
ABSTRACT
Age-related DNA methylation is a potential mechanism contributing to breast cancer development. Studies of primarily Caucasian women have identified many CpG sites of age-related methylation in non-diseased breast tissue possibly driving cancer development over time. There is a paucity of studies involving Asian women whose ages at breast cancer onset are usually younger than Caucasians. We identified the 181 most consistent age-related methylation events in non-diseased breast tissue across published studies. Age-related methylation events were measured in adjacent normal and breast tumour tissue in an exclusively Asian population at the previously identified age-related methylation sites. Age-related methylation was found in 118 probes in adjacent normal breast tissue. Methylation of 99% of these sites was increased with age and predominantly located on CpG islands in promoter regions. To ascertain biological relevance to breast cancer, we focused on the 37 sites with overall higher methylation in tumour compared to adjacent normal samples. Some sites positively related to age, including AQP5 and CORO6, inversely correlated with gene expression. Several others have known involvement in suppression of carcinogenesis including GPC5 and SST, suggesting that perturbation of epigenetic regulation at these sites due to ageing may contribute to the progression of carcinogenesis. This study highlights an age-related methylation landscape in non-tumour tissue, consistent not just across studies, but also across different populations. We present candidate age-related methylation sites warranting further investigation as potential epigenetic drivers of breast cancer. They may serve as potential targets of site-specific demethylation intervention strategies for the prevention of age-related breast cancer.
KEYWORDS:
Introduction
Breast cancer is the most commonly diagnosed cancer and a leading cause of cancer death in females worldwide [Citation1]. Age is a well-established risk factor for breast cancer suggesting that the biological ageing process is a direct driver of breast cancer aetiology. In the United States (US), the probability of developing an invasive breast cancer increases steadily from 2.3% for women aged between 50 and 59 years to 6.7% for women aged >70 years [Citation2]. However, the biological mechanisms through which age contributes to carcinogenesis have not been fully elucidated.
DNA methylation has been investigated as an epigenetic process driving age as a breast cancer risk factor. DNA methylation is a stable but modifiable epigenetic process where a methyl group is transferred onto the C5 position of a cytosine, forming 5-methylcytosine. Increased methylation at CpG sites in promoter regions is associated with gene inactivation and transcriptional repression. DNA methylation has previously been shown to be associated with breast carcinogenesis [Citation3,Citation4]. DNA methylation is reversible however, suggesting it could be a targetable intervention strategy in cancer prevention.
Methylation changes are hypothesized to occur very early on in the carcinogenic process. According to this hypothesis, a subpopulation of normal cells undergoes methylation changes due to normal ageing which may serve as preferred targets for oncogenic transformation [Citation5]. Those clones that acquire cancer-promoting epigenetic alterations due to ageing may outgrow other clones. Genome-wide studies have been published in the past 5 years reporting increased age-related methylation in normal breast tissue. These studies have identified changes occurring in tissue long before any detectable breast cancer may develop which might be contributing to the carcinogenic process [Citation4,Citation6,Citation7]. Participants in these studies were mostly Caucasians with small numbers of Hispanic [Citation5] and African-American participants [Citation5,Citation6] and no reported Asian participants.
Patterns of genome-wide methylation have been found to differ between African-American (AA) women and European-American (EA) women in both healthy and cancerous breast tissue [Citation8,Citation9]. Hypermethylation of genes related to breast cancer in healthy tissue is associated with previous family history but also with race [Citation8]. In the tumour, hypermethylation of sites on genes coding for certain tumour suppressors, e.g., HIN1, RASSF1A, and RAR-β occur more commonly in AA women compared to EA women and decreased expression of these genes potentially contributes to the disproportionately negative outcomes experienced by AA women with breast cancer (reviewed extensively in [Citation6]). However, there is a paucity of studies involving Asian participants in this regard.
Age-standardized incidence rates of female breast cancer in many Asian countries including Hong Kong are lower compared to Northern America and Europe but it is still the most common cancer among Asian women [Citation1,Citation2]. The lower incidence has been linked to the lower prevalence of known breast cancer risk factors in Asian populations such as obesity and differing screening practices between countries. However, incidence rates have been increasing sharply in recent decades and this increase has been mostly attributed to the adoption of a more westernized lifestyle in many Asian countries [Citation1]. Nevertheless, breast cancer among Asian women seems to have earlier age at breast cancer onset, which may largely be driven by the cohort effect [Citation10], but it is also possible that there is a distinct age-related aetiology in Asian women. Identification of age-related methylation changes in an Asian population may improve our understanding of age disparities as well as uncover important, targetable molecular drivers of breast cancer.
In this study, we identified age-related methylation sites that were consistently reported in normal breast tissue across at least two previously published studies [Citation4,Citation6,Citation7]. We confirm that previously reported age-related methylation in normal breast tissue is also present to a large degree in tumour-adjacent normal tissue and among Asian women. We used these age-related methylation events to identify candidates that warrant further investigation as potential molecular drivers underlying ageing that may be viable targets for intervention.
