
ABSTRACT
Latinos are heavily affected with childhood asthma. Little is known about epigenetic mechanisms of asthma in Latino youth. We conducted a meta-analysis of two epigenome-wide association studies (EWAS) of asthma, using DNA from white blood cells (WBCs) from 1,136 Latino children and youth aged 6 to 20 years. Genes near the top CpG sites in this EWAS were examined in a pathway enrichment analysis, and we then assessed whether our results replicated those from publicly available data from three independent EWAS conducted in non-Latino populations. We found that DNA methylation profiles differed between subjects with and without asthma. After adjustment for covariates and multiple testing, two CpGs were differentially methylated at a false discovery rate (FDR)-adjusted P < 0.1, and 193 CpG sites were differentially methylated at FDR-adjusted P < 0.2. The two top CpGs are near genes relevant to inflammatory signalling, including CAMK1D (Calcium/Calmodulin Dependent Protein Kinase ID) and TIGIT (T Cell Immunoreceptor With Ig And ITIM Domains). Moreover, 25 genomic regions were differentially methylated between subjects with and without asthma, at Šidák-corrected P < 0.10. An enrichment analysis then identified the TGF-beta pathway as most relevant to asthma in our analysis, and we replicated some of the top signals from publicly available EWAS datasets in non-Hispanic populations. In conclusion, we have identified novel epigenetic markers of asthma in WBCs from Latino children and youth, while also replicating previous results from studies conducted in non-Latinos.
Introduction
Asthma is the most common chronic respiratory disease of childhood, affecting nearly 6 million children in the United States (U.S.). In this country, the estimated prevalence of current childhood asthma is 13.6% in Puerto Ricans, 6.8% in non-Hispanic whites, 6.6% in Mexicans, and 7.6% in other Latino subgroups [Citation1].
Asthma is a complex, multifactorial disease, whose genetic component has been studied in several genome-wide association studies (GWASs). A large meta-analysis of GWASs of asthma including members of multiple racial or ethnic groups, identified 878 single nucleotide polymorphisms (SNPs) at 18 genomic loci as associated with asthma. However, such loci explained less than 5% of the variability in asthma risk, supporting a substantial contribution of environmental exposures to disease pathogenesis [Citation2]. Such exposures could increase asthma risk by altering the expression of disease-susceptibility genes through epigenetic mechanisms such as DNA methylation [Citation3].
Epigenome-wide association studies (EWASs) using DNA from whole blood or white blood cells (WBCs) have identified differential methylation of CpG sites or genomic regions by asthma status [Citation4–8]. The first EWAS of childhood asthma assessed methylation in DNA from peripheral blood mononuclear cells from 194 African American, Dominican, and Haitian school-aged children with (n = 97) and without (n = 97) atopic asthma [Citation8]. Of 81 genomic regions that were differentially methylated in atopic asthma, 10 were selected for replication in a cohort of 101 African American children with atopic asthma. Of those 10 loci, 5 (RUNX3, IL4, CAT, KLF6, and NBR2) were associated with atopic asthma in the replication cohort, (P < 0.05 in a one-tailed t-test). More recently, a meta-analysis of whole-blood DNA methylation in 2,862 children with (n = 631) and without (n = 2231) asthma identified 179 CpGs differentially methylated in asthma, of which only 2 were nominally associated with asthma (in the same direction of association) in a parallel meta-analysis of whole-blood DNA methylation in newborns and asthma (cg16409452 in EVL and cg09423651 in NCK1) [Citation6]. Despite efforts to include multi-ethnic populations in this study, 87% of the participants were of European ancestry.
To date, no EWAS of asthma assessing DNA methylation in WBCs has been conducted solely in Latino children and young adults. We report the results of a meta-analysis of two epigenome-wide studies of DNA methylation in WBCs from Latino children and youth. Moreover, we examined whether the top signals from this analysis replicate findings from previous studies in other cohorts.
Methods
Please see the Supplementary Appendix for additional details.
