R. Lee Clark Fellow award. Cancer Prevention Research Institute (R120501) and Welch Foundation’s Robert A. Welch Distinguished University Chair Awards (G-0040) were given to PAF. An NIH R01 grant (1R01AI121403-01A1) was awarded to TC. AML samples were a gift from Hui Yang and Carlos Bueso-Ramos; CMML samples were given by K.P. Patel). These sponsors were public and nonprofit organizations who did not take part in collecting, processing, or interpreting the data. The results in the discussion on IDH2 frequencies are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Disclosure statement
The authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Furutani E, Shimamura A. Germline genetic predisposition to hematologic malignancy. J Clin Oncol. 2017;35:1018–1028.
- Godley LA, Shimamura A. Genetic predisposition to hematologic malignancies: management and surveillance. Blood. 2017;130:424–432.
- Churpek JE, Pyrtel K, Kanchi KL, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015;126:2484–2490.
- Robertson KD, Uzvolgyi E, Liang G, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298.
- Im AP, Sehgal AR, Carroll MP, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28(9):1774–1783.
- Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. .
- Papaemmanuil E, Dohner H, Campbell PJ. Genomic classification in acute myeloid leukemia. N Engl J Med. 2016;375:900–901.
- Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498.
- Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487.
- Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23–31.
- Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152–165.
- Qu Y, Lennartsson A, Gaidzik VI, et al. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 2014;9:1108–1119.
- Russler-Germain DA, Spencer DH, Young MA, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442–454.
- Information NCfB. dbSNP: database of single nucleotide polymorphisms.
- Auton A, Brooks LD, Durbin RM, et al.; Genomes Project C. A global reference for human genetic variation. Nature. 2015;526:68–74.
- National Heart L, and Blood Institute. EXP: grand opportunity exome sequencing project.
- Kim SJ, Zhao H, Hardikar S, et al. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013;122:4086–4089.
- Gaidzik VI, Schlenk RF, Paschka P, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML study group (AMLSG). Blood. 2013;121:4769–4777.
- Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433.
- Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem. 2012;287:30941–30951.
- Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567.
- Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221.
- Chen C, Liu Y, Lu C, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27:1974–1985.
- DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90:732–736.
- Ley TJ, Miller C, Ding L, et al.; Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074.
- Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234.
- Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478.
- Online mendelian inheritance in man OJHU. Baltimore, MD. (MIM Number: 615879).
- Kosaki R, Terashima H, Kubota M, et al. Acute myeloid leukemia-associated DNMT3A p.Arg882His mutation in a patient with Tatton-Brown-Rahman overgrowth syndrome as a constitutional mutation. Am J Med Genet A. 2017;173:250–253.
- Tatton-Brown K, Seal S, Ruark E, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46:385–388.
- Xin B, Cruz Marino T, Szekely J, et al. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin Genet. 2016; 91:623–628.
- Shen W, Heeley JM, Carlston CM, et al. The spectrum of DNMT3A variants in Tatton-Brown-Rahman syndrome overlaps with that in hematologic malignancies. Am J Med Genet A. 2017;173:3022–3028.
- Hollink I, van den Ouweland AMW, Beverloo HB, et al. Acute myeloid leukaemia in a case with Tatton-Brown-Rahman syndrome: the peculiar DNMT3A R882 mutation. J Med Genet. 2017;54:805–808.
- Tatton-Brown K, Zachariou A, Loveday C, et al. The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018;3:46.
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v1. 2013.
- Rimmer A, Phan H, Mathieson I, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–918.
- Olshen AB, Venkatraman ES, Lucito R, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572.
- Ha G, Roth A, Lai D, et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012;22:1995–2007.
- Takahashi K, Patel K, Bueso-Ramos C, et al. Clinical implications of TP53 mutations in myelodysplastic syndromes treated with hypomethylating agents. Oncotarget. 2016;7:14172–14187.
- Estecio MR, Gharibyan V, Shen L, et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One. 2007;2:e399.
- Gertz J, Varley KE, Reddy TE, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7:e1002228.
- Gu H, Smith ZD, Bock C, et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6:468–481.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27:1571–1572.
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404.
- Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
- Wang Y, Geer LY, Chappey C, et al. Cn3D: sequence and structure views for Entrez. Trends Biochem Sci. 2000;25:300–302.