Materials and methods
Study population
Data and biospecimens were collected from a hospital-based breast cancer case–control study in Hong Kong (HK) as previously described [Citation11]. Briefly, fresh frozen breast tumours and paired normal tissues were collected from incident breast cancer patients (aged 20–84 years) in two HK hospitals diagnosed between 2013 and 2016. Patients with pre-surgery treatment were excluded from the study. Clinical characteristics and risk factors were obtained from medical records and questionnaires. The study protocol was approved by ethics committees of the Joint Chinese University of Hong Kong-New Territories East Cluster, the Kowloon West Cluster, and the National Cancer Institute (NCI). Written informed consent was obtained prior to the surgery for all participants.
Methylation (adjacent normal: n = 97 and tumour: n = 559) and gene expression data from Caucasian women (adjacent normal: n = 111 and tumour: n = 559) were extracted from The Cancer Genome Atlas (TCGA) and were used to compare results.
DNA methylation analysis
Paired tumour and histologically normal breast tissue samples were processed for pathology review at the Biospecimen Core Resource, Nationwide Children’s Hospital, using modified TCGA criteria [Citation12]. Specifically, only tumours with >50% tumour cells and normal tissue with 0% tumour cells were included for DNA/RNA extraction. We subsequently performed principle component analysis and further excluded tumour samples that grouped with normal samples and normal samples that grouped with tumour samples. Dual DNA and RNA Extraction was performed utilizing the AllPrep™ DNA/RNA Mini Kit (Qiagen) for DNA and mirVana™ miRNA Isolation Kit (Applied Biosystems) for total RNA and small RNA. RNA was tested for quality and quantity utilizing the Agilent 2100 Bioanalyzer with the RNA 6000 NanoChip (Agilent). DNA was quantified with the Quant-iT™ PicoGreen™ dsDNA Assay Kit (ThermoFisher). DNA molecular weight was evaluated by E-Gel™ 48 Agarose Gels, 1% (ThermoFisher). Genome-wide methylation profiling was analysed on tumour and paired histologically normal breast tissue from 139 patients using the Infinium MethylationEPIC BeadChip Kit (Illumina) at the Cancer Genomics Research Laboratory, NCI. Quality control (QC) was performed using the basic intensity R package minfi. Raw methylated and unmethylated intensities were background corrected and dye-bias-equalized to correct for technical variation in signal between arrays. Probes located on chrX and chrY, probes including common SNPs, probes annotated in repetitive genomic regions (repeatmask hg19 database) and probes with missing rate >5% were removed. Probes with detection p-value >0.01 were labelled as missing. No probes overlapped with GWAS loci. Test samples with >4% missing genotyping data and QC samples were removed. A functional normalization algorithm was employed for normalization [Citation13]. Functional genomic locations, gene names, and chromosomal location of CpG probes were determined using the Illumina annotation file.
Gene expression
RNA sequencing data were generated in 139 tumours and 92 histologically normal breast tissue samples that passed all quality control metrics at Macrogen Corporation on Illumina HiSeq4000 using TruSeq stranded RNA kit with Ribo-Zero for rRNA depletion and 100-bp paired-end method. Gene expression was quantified as TPM (transcript per million) using RSEM [Citation14] and log2TPM was used for statistical analyses. PAM50 subtype was defined by an absolute intrinsic subtyping (AIMS) method [Citation15]. The Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data (ESTIMATE) tool was employed to predict tumour purity [Citation16].
Statistical analysis
Linear regression models were used to assess associations between methylation beta values and age. Age was used as a continuous variable with the adjustment for sample composition as a continuous variable. Sample composition is a measurement of percentage fat per sample in adjacent normal tissue and tumour purity (ESTIMATE and tumour purity data based on RNA sequencing data) for tumour tissue. Further adjustment of BMI and parity did not significantly change the results and we, therefore, did not include them in the final model since risk factor data were not available for all patients. A false discovery rate (FDR) (q < 0.05) was applied to account for multiple testing.
Spearman’s correlation was used to assess the correlation of methylation (beta value) with an expression of the corresponding gene. Samples were only included in the correlation analysis if both methylation and gene expression data were available for the same patients (HK cohort-adjacent normal n = 55, tumour n = 54; TCGA-adjacent normal n = 83, tumour n = 71) and sample composition was adjusted for. Wilcoxon signed-rank tests were used to compare expression levels of genes in adjacent normal and tumour tissue. ANOVA with post-hoc Bonferroni correction was used to compare methylation and gene expression by molecular subtype. Disease-free survival for CORO6 was determined using BreastMark (http://glados.ucd.ie/BreastMark/) [Citation17].
Linear regression models were also used to assess genome-wide associations between methylation beta values and age. Post-hoc Bonferroni correction (p-value = (0.05/486910 = 0.0000001026)) was applied to account for multiple testing. All statistical tests were two-sided unless otherwise stated. Stata SE 15 was used for all analyses.
Ingenuity pathway analysis (IPA)
The gene list associated with age-related methylation probes was uploaded to IPA (Ingenuity Systems, www.ingenuity.com) Qiagen, 2019. Based on this list, IPA computed a score [score = -log10(p-value)] as a measure of the probability of finding these genes in a list of biological functions and diseases stored in the IPA database.
