
ABSTRACT
Targeted analysis of DNA methylation patterns based on bisulfite-treated genomic DNA (BT-DNA) is considered as a gold-standard for epigenetic biomarker development. Existing software tools facilitate primer design, primer quality control or visualization of primer localization. However, high-throughput design of primers for BT-DNA amplification is hampered by limits in throughput and functionality of existing tools, requiring users to repeatedly perform specific tasks manually. Consequently, the design of PCR primers for BT-DNA remains a tedious and time-consuming process. To bridge this gap, we developed AmpliconDesign, a webserver providing a scalable and user-friendly platform for the design and analysis of targeted DNA methylation studies based on BT-DNA, e.g. deep amplicon bisulfite sequencing (ampBS-seq) or EpiTYPER MassArray. Core functionality of the web server includes high-throughput primer design and binding site validation based on in silico bisulfite-converted DNA sequences, prediction of fragmentation patterns for EpiTYPER MassArray, an interactive quality control as well as a streamlined analysis workflow for ampBS-seq.
Introduction
DNA methylation is an epigenetic mark that is involved in tissue specification, developmental homoeostasis and the formation of an epigenetic memory [Citation2]. In eukaryotes, 5-methylcytosine (5mC) is mainly found in the context of CpG dinucleotides [Citation3,Citation4].
Aberrant DNA methylation patterns are present in a variety of human cancers, thereby serving as powerful biomarkers for diagnosis, patient stratification and for prediction of treatment response [Citation5–9]. Many methods for the quantification of DNA methylation are based on bisulfite conversion, during which unmethylated cytosine residues are deaminated to uracil while methylated cytosines remain unconverted. Targeted amplicon bisulfite sequencing (ampBS-seq) and EpiTYPER MassARRAY (MassArray) are cost- and time-efficient methods to reliably measure DNA methylation levels across different laboratories which makes these technologies well-suited for implementation into routine clinical diagnostics [Citation10]. Both methods require the design, validation and analysis of dozens to hundreds of PCR amplicons from bisulfite-treated DNA (BT-DNA). This process can be challenging, as DNA loses complementarity and sequence complexity after bisulfite conversion. Several software tools have been developed to facilitate primer design [Citation11–15], quality control [Citation16] and visualization of primer localization [Citation17]. Nevertheless, primer design for targeted DNA methylation analysis remains a time-consuming process, as many of these tools are restricted in throughput and users are forced to manually switch between different software packages.
To overcome these limitations, we have developed AmpliconDesign, an integrative web-server, featuring the design, visualization and quality control of primers for ampBS-seq and MassArray, as well as a user-friendly interactive analysis workflow for ampBS-seq ().
Figure 1. Schematic representation of the AmpliconDesign web-server
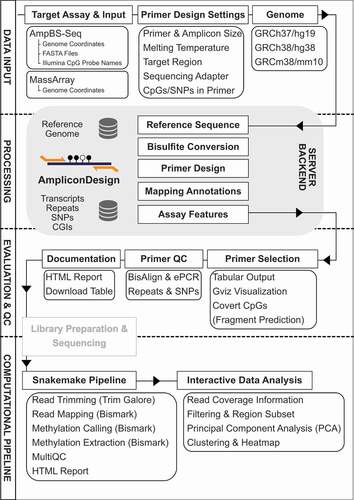
Implementation and methods
The AmpliconDesign graphical user interface has been implemented using the R Shiny [Citation1] framework. The source code is publicly available under the GNU General Public Licence v3.0 (https://github.com/MaxSchoenung/AmpliconDesign).
MassArray
After input of the genomic coordinates, sequences are extracted from common reference genome builds (GRCh38/hg38, GRCh37/hg19, GRCm38/mm10). Single nucleotide polymorphisms (hg38: dbSNP 151; hg19: dbSNP 151; mm10: dbSNP 142), repeats and CpG dinucleotides are annotated. The genomic sequence and the reverse complement are bisulfite-converted in silico. MassArray fragmentation patterns and amplicon prediction plots are calculated using the Bioconductor MassArray package [Citation18]. Genomic features of the selected regions are plotted with the Bioconductor Gviz package [Citation19]. Primers can be designed either manually or automatically using primer3 [Citation20].