Study populations
Puerto Rico cohorts: 1) The Epigenetic Variation and Childhood Asthma in Puerto Ricans Study (EVA-PR): Subject recruitment and the study protocol of EVA-PR have been previously described [Citation9]. In brief, EVA-PR is a case-control study of asthma in 543 Puerto Ricans aged 9 to 20 years who were recruited in the metropolitan area of San Juan and Caguas (Puerto Rico) from February 2014 to May 2017, using the same multistage probability sample design as in a prior study (PR-GOAL, see below). Of the 543 participants, 487 had data on genome-wide DNA methylation in WBCs and relevant covariates and were thus included in the current analysis. 2) The Puerto Rico Genetics of Asthma, Obesity, and Lifestyle (PR-GOAL) Study: Subject recruitment and study protocol for PR-GOAL have been described elsewhere [Citation10,Citation11]. In brief, 678 subjects aged 6 to 14 years were recruited in the metropolitan area of San Juan and Caguas (PR) from March 2009 to June 2010, using a multistage probabilistic sampling approach. Of the 562 participants who had sufficient DNA for genome-wide genotyping, 114 also had a genome-wide study of methylation using DNA from WBCs and were thus included in this analysis.
In both PR-GOAL and EVA-PR, all participants had to have four Puerto Rican grandparents. In both studies, cases were children who had physician-diagnosed asthma and at least one episode of wheeze in the previous year, and control subjects were children who had neither physician-diagnosed asthma nor wheeze in the previous year. For the current genome-wide methylation analysis of asthma, we combined data for the 601 participants in PR-GOAL (n = 114) and EVA-PR (n = 487). Written parental consent and assent were obtained from all participants. The studies were approved by the institutional review boards of the University of Puerto Rico (San Juan, PR) and the University of Pittsburgh (Pittsburgh, PA).
The Genes-environments & Admixture in Latino Americans (GALA II) study is a multicenter case-control study of asthma in 5,147 Latino children and youth [Citation12,Citation13]. Cases and healthy control subjects were recruited from five centres throughout the United States (Chicago, the Bronx, Houston, the San Francisco Bay Area, and Puerto Rico). Cases had physician-diagnosed asthma and reported symptoms (wheeze or shortness of breath) and medication use within the two years prior recruitment. Controls had no reported history of asthma or allergies and no reported symptoms. To be eligible for the study, the parents and all four grandparents of the participants had to have self-identified as Latino. A subset of 535 participants (206 Puerto Ricans, 248 Mexicans, and 81 other Latinos) had available data for genome-wide methylation in DNA from WBCs and relevant covariates and were thus included in the current analysis. The study was approved by the Institutional Review Boards of the University of California at San Francisco and all other participating centres. All children and their parents provided written informed assent and consent, respectively.
Assessment of genome-wide DNA methylation
In both the Puerto Rican cohorts and in GALA II, whole-genome DNA methylation was measured using the HumanMethylation450KBeadChip system (Illumina, San Diego, CA), as previously described [Citation14]. In both studies, preprocessing of methylation data began by removing poor-quality CpG probes (with detection P values greater than 0.01 in at least 10% of the samples). The methylation β-value for each CpG in each sample was calculated as a percentage: β = M/(M + U + α), where M and U represent methylated and unmethylated signal intensities, respectively, and α is an arbitrary offset (usually 100) intended to stabilize β-values where fluorescent intensities are low.
The R package ENmix was used to perform background correction and normalization of data from the Puerto Rican cohorts [Citation15]. Multimodal distributed, cross-reactive, SNP-containing CpG probes and sex chromosomal probes were also removed. We further filtered ‘noise’ CpGs (mean β-value <0.1 or >0.9, or with extreme β-values in >80% samples) to reduce multiple testing [Citation14]. The preprocessing of genome-wide DNA methylation in GALA II has been described elsewhere [Citation15].
After quality control measures were implemented in both the Puerto Rico and GALA II cohorts, 153,772 probes were available for the EWAS of asthma. β-values were transformed to M-values as log2(β/(1-β)), with M-values used in all downstream association analyses. We then applied R package sva to estimate significant surrogate variables, representing unknown latent factors (LFs) that capture heterogeneity in the data [Citation16].