Results
Demographic and clinical features of study participants are outlined in . All participants were of Chinese ethnicity and had lived in HK for 5 or more years before breast cancer diagnoses. The mean age, BMI, and number of children in this patient cohort were 58 years, 24.8 (kg/m2), and 2, respectively. The number of patients with methylation and gene expression data is shown in ) (HK) and ) (TCGA).
Table 1. Demographic and clinical features of study participants
Figure 1. Number of samples analysed from Asian women in the Hong Kong study (HK) and Caucasian women The Cancer Genome Atlas (TCGA) with available methylation and gene expression data
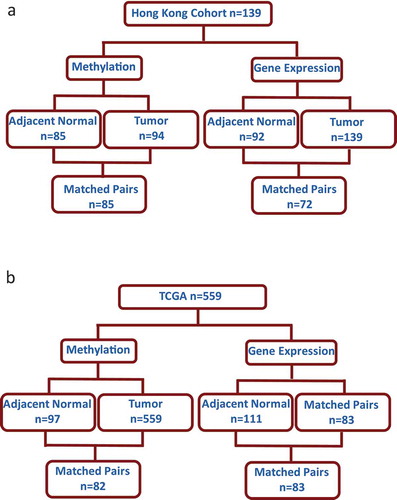
Consistent age-related methylation events observed in HK women with breast cancer
Comparison of age-related methylation in normal breast tissue reported across three previously published genome-wide studies identified 181 probes that consistently had age-related methylation changes in at least two of the studies [Citation4,Citation6,Citation7] ()). The full list of the 181 probes can be found in Supplementary Table 1. Data were available in the HK dataset for 165 of these probes. After adjusting for percentage fat in normal tissue samples, 118/165 (72%) of previously identified age-related methylation changes were also significant (FDR q < 0.05) in adjacent normal tissue of HK patients (n = 84) (Supplementary Table 1). Among the 118 probes, 116 showed a positive relationship to age, while only two probes had decreased methylation (Supplementary Table 2). A predominantly positive relationship between methylation and age is consistent with previously published effect size and direction of association for Caucasian women (Supplementary Table 1) [Citation4,Citation6,Citation7]. The positive association between age and methylation was less significant in tumour tissue (n = 94), with just 18/118 probes showing significant positive relation to age ()) and the results did not vary significantly by ER status (Supplementary Table 3). Similar results were seen in TCGA despite the cohort being from a very different demographic (100% white) (demographic and clinical features of TCGA tumour samples are provided in Supplementary Table 4). Data for just 106/118 of the probes were available in TCGA but 105/106 of those probes had significant positive relationships to age in adjacent normal tissue and only one probe had decreased age-related methylation (n = 82, matched adjacent normal and tumour) ()).
Figure 2. Consistent age-related methylation events across different studies in non-diseased breast tissue and in the adjacent normal tissue of breast cancer patients from Hong Kong (HK) and The Cancer Genome Atlas (TCGA). (a) Venn diagram comparing the number of age-related methylation events in three previously published genome-wide studies. (b) Box plot of beta coefficient values for both adjacent normal and tumour tissue for the 118 probes with increased age-related methylation identified in the HK study (n = 84). (c) Box plot of beta coefficient values for both adjacent normal and tumour tissue for the 105 probes with increased age-related methylation in TCGA (n = 82) (d) Pie chart with the location (left) and functional genomic location (right) of statistically significant age-related methylation probes from the HK study. (e) Ingenuity Pathway Analysis of 112 genes corresponding to probes with increased age-related methylation sites in the HK cohort
Most age-related methylation sites were in CpG islands (72%) with probes also located in N_Shore (13%), S_Shore (6%), OpenSea (5%), and S_Shelf (1%) ()). The age-related methylation was enriched in core promoter regions [transcription start site (TSS200) (42%) and TSS1500 (30%)] ()). IPA was used to identify the top diseases associated with the CpG sites that showed age-related methylation in the HK cohort. Genes associated with the 118 significant age-related probes were uploaded to IPA of which 112 gene names were recognized. The top associated disease with this list of genes was cancer with 105/112 genes previously linked to cancer in the IPA network and the top molecular function was cell to cell signalling and interaction ()).
Potential biological relevance of the age-related methylation events to breast cancer
To identify some key targets from this list of age-related methylation sites which may have potential biological relevance to breast cancer, we focused on probes with higher overall methylation, i.e. higher average beta value across all subjects, in tumour compared to normal tissue (37 probes – see Supplementary Table 1) and restricted even further only to probes located in the promoter region of the gene which we defined here to be TSS200 or TSS1500. This resulted in a list of 20 probes ().