Amplicon bisulfite sequencing
As input, genomic coordinates, Illumina CpG identifiers or FASTA files can be used. DNA sequences are extracted from the selected reference genome build and primers are designed using primer3 [Citation20]. By default, CpG dinucleotides are excluded from the primer sequence, but this option can be overruled by the user if needed. This option should only be used by experienced users to facilitate amplification of regions-of-interest with high CpG-content for which primer design fails otherwise. Users need to be aware of the fact that allowing CpGs in primer sequences will likely introduce amplification biases that will affect the DNA methylation measurements. Additionally, common SNPs can be excluded from the primer sequence. Localization of primers is visualized using the Bioconductor Gviz package. Potential off-target primer binding sites and resulting PCR amplicons are determined using Bowtie with default parameters [Citation21].
Analysis pipeline
AmpliconDesign allows high-throughput quality-controlled experimental design and a scalable computational pipeline. The provided Snakemake pipeline includes adapter trimming (Trim Galore!), alignment and methylation calling using Bismark [Citation22]. Bismark coverage files can be uploaded to the AmpliconDesign web-tool together with target regions and sample annotation, to facilitate an interactive exploratory analysis. Users can choose between coverage and quality control plots, read filtering, principal component analysis and a heatmap visualization of target regions. Selected plots and coverage filtered beta-value matrices can be downloaded.
Benchmarking
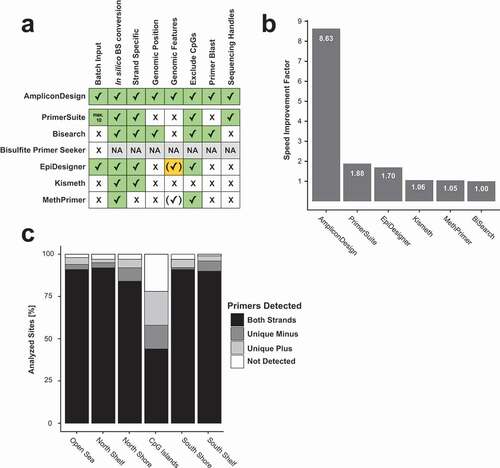
The genomic regions of 16 imprinted regions and 4 control regions [Citation23] were expanded by 100 base pairs (bp) up- and downstream (Supplementary Table 1). For AmpliconDesign genomic coordinates were used as an input whereas for all other tools DNA sequences were retrieved as FASTA files using SeqTailor [Citation24]. Primer pairs for each region were designed by adjusting the default parameters of each web server to an amplicon size of 150 bp to 300 bp, melting temperatures between 50°C and 62°C with and optimum of 55°C and a primer size between 15 bp and 24 bp with an optimum of 20 bp. The designed primer pairs were collected as a table including genomic coordinates and size of each amplicon, primer sequences, melting temperatures and the number of covered CpG sites. The time spent, starting from the input of genomic coordinates until obtaining the table with ready-to-order primer sequences, was documented (Supplementary Table 1).
For benchmarking the primer design efficacy of AmpliconDesign in different genomic contexts, promoter-overlapping CpG sites with different CpG-island (CGI) contexts (i.e. open sea, CGI, CGI shores and CGI shelves) were randomly selected from the probes present on the Infinium MethylationEPIC BeadChip array (Illumina). Primers were designed using the above-mentioned parameters (Supplementary Table 2). The percentage of sites where primers could be detected on one or both DNA strands was calculated and plotted using ggplot2 [Citation25].