Statistical analysis
The EWAS of asthma was first conducted separately in each of the two main study cohorts (Puerto Rico cohorts and GALA II), using multivariable linear regression with the R package limma. Measured percentages of WBC types (neutrophils, eosinophils, lymphocytes, and monocytes, since the sum of the percentages of these four WBC types determines the percentage of the fifth possible type) were included as covariates in the analysis of the Puerto Rico cohorts. In GALA II, percentages of WBC types were imputed using the method described by Houseman et al [Citation17]. Principal components (PCs) were derived from available genome-wide genotypic data, using EIGENSTRAT [Citation18]. Additional covariates to adjust included age, sex, cohort-specific covariates and estimated latent factors. Next, we performed a meta-analysis to combine summary statistics from the analyses in the Puerto Rico cohorts and in GALA II, by using inverse variance weighting to generate the combined effect estimate and P value for each CpG site; the Benjamini-Hochberg procedure was utilized to adjust for multiple testing.
We then conducted a pathway analysis with Ingenuity Pathway Analysis (IPA), which included the genes containing the top CpG sites associated with asthma in the meta-analysis. In this analysis, a false discovery rate (FDR) approach was used to adjust for multiple testing. Next, differentially methylated regions (DMRs) were analysed using Python tool comb-p [Citation19] to identify genomic regions involved in gene transcriptional regulation. We used a seed p-value of 0.1 and a maximum extendable distance of 500 base pairs (bp).
To assess whether we replicated findings from prior studies, we compared our top signals in the meta-analysis of Latino children to those reported in previously published EWAS of asthma [Citation4,Citation6,Citation7]. Given small sample size, we report a CpG as replicated in our study if it was associated with asthma at one-sided P < 0.05, in the same direction of association as in a previous study.
Results
A total of 599 subjects with asthma (cases, 302 in the Puerto Rico cohorts and 297 in GALA-II) and 537 control subjects (299 in the Puerto Rico cohorts and 238 in GALA-II) were included in the current analysis. The main characteristics of study participants are shown in . In the Puerto Rico cohorts, cases were more likely to be male and atopic, and to have higher total immunoglobulin E (IgE) than control subjects. In GALA II, cases were more likely to be Puerto Rican and to have higher total IgE than control subjects.
Table 1. Characteristics of the study populations included in EWAS of asthma in Hispanic children
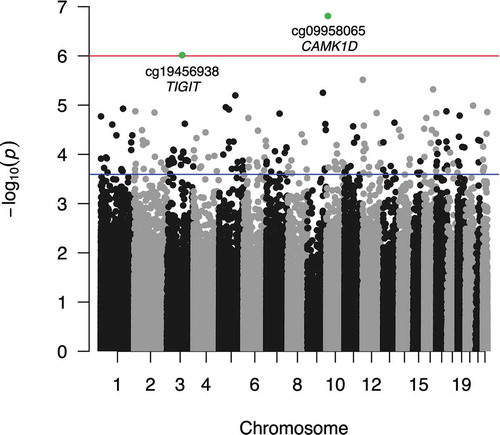
Figure S1 illustrates the schematic procedure of the EWAS of asthma. No probe reached significance at FDR-adjusted P (FDR-P) < 0.1 in the Puerto Rico cohorts, but 787 CpG sites were associated with asthma at FDR-P < 0.1 in GALA II. In the meta-analysis of the two study cohorts, we identified two CpG sites associated with asthma at FDR-P < 0.1 and 193 CpG sites associated with asthma at FDR-P < 0.2 (). Of the 193 CpG sites associated with asthma at FDR-P < 0.2, 190 had the same direction of association in the two study cohorts. Of these 190 CpGs, 73 were located within gene enhancer regions and 27 were located in genomic regions related to transcriptional activity, identified as DNase I hypersensitive sites. The top 20 CpG sites associated with asthma in the meta-analysis of the two study cohorts are listed in .