Table 2. Age-related methylation probes with higher overall methylation in tumor compared to normal restricted to promoter region
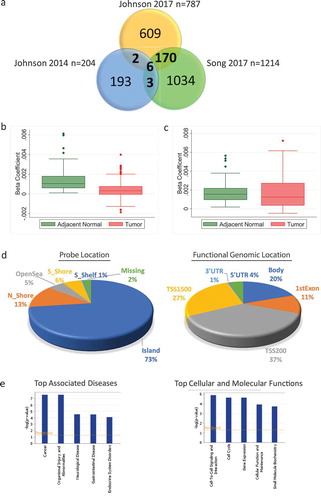
Potential functional relevance was ascertained by examining the correlation of the increased age-related methylation of these probes with an expression of the corresponding gene. Five probes with higher overall methylation in tumour compared to adjacent normal and located in the promoter region had an inverse correlation with gene expression in adjacent normal tissue (probes associated with genes, AQP5, CPNE4, CORO6, KCNC3, and RAB4A) (Supplementary Table 5). Methylation of probes associated with AQP5, CORO6 and RAB4A also inversely correlated with gene expression in tumour tissue (Supplementary Table 6). AQP5 is the only candidate from this list where age-related methylation of the probe co-occurred with age-related decreased expression of the gene in both adjacent normal and tumour tissue. This was fully confirmed in TCGA data ( and Supplementary Figure 1). The probe associated with AQP5, cg03020208, had increased age-related methylation and decreased gene expression in adjacent normal in both HK and TCGA cohorts (), 3(b)). Overall methylation of this probe was higher in tumour (), while the overall expression of AQP5 was lower in the tumour compared to normal tissue in both cohorts ()). Interestingly, the methylation and expression of AQP5 appeared to vary by molecular subtype. Methylation was lower in basal tumours compared to other subtypes although the differences did not reach statistical significance in either dataset ()). Consistently, the highest expression levels of AQP5 were seen in basal tumour samples in both HK and TCGA datasets and this reached statistical significance when basal patients were compared to luminal A patients in TCGA ().
Figure 3. Methylation and expression of AQP5 in tumour and adjacent normal tissue of breast cancer patients in Hong Kong (HK) and The Cancer Genome Atlas (TCGA) datasets. (a) Methylation increases with increasing age for the probe cg03020208 corresponding to the gene AQP5 in adjacent normal tissue in HK (n = 84) (left) and TCGA (n = 97) (right). (b) Gene expression decreases as age increases for AQP5 in HK (n = 92) (left) and TCGA (n = 83) (right). (c) Overall methylation levels in adjacent normal and tumour tissue for the AQP5 probe cg03020208 in HK (n = 85) (left) and TCGA (n = 82) (right). (d) Overall AQP5 expression levels in adjacent normal and tumour tissue in HK (n = 72) (left) and TCGA (n = 83) (right). (e) Methylation of AQP5 in tumour tissue by molecular subtype in HK (n = 70) (left) and TCGA (n = 63) (right) (f) Expression of AQP5 in tumour tissue by molecular subtype in HK (n = 116) (left) and TCGA (n = 505) (right)
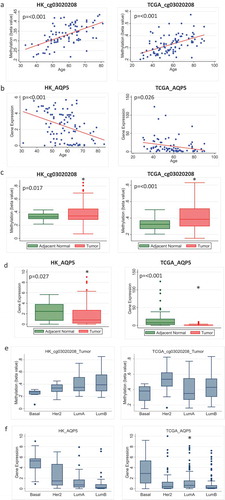
The probe cg00516222, which is associated with the gene CORO6, also showed increased age-related methylation and higher overall methylation in tumour compared to adjacent normal tissue in both HK and TCGA (). Although we report no corresponding age-related decrease in gene expression ()), inverse correlations between methylation and gene expression for this probe and corresponding gene were seen in both adjacent normal and tumour (Supplementary Figure 2). In addition, tumours had a significantly lower expression level of this gene compared to adjacent normal tissue in the HK and TCGA datasets ()). Publicly available survival data show that lower expression of CORO6 was associated with decreased disease-free survival in breast cancer patients (n = 1178, number of events = 625, Hazard ratio = 0.83 (95% CI: 0.71–0.97) ()).
Figure 4. Methylation and expression of CORO6 in tumour and adjacent normal tissue of breast cancer patients in Hong Kong (HK) and The Cancer Genome Atlas (TCGA) datasets. (a) Scatterplot of increasing methylation with increasing age for the probe cg00516222 corresponding to the gene CORO6 in adjacent normal tissue in HK (n = 84) (left) and TCGA (n = 97) (right). (b) Gene expression doesn’t decrease as age increases for CORO6 in HK (n = 92) (left) and TCGA (n = 83) (right). (c) Overall methylation levels in adjacent normal and tumour tissue for the CORO6 probe cg00516222 in HK (left) and TCGA (right). (d) Overall CORO6 expression levels in adjacent normal and tumour tissue in HK (n = 72) (left) and TCGA (n = 83) (right). (e) Overall survival for patients with high versus low expression of CORO6.