Application
Analysis of DNA methylation using ampBS-seq or MassArray is a multi-step process, starting with the extraction of DNA sequences from reference genomes, followed by in silico bisulfite-conversion, primer design, quality control and finally downstream computational analysis. We have compared AmpliconDesign to five previously published web-tools (PrimerSuite, BiSearch, EpiDesigner, Kismeth and MethPrimer) supporting at least one of these steps with respect to usability and throughput (Supplementary Table 1; ). All tools except AmpliconDesign required users to manually extract FASTA sequences from reference genomes and only EpiDesigner and PrimerSuite allowed a batch processing of multiple sequences. Furthermore, only BiSearch aligned the retrieved primer sequences to a reference genome and reported the genomic location of primer binding sites. The time from the input of genomic coordinates until ready-to-order primer sequences were obtained, was benchmarked for all tools by designing primer pairs for 20 imprinted regions [Citation23]. There was a strong increase in speed for tools that feature batch processing (mean: 512.3 sec) in comparison to single FASTA input tools (mean: 1202.0 sec; ). Among all tools, AmpliconDesign allowed the fastest primer design (144 sec) and was 8.6 times faster than the slowest tool BiSearch. To assess the efficacy of AmpliconDesign in identifying primer pairs for CpG-rich regions, we designed primers for 600 promoter CpG sites selected from the probes present on the Infinium MethylationEPIC BeadChip array. Probes were randomly selected stratified for their genomic context (CGI, CGI shore, CGI shelf & open sea; Supplementary Table 2). Overall, AmpliconDesign identified primer pairs for 94% of all queried CpG sites, 98% of non-CGI and 78% of CGI promoters ().
Figure 2. Benchmarking of publicly available BT-DNA primer design web-servers
Hence, AmpliconDesign overcomes limitations of existing tools by providing a reproducible all-in-one workflow for efficient design and analysis of targeted DNA methylation assays:
1.) A complete primer design workflow. AmpliconDesign integrates all steps of the design process and thus provides a turnkey solution from genomic coordinates to ready-to-order primer sequences for targeted DNA methylation analysis. This eliminates the need for manual integration of separate tools, leveraging a significant simplification, speedup, and increase in throughput.
2.) User-friendly input for fast and efficient primer design. Primers can be designed for MassArray and ampBS-seq based on genomic coordinates. Currently, human and mouse reference genomes are supported. For ampBS-seq there are two additional input formats possible: a) Illumina 450 k & EPIC Methylation BeadChip array CpG identifiers and b) FASTA files to enable primer design from non-supported organisms. Thus, AmpliconDesign provides a flexible user interface for data input and overcomes the time-consuming input restrictions of other primer design tools.
3.) Visualization of primer binding sites in the context of genomic regions. The web-tool automatically generates a graphical display of primer binding sites in the context of genomic annotations (repeat regions, CGIs, SNPs and CpG sites). Primers are aligned to the bisulfite-converted reference genome (BisAlign and ePCR mode) to prevent the selection of primers showing multiple binding sites in the genome or having a high potential to show off-target amplification.
4.) Batch input for ampBS-seq primer design. Users can upload a list of up to 250 genomic regions, FASTA files or Illumina 450 k/EPIC CpG identifiers to design primers.
5.) Assay-specific primer design. For MassArray, AmpliconDesign predicts fragments for both T- and C-cleavage reactions to automatically evaluate which CpGs from the regions of interest can be analysed and whether a spectral overlap between fragments is to be expected.
6.) Complete ampBS-seq analysis pipeline. AmpliconDesign offers a Snakemake-based pipeline for processing (adapter trimming, alignment, methylation calling and extraction) of ampBS-seq data which can be installed with a single command on local instances. Bismark coverage files (.cov) can be further processed by an interactive online quality control and analysis pipeline which is available on the AmpliconDesign website.
Conclusion
AmpliconDesign is a fully integrated primer design web-tool for targeted DNA methylation assays. Starting from commonly used data formats, users can design and review primers for ampBS-seq or MassArray in a single step. This includes quality control, visualization of primer binding sites, annotation of genomic regions and primer documentation. AmpliconDesign facilitates time-efficient high-throughput design and analysis of targeted DNA methylation assays.