Table 2. Top 20 CpG sites associated with asthma in a meta-analysis of 1,136 Latino children
Figure 1. Manhattan plot of meta-EWAS of asthma in Latino children Manhattan plot shows the two CpG sites (green dots) associated with asthma at FDR-adjusted P < 0.1 (red line) and 193 CpG sites associated with asthma at FDR-adjusted P < 0.2 (blue line)
Pathway enrichment analysis of genes closest to marginally significant CpG sites (FDR-P < 0.2 in the meta-analysis) revealed 25 pathways enriched at P < 0.1 (), including TGF-β signalling, interferon signalling, and Th1 and Th2 activation pathways.
Table 3. Pathway analysis of genes near top CpG sites associated with asthma in the meta-analysis of data from 1,136 Latino children
lists the 25 DMRs (distance within 500 bp) associated with asthma at Šidák-corrected P < 0.1 in the meta-analysis. The top DMRs included several biological plausible genes, such as mucin 6, oligomeric mucus/gel-forming (MUC6) and a few inflammation-related genes, including cystatin A (CSTA) and the interferon induced transmembrane protein 3 (IFITM3).
Table 4. Differentially methylated regions by asthma among 1,136 Latino children
Previously published EWAS using DNA from whole blood or WBCs have identified differential methylation by asthma status at some genes or genomic regions, as summarized in Table S1 [Citation4,Citation6–8]. Of the five publications listed, two were performed using DNA from peripheral blood monocytes or peripheral blood mononuclear cells and are thus not comparable to findings from the current analysis of WBCs [Citation8,Citation20]. We examined whether findings from the other three EWAS, conducted using DNA from whole blood or WBCs, were replicated in the current meta-analysis, in the same direction of association and at one-sided P < 0.05.
The most recent and largest meta-analysis of childhood asthma by Reese et al. was performed in newborns (8 cohorts, 668 cases, 87% European) and school-aged children (9 cohorts, 631 cases, 69% European) [Citation6]. None of the 9 CpGs shown to be differentially methylated among newborns in that study passed quality control in our study of children and youth. Of the 179 CpGs associated with asthma among school-aged children in the same study [Citation6], 130 were available in our meta-analysis; 42 of these 130 CpGs were associated with asthma at one-sided P < 0.05 with consistent direction of association (Table S2). It should be noted that 193 individuals (106 cases and 87 controls) in GALA II were also included in the study by Reese et al.
A large-scale EWAS in the European MeDALL project by Xu et al. was conducted in whole blood from 207 children with asthma and 610 controls aged 4 to 5 years, and in 185 children with asthma and 546 controls at age 8 years [Citation7]. Of 27 CpGs identified in a discovery analysis, 14 were both replicated and achieved genome-wide significance in a meta-analysis. Although that analysis was not adjusted for WBC types, all 14 CpGs remained significantly associated with asthma in an analysis conducted in purified eosinophils. Of those 14 CpGs, 12 were available in our meta-analysis and 4 were replicated at one-sided P < 0.05 (Table S3).
An early European EWAS (ALSPAC) by Arathimos et al. identified 302 CpG sites associated with current asthma at age 7.5 years, but all associations were attenuated after adjustment for estimated eosinophil and neutrophil cell counts [Citation4]. At age 16.5 years, two CpG sites were associated with current asthma after adjustment for cell counts. Of those two CpGs, one (cg10159529) was available in our study; this CpG was associated with asthma at one-sided P < 0.05 in the same direction of association in our meta-analysis of Latino children.
We compared the findings from our current meta-analysis of EWAS of asthma using DNA from WBCs with those from a recent EWAS of atopic asthma using DNA from nasal epithelial cells, which was conducted in the EVA-PR cohort [Citation9]. Of the top 20 CpG sites listed in were significantly associated with atopic asthma in nasal epithelium at one-sided P < 0.05 (Table S4).
Discussion
A meta-analysis of EWAS in WBCs from Latino children identified 25 DMRs and two CpG sites associated with asthma at FDR-P < 0.10. In this analysis, we replicated findings from previous EWAS of asthma in non-Latino children, which used DNA from whole blood or WBCs.