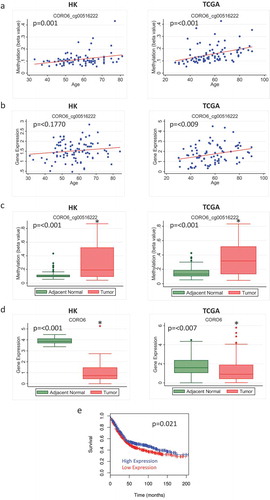
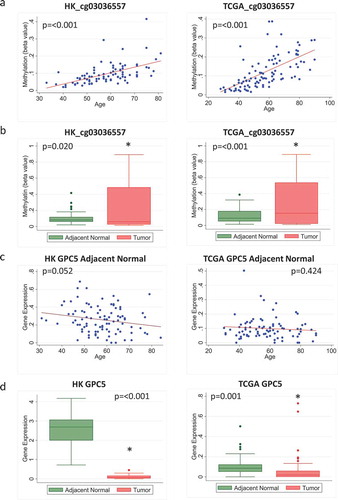
In contrast, some other probes did not show negative correlations between methylation and expression levels of the corresponding genes. For example, the probe cg03036557, which is associated with the gene GPC5, showed increased age-related methylation and higher overall methylation in tumour compared to adjacent normal tissue in both HK and TCGA (). Although the corresponding age-related decrease in gene expression was not significant ()), and there was no negative correlation between methylation and gene expression for this probe in adjacent normal or tumour tissue in HK, there was a significant correlation between methylation and expression in tumour tissue from TCGA (Supplementary Figure 3). Further, the overall expression of this gene was decreased in tumour compared to adjacent normal tissue in both HK and TCGA ()), supporting the link of age-related methylation of this probe with cancer-suppressing activities of this gene.
Figure 5. Methylation and expression of GPC5 in tumour and adjacent normal tissue of breast cancer patients in Hong Kong (HK) and The Cancer Genome Atlas (TCGA) datasets. (a) Scatterplot of increasing methylation with increasing age for the probe cg03036557 corresponding to the gene GPC5 in adjacent normal tissue in HK (n = 84) (left) and TCGA (n = 97) (right). (b) Overall methylation levels in adjacent normal and tumour tissue for the GPC5 probe cg03036557 in HK (left) and TCGA (right). (c) Scatterplot of GPC5 expression as age increases in HK (n = 92) (left) and TCGA (n = 83) (right). (d) Overall GPC5 expression levels in adjacent normal and tumour tissue in HK (n = 72) (left) and TCGA (n = 83) (right)
Age-related methylation events observed only in HK women with breast cancer
Taking advantage of the genome-wide methylation data, we performed an agnostic search for age-related methylation changes in adjacent normal tissue in the HK dataset. After adjusting for multiple comparisons and removing probes that have been previously identified in Caucasian studies and those that overlapped with either the Horvath or breast tissue-specific epigenetic clocks, we identified 154 CpG probes corresponding to 119 unique genes as potential age-related methylation sites that are exclusive in the HK dataset (Supplementary Table 7). Many of these genes play a role in breast cancer. Some have been investigated as potential tumour suppressors, e.g., PDLIM2 in breast [Citation18] and PIK3IP1 in hepatocellular carcinoma [Citation19] or promoters of metastatic processes (ZEB1) [Citation20] with others reported as having dual tumour suppressor and promoter roles depending on context, e.g., RUNX1 [Citation21] and SMAD3 [Citation22,Citation23].
Discussion
Age is well established as a major breast cancer risk factor. Identifying age-related molecular changes associated with breast cancer development could be key to early risk stratification and prevention strategies.
Our study compiles the most consistent age-related methylation changes in non-diseased breast tissue reported across several genome-wide studies. We progressed this work by reporting age-related methylation changes in the adjacent normal breast tissue of an independent cohort consisting of an Asian population which had not been included in previous studies. Our study confirms the most significant age-related methylation events and showed that these findings are generalizable across different population groups. It confirms that age-related methylation occurs before the development of disease or very early in the carcinogenic process and remains present after the development of disease in tissue adjacent to the tumour. We sought to identify which of these age-related methylation changes are playing a role in driving carcinogenesis.
In this study, we report consistent sites of age-related methylation changes in non-cancer breast tissue with the vast majority showing increased methylation with age. These age-related relationships are not reported as frequently in the tumour in our dataset however that is not a big surprise as the age-related effect could be masked by the other signalling events or disruptions that are common in the chaotic cancer environment. To examine the potential biological relevance of these age-related methylation events in driving the cancer process, we focused on probes with higher overall methylation in tumour compared to normal tissue, i.e., probes that were related to ageing and cancer in the same manner. This was motivated by the hypothesis that due to ageing, a subpopulation of normal cells undergo methylation changes which may serve as preferred targets for oncogenic transformation. While tumour-initiating mutations can occur in any cell, those with high levels of methylation are predisposed to cancer formation. The hypothesis follows that most of the de novo methylation seen in cancer actually occurs prior to transformation and is then clonally maintained in the tumour together with the mutation. In addition, we further restricted our analysis to probes located in the promoter region and examining the correlation of methylation with an expression of the corresponding genes. Our final list of 20 genes includes some candidates already linked to cancer-causing processes as well as genes with unclear relevance to cancer. While this hypothesis-driven approach facilitated the generation of a focused list of targets with a strong biological rationale, it has to be noted that the other age-related genes that were not highlighted using this approach may also be important and relate to carcinogenesis through mechanisms that are not straightforward for us to evaluate.