List of abbreviations
| 5mC = 5-methylcytosine | = | |
| ampBS-seq = amplicon bisulfite sequencing | = | |
| BT-DNA = bisulfite-treated DNA | = | |
| CGI = CpG-island | = | |
| DNA = Deoxyribonucleic acid | = | |
| EPIC array = Illumina Infinium MethylationEPIC array | = | |
| MassArray = EpiTYPER MassARRAY | = | |
| PCR = Polymerase chain reaction | = | |
| SNPs = single nucleotide polymorphisms | = |
Authors’ contributions
MS, JaH, PB, YA, DW, PL and DBL designed the study. MS and JaH have written the AmpliconDesign source code. PB implemented the genome annotation database. JL performed experiments. MS, SS, JL and JoH performed data analysis and data interpretation. PL & DBL jointly coordinated and supervised the study. MS, PL and DBL wrote the first draft of the manuscript. All coauthors contributed to the final version of the manuscript.
Availability of data and materials
The source code of the AmpliconDesign software is publicly available under the GNU General Public License v3.0 (https://github.com/MaxSchoenung/AmpliconDesign).
Supplemental Material
Download MS Excel (157 KB)Supplemental Material
Download MS Excel (42.9 KB)Acknowledgments
We thank all members of the Division of Cancer Epigenomics (DKFZ) for thoughtful discussions related to this study and we especially thank Christoph Plass for supporting this project. We also thank Johannes Beisiegel and the IT Core Facility (DKFZ) for excellent support and technical service related to setting up and hosting the web server.
Disclosure statement
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Chang W, Cheng J, Allaire JJ, et al. shiny: web application framework for R. CRAN R Package. 2018.
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21.
- Jang HS, Shin WJ, Lee JE, et al. CpG and Non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel). 2017;8:148.
- Ramsahoye BH, Biniszkiewicz D, Lyko F, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. 2000;97:5237–5242.
- Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474.
- Lipka DB, Witte T, Toth R, et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat Commun. 2017;8:2126.
- Oakes CC, Seifert M, Assenov Y, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48:253–264.
- Sahm F, Schrimpf D, Stichel D, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18:682–694.
- Sill M, Plass C, Pfister SM, et al. Molecular tumor classification using DNA methylome analysis. Hum Mol Genet. 2020. DOI:https://doi.org/10.1093/hmg/ddaa147
- Bock C, Halbritter F, Carmona FJ, et al. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34:726–737.
- Arányi T, Váradi A, Simon I, et al. The BiSearch web server. BMC Bioinformatics. 2006;7(1):431.
- Gruntman E, Qi Y, Slotkin RK, et al. Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics. 2008;9:371.
- Kovacova V, Janousek B. Bisprimer—A program for the design of primers for bisulfite-based genomic sequencing of both plant and mammalian DNA samples. J Hered. 2012;103:308–312.
- Li L-C, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431.
- Lu J, Johnston A, Berichon P, et al. PrimerSuite: A high-throughput web-based primer design program for multiplex bisulfite PCR. Sci. Rep. 2017;7:41328.
- Pattyn F, Hoebeeck J, Robbrecht P, et al. methBLAST and methPrimerDB: web-tools for PCR based methylation analysis. BMC Bioinformatics. 2006;7:496.
- Lefever S, Hoebeeck J, Pattyn F, et al. methGraph: a genome visualization tool for PCR-based methylation assays. Epigenetics. 2010;5:159–163.
- Thompson RF, Suzuki M, Lau KW, et al. A pipeline for the quantitative analysis of CG dinucleotide methylation using mass spectrometry. Bioinformatics. 2009;25:2164–2170.
- Hahne F, Ivanek R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol Biol. 2016;1418:335-51.
- Untergasser A, Cutcutache I, Koressaar T, et al. Primer3—new capabilities and interfaces. Nucleic Acids Research. 2012;40:e115–e115.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572.
- Klobučar T, Kreibich E, Krueger F, et al. IMPLICON: an