The only CpG site associated with asthma at FDR-P < 0.05 in the current analysis was near the calcium/calmodulin dependent protein kinase ID gene (CAMK1D), which is located in one of our top 10 DMRs and encodes for a component of the calcium-regulated calmodulin-dependent protein kinase cascade and has been associated with multiple processes, including regulation of granulocyte function and chronic obstructive pulmonary disease [Citation21,Citation22]. Among the top ten genes mapped to the differentially methylated regions, MUC6 encodes for mucin protein, a protein family identified as associated with bronchial hypersecretion [Citation23] and moderate to severe asthma [Citation24]. The proline rich 5 gene (PRR5) has been previously identified as part of a gene set that may influence asthma through ILC3-mediated pathways [Citation25].
Our pathway enrichment analysis identified potential associations between epigenetic regulation in WBCs and biological processes. The most significantly enriched pathway was transforming growth factor-beta (TGF-β) signalling. TGF-β is an important fibrogenic and immunomodulatory factor that has been associated with asthma and may play a role in airway remodelling in asthma [Citation26].
We replicated findings from previous large-scale EWAS in our cohorts of Latino children. Of the available probes, 32.3% were replicated from the study of Reese et al., while 41.7% and 19.4% were replicated from the studies of Xu et al. and Arathimos et al. respectively. This suggests that some methylation changes in WBCs have cosmopolitan effects on asthma across diverse racial or ethnic groups. The effect estimates vary across studies, probably due to differences in sample size, ethnic background, and analytical methods. The probe cg01445399 was significant in the first two published studies above, by Reese et al. and Xu et al., and was further validated in our data. The corresponding gene long intergenic non-protein coding RNA 1140 (LINC01140) may negatively regulate oxidized low-density lipoprotein-induced inflammatory responses [Citation27].
An enrichment analysis for cell type specificity of the top 20 CpG sites was performed with eFORGE [Citation28]. This analysis revealed primary T cells and NK cells as marginally significant (Figure S2), further supporting that these two cell populations are relevant to asthma, along with eosinophils. Indeed, T cell subsets are central to regulation of immune responses, while NK cells have been shown to enhance eosinophilic inflammation and airway hyperresponsiveness [Citation29,Citation30].
Our study has several limitations. First, our sample size is relatively small compared with that of other studies, which could impair statistical power for discovery of associations. Second, our study is cross-sectional, and we cannot thus assess methylation changes over time. Third, WBC percentages were imputed rather than measured in the GALA II study, which may have reduced our statistical power in the meta-analysis.
In summary, we identified methylation markers associated with asthma using DNA from WBCs from Latino children and youth, validated previous findings in non-Latino populations, and showed potential mechanistic pathways for asthma. Functional studies are needed to further validate and better understand the effects of our top methylation markers and potential pathways on asthma.
Supplemental Material
Download MS Word (396.5 KB)Acknowledgments
The authors thank the children, youth, and families who participated in the PR-GOAL, EVA-PR, and GALA II studies. The authors thank all study collaborators in GALA II: Shannon Thyne, UCSF; Harold J. Farber, Texas Children’s Hospital; Denise Serebrisky, Jacobi Medical Centre; Rajesh Kumar, Lurie Children’s Hospital of Chicago; Emerita Brigino-Buenaventura, Kaiser Permanente; Michael A. LeNoir, Bay Area Paediatrics; Kelley Meade, Children’s Hospital, Oakland; William Rodriguez-Cintron, VA Hospital, Puerto Rico; Pedro C. Avila, Northwestern University. The authors thank the health care providers and community clinics who participated and supported GALA II. In particular, the authors thank study coordinator Sandra Salazar; the recruiters who obtained the data: Duanny Alva, MD, Gaby Ayala-Rodriguez, Lisa Caine, Elizabeth Castellanos, Jaime Colon, Denise DeJesus, Blanca Lopez, Brenda Lopez, MD, Louis Martos, Vivian Medina, Juana Olivo, Mario Peralta, Esther Pomares, MD, Jihan Quraishi, Johanna Rodriguez, Shahdad Saeedi, Dean Soto, and Ana Taveras.
Disclosure statement
Dr. Celedon has received research materials from GSK and Merck (inhaled steroids) and Pharmavite (vitamin D and placebo capsules) in order to provide medications free of cost to participants in NIH-funded studies, unrelated to the current work. The other authors report no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Center for Disease Control and Prevention. Asthma. Data, statistics, and surveillance. [ cited 2020 Apr 9].