We highlight three genes from the list, AQP5, CORO6, and GPC5 for different reasons. AQP5 is a water channel protein and is the only candidate from our list which showed significant increased age-related methylation in adjacent normal and tumour in both HK and TCGA. Further, we observed a decreased age-related AQP5 expression, suggesting that the age-related methylation had a silencing effect on the expression of the gene. Indeed, the inverse correlation between methylation and gene expression was found in both HK and TCGA datasets. Interestingly, lower methylation and higher expression levels of AQP5 were seen in basal compared to luminal tumours. This could be important as high expression of AQP5 has been associated with poor prognosis [Citation24,Citation25]. Differential expression of this gene by subtype may have important implications in methylation-based interventions involving this gene, in that we may need to think by subtype and not just at breast cancer overall.
CORO6 has previously been identified as a potential tumour suppressor in renal cancer [Citation26]. It was found frequently methylated in primary clear cell renal cell carcinoma and promoter hypermethylation of CORO6 resulted in a significant reduction of its expression level. In our study, methylation increased with increasing age in adjacent normal tissue. Consistently, overall expression of CORO6 was at a much lower level in tumour compared to adjacent normal tissue (although the difference was not statistically significant in TCGA), and there was an inverse correlation between methylation and gene expression in the HK cohort and in the tumour tissue of the TCGA cohort. So, as methylation increases, we see a decrease in gene expression in both adjacent normal and tumour samples, suggesting that the expression of this tumour suppressor gene might be regulated by age-related methylation. In contrast to AQP5, expression of CORO6 in the tumour did not differ by molecular subtype.
The short follow-up time in the HK study prohibited us from assessing the prognostic outcome in relation to methylation/gene expression. We, therefore, looked at disease-free survival data for CORO6 in a publicly available dataset and found that low expression was associated with worse breast cancer overall survival. This suggests that targeting CORO6 with strategies to demethylate its methylation sites may have utility in increasing expression of CORO6 which may help prolong survival.
Inverse correlation between methylation and gene expression for probes located in the promoter region of the gene provides a straightforward and useful biological rationale, yet it may not be the only criteria for a potential target of intervention. Expression of a gene in tumours may be influenced by multiple mechanisms such as tumour purity, copy number alterations, methylation, somatic mutations, or other signalling pathways, and so the correlation between methylation and expression may not always be found. Those relationships that were preserved in cancer tissue, such as AQP5, are of particular interest as potential intervention targets. However, this does not mean that those not seen in cancer tissue are not important since these age-associated signatures may be disrupted during tumour progression.
Several genes from our probe list were located inside or outside the promoter region, which did not show significant correlations with expression levels of the corresponding genes, but they have been suggested as tumour suppressors in other cancer types. Thus, methylation of sites on those genes may still regulate gene expression in tumours and interfere with the tumour suppressing abilities of the proteins. For example, GPC5 was investigated as a metastasis suppressor gene in non-small cell lung cancer [Citation27]. Expression of GPC5 was lower in lung cancer tissues compared with adjacent noncancerous tissues and overall 5-year survival was higher for adenocarcinoma patients that tested positive for GPC5 compared to those tested negative. Although there was no direct correlation between methylation and expression of GPC5 in the HK dataset or age-related decrease in gene expression, the age-related methylation increase was accompanied by higher overall methylation and decreased expression of the gene in tumour compared to normal in both HK and TCGA. Probes corresponding to genes such as GPC5 in our analysis showed age- and cancer-related increased methylation despite the lack of correlation between methylation and expression, suggesting that age-related methylation in these genes might be driving pro-carcinogenesis processes through alternative mechanisms.
Location of DNA methylation is thought to be important to the complex relationship between methylation and gene expression. Methylation does not just occur within the promoter region but is also common in the gene body. While methylation surrounding the TSS and the region of the first exon is strongly linked with transcriptional silencing, methylation further downstream of the promoter region in the gene body does not have this same close association [Citation28,Citation29]. Differential gene-body methylation has been shown to have implications in leukaemogenesis and as a potential drug target in colon cancer suggesting potential clinical value for targeting methylation in this region of the gene [Citation30,Citation31].
Methylation of probes from our list located outside of the promoter region may also have biological relevance. For example, UNC13A has been included in a panel of 21 DNA hypermethylation hotspots in a signature developed to detect breast cancer patients at high risk of recurrence [Citation32]. Somatostatin (SST) is a neuroendocrine peptide, shown to act as a tumour suppressor through suppression of growth factor secretion, controlling cell proliferation and inhibition of cell invasion [Citation33]. Hypermethylation of SST leading to gene silencing has been suggested to be involved in colorectal and oesophageal and gastric tumorigenesis [Citation34–36]. An intervention to demethylate SST has already been demonstrated which triggered up-regulation of SST expression [Citation36].
DNA methylation-based epigenetic age-estimators or ‘Epigenetic clocks’ are sets of CpG probes coupled with a mathematical algorithm to estimate the age of a DNA source such as tissue or cells [Citation37–40], reviewed in [Citation41]. Chronological age has a substantial effect on patterns of methylation in a tissue but biological age increases due to environmental exposures and can affect the methylation landscape [Citation42]. Breast cancer patients have also been shown to exhibit epigenetic age acceleration in their normal breast tissue when compared to non-diseased counterparts so the use of the clocks as measures of biological age may have utility as predictors of breast cancer risk [Citation43]. The Horvath clock was developed using multiple tissue types so may have wide utility as a multi-tissue predictor of biological age [Citation38]. We report very little overlap (10 CpG sites) between the CpG sites used to estimate epigenetic age in the Horvath clock and in our set of age-related methylation sites that are present in at least 3 separate studies including our own Hong Kong dataset (Supplementary Table 8 for list of overlapping CpGs). This is not surprising since it has been shown that DNA methylation age was poorly calibrated in breast tissue [Citation38]. However, we also report very little overlap (3 CpG sites) between our sites and the CpG sites included in a recently developed breast tissue-specific epigenetic clock (Supplementary Table 9 for list of overlapping CpGs) [Citation40]. Our results suggest that in breast cancer specifically, other methylation sites separate to what has been included in existing epigenetic clocks are consistent in the ageing process of breast tissue and may have biological relevance in the progression to breast cancer.
The age-related methylation sites highlighted in this study are potential epigenetic drivers of age as a breast cancer risk factor. Investigating these potential targets using site-specific demethylation intervention strategies could contribute to the prevention of age-related breast cancer. Targeting methylation to date has mostly focused on the removal of DNA epigenetic modifier enzymes and the use of pan DNA methylation inhibitors [Citation44]. Targeted site-specific manipulation of DNA methylation as an intervention strategy in disease is a very recent area of research with few studies. It is recognized that DNA demethylation is technically challenging as there is no single mechanism to remove methylation directly in mammals and off-target effects are a concern (reviewed in [Citation44]). However, it is possible. A recent, promising study reported promoter reactivation of the epigenetically silenced FMR1 gene which causes fragile X syndrome by demethylation of CGG repeats in Fragile X syndrome-induced pluripotent stem cells using a fusion dCas9–TET1CD tool [Citation45]. The induced reactivation was sustainable in a human–mouse chimeric model. This success indicates potential applications for targeted DNA demethylation in disease states.
The strengths of our study include a comprehensive collection of clinical and exposure information and a detailed profiling of both gene expression and methylation in both tumours and paired normal tissue in an Asian population, and the replication of findings in independent datasets. However, there are some limitations to our study. First, we did not have breast tissue from non-diseased people available so we could not directly replicate the work done in other studies in our Asian population. It is recognized that tumour adjacent normal tissue can have methylation changes as a result of nearby tumour influence [Citation46]. However, the concordance between results shows that the age-related methylation landscape is very similar between both normal and adjacent normal tissue regardless of race. Second, our sample size is small; however, we validated all major findings in TCGA. Further, the short follow-up time in the HK study prohibited us from assessing the prognostic outcome in relation to methylation/gene expression.
In conclusion, this study has identified the most consistent age-related methylation events in non-cancer breast tissue across different populations. We have generated a focused list of candidates where methylation changes due to ageing may be driving the development of breast cancer. These candidate genes serve as a launchpad to explore how targeted intervention at certain sites of epigenetic change may be beneficial in determining the role ageing is playing in breast cancer.
Supplemental Material
Download MS Excel (13.5 KB)Supplemental Material
Download MS Power Point (180.1 KB)Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Division of Cancer Epidemiology and Genetics, and Research Grants Council (grant number 474811 to Dr. Tse), Hong Kong SAR.
Supplementary material
Supplemental data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
- Kresovich JK, Xu Z, O’Brien KM, et al. Methylation-based biological age and breast cancer risk. JNCI. 2019;111(10):1051–1058.
- Johnson KC, Koestler D, Cheng C et al. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics. 2014;9(2):268–275.
- Klutstein M, Nejman D, Greenfield R, et al. DNA methylation in cancer and aging. Cancer Res. 2016;76(12):3446–3450.
- Johnson KC, Houseman EA, King JE, et al. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res Treat. 2017;19(1):81.
- Song M-A, Brasky TM, Weng DY, et al. Landscape of genome-wide age-related DNA methylation in breast tissue. Oncotarget. 2017;8(70):114648.
- Dumitrescu R, Marian C, Krishnan SS, et al. Familial and racial determinants of tumour suppressor genes promoter hypermethylation in breast tissues from healthy women. J Cell Mol Med. 2010;14(6b):1468–1475.
- Song M-A, Brasky TM, Marian C, et al. Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics. 2015;10(12):1177–1187.