- Demenais F, Margaritte-Jeannin P, Barnes KC, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet. 2018;50(1):p. 42–53.
- Forno E, Celedon JC. Epigenomics and transcriptomics in the prediction and diagnosis of childhood asthma: are we there yet? Front Pediatr. 2019;7:115.
- Arathimos R, Suderman M, Sharp GC, et al. Epigenome-wide association study of asthma and wheeze in childhood and adolescence. Clin Epigenetics. 2017;9:112.
- Davidson EJ, Yang IV. Role of epigenetics in the development of childhood asthma. Curr Opin Allergy Clin Immunol. 2018;18(2):132–138.
- Reese SE, Xu CJ, Herman T, et al. Epigenome-wide meta-analysis of DNA methylation and childhood asthma. J Allergy Clin Immunol. 2019;143(6):2062–2074.
- Xu CJ, Söderhäll C, Bustamante M, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med. 2018;6(5):379–388.
- Yang IV, Pedersen BS, Liu A, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. 2015;136(1):69–80.
- Forno E, Wang T, Qi C, et al. DNA methylation in nasal epithelium, atopy, and atopic asthma in children: a genome-wide study. Lancet Respir Med. 2019;7(4):336–346.
- Rosas-Salazar C, Ramratnam SK, Brehm JM, et al. Prematurity, atopy, and childhood asthma in Puerto Ricans. J Allergy Clin Immunol. 2014;133(2):357–362.
- Brehm JM, Acosta-Pérez E, Klei L, et al. African ancestry and lung function in Puerto Rican children. J Allergy Clin Immunol. 2012;129(6):1484–90 e6.
- Oh SS, Tcheurekdjian H, Roth LA, et al. Effect of secondhand smoke on asthma control among black and Latino children. J Allergy Clin Immunol. 2012;129(6):1478–83 e7.
- Nishimura KK, Galanter JM, Roth LA, et al. Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188(3):309–318.
- Chen W, Wang T, Pino-Yanes M, et al. An epigenome-wide association study of total serum IgE in hispanic children. J Allergy Clin Immunol. 2017;140(2):571–577.
- Xu Z, Niu L, Li L, et al. ENmix: a novel background correction method for illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44(3):e20.
- Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13(1):86.
- Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909.
- Pedersen BS, Schwartz DA, Yang IV, et al. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–2988.
- Gunawardhana LP, Gibson PG, Simpson JL, et al. Characteristic DNA methylation profiles in peripheral blood monocytes are associated with inflammatory phenotypes of asthma. Epigenetics. 2014;9(9):1302–1316.
- Kourepini E, Paschalidis N, Simoes DC, et al. TIGIT enhances antigen-specific Th2 recall responses and allergic disease. J Immunol. 2016;196(9):3570–3580.
- Yuan YP, Shi YH, Gu WC. Analysis of protein-protein interaction network in chronic obstructive pulmonary disease. Genet Mol Res. 2014;13(4):8862–8869.
- Crespo-Lessmann A, Mateus E, Torrejón M, et al. Asthma with bronchial hypersecretion: expression of mucins and toll-like receptors in sputum and blood. J Asthma Allergy. 2017;10:269.
- Shrine N, Portelli MA, John C, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med. 2019;7(1):20–34.
- Kuo C-HS, Pavlidis S, Loza M, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. 2017;49(2):1602135.
- Duvernelle C, Freund V, Frossard N. Transforming growth factor-beta and its role in asthma. Pulm Pharmacol Ther. 2003;16(4):181–196.
- He L, Zhao X, He L. LINC01140 alleviates the oxidized low-density lipoprotein-induced inflammatory response in macrophages via suppressing miR-23b. Inflammation. 2020;43(1):66–73.
- Breeze CE, Paul DS, van Dongen J, et al. eFORGE: a tool for identifying cell type-specific signal in epigenomic data. Cell Rep. 2016;17(8):2137–2150.
- Gorska MM. Natural killer cells in asthma. Curr Opin Allergy Clin Immunol. 2017;17(1):50–54.
- Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10(12):838–848.