- Sung H, Rosenberg PS, Chen W-Q, et al. Female breast cancer incidence among Asian and Western populations: more similar than expected. JNCI. 2015;107(7):djv107.
- Li M, Tse LA, Chan W-C, et al. Nighttime eating and breast cancer among Chinese women in Hong Kong. Breast Cancer Res Treat. 2017;19(1):31.
- Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61.
- Fortin J-P, Labbe A, Lemire M, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(11):503.
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323.
- Paquet ER, Hallett MT. Absolute assignment of breast cancer intrinsic molecular subtype. J Natl Cancer Inst. 2014;107(1):dju357.
- Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4(1):2612.
- Madden SF, Clarke C, Gaule P, et al. BreastMark: an integrated approach to mining publicly available transcriptomic datasets relating to breast cancer outcome. Breast Cancer Res Treat. 2013;15(4):R52.
- Deevi RK, Cox OT, O’Connor R. Essential function for PDLIM2 in cell polarization in three-dimensional cultures by feedback regulation of the β1-integrin–RhoA signaling axis. Neoplasia. 2014;16(5):422–431.
- Lee K, Kitagawa M, Liao P, et al. A Ras-LSD1 axis activates PI3K signaling through PIK3IP1 suppression. Oncogenesis. 2020;9(1).
- Kikuchi M, Yamashita K, Waraya M, et al.. Epigenetic regulation of ZEB1-RAB25/ESRP1 axis plays a critical role in phenylbutyrate treatment-resistant breast cancer. Oncotarget. 2016;7(2):1741.
- Chimge N-O, Ahmed-Alnassar S, Frenkel B. Relationship between RUNX1 and AXIN1 in ER-negative versus ER-positive breast cancer. Cell Cycle. 2017;16(4):312–318.
- Rypens C, Marsan M, Van Berckelaer C, et al. Inflammatory breast cancer cells are characterized by abrogated TGFβ1-dependent cell motility and SMAD3 activity. Breast Cancer Res Treat. 2020;180(2):385–395.
- Singha PK, Pandeswara S, Geng H, et al. Increased Smad3 and reduced Smad2 levels mediate the functional switch of TGF-β from growth suppressor to growth and metastasis promoter through TMEPAI/PMEPA1 in triple negative breast cancer. Genes Cancer. 2019;10(5–6):134.
- Lee SJ, Chae YS, Kim JG, et al. AQP5 expression predicts survival in patients with early breast cancer. Ann Surg Oncol. 2014;21(2):375–383.
- Lee SJ, Kang BW, Kim JG, et al. AQP5 variants affect tumoral expression of AQP5 and survival in patients with early breast cancer. Oncology. 2017;92(3):153–160.
- Morris MR, Ricketts CJ, Gentle D, et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30(12):1390. DOI:10.1038/onc.2010.525.
- Yang X, Zhang Z, Qiu M, et al. Glypican-5 is a novel metastasis suppressor gene in non-small cell lung cancer. Cancer Lett. 2013;341(2):265–273.
- Jjingo D, Conley AB, Yi SV, et al. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3(4):462.
- Brenet F, Moh M, Funk P, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PloS One. 2011 Jan
- Blagitko-Dorfs N, Schlosser P, Greve G, et al. Combination treatment of acute myeloid leukemia cells with DNMT and HDAC inhibitors: predominant synergistic gene downregulation associated with gene body demethylation. Leukemia. 2019;33(4):945–956.
- Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236.
- Legendre C, Gooden GC, Johnson K, et al. Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clin Epigenetics. 2015;7(1):100.
- Pyronnet S, Bousquet C, Najib S, et al. Antitumor effects of somatostatin. Mol Cell Endocrinol. 2008;286(1–2):230–237.
- Jin Z, Mori Y, Hamilton JP, et al. Hypermethylation of the somatostatin promoter is a common, early event in human esophageal carcinogenesis. Cancer. 2008;112(1):43–49.
- Mori Y, Cai K, Cheng Y, et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology. 2006;131(3):797–808.
- Jackson K, Soutto M, Peng D, et al. Epigenetic silencing of somatostatin in gastric cancer. Dig Dis Sci. 2011;56(1):125–130.
- Hannum G, Guinney J, Zhao L, et al.. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367.
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):3156.
- Levine ME, Lu AT, Quach A, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573.
- Castle JR, Lin N, Liu J, et al. Estimating breast tissue-specific DNA methylation age using next-generation sequencing data. Clin Epigenetics. 2020;12(1):1–14.
- Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371.
- White AJ, Kresovich JK, Xu Z, et al. Shift work, DNA methylation and epigenetic age. Int J Epidemiol. 2019;48(5):1536–1544.
- Hofstatter EW, Horvath S, Dalela D, et al. Increased epigenetic age in normal breast tissue from luminal breast cancer patients. Clin Epigenetics. 2018;10(1):112.
- Lei Y, Huang Y-H, Goodell MA. DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 2018;19(1):187.
- Liu XS, Wu H, Krzisch M, et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172(5):979–992.
- Yan PS, Venkataramu C, Ibrahim A, et al. Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res. 2006;12(22):6626–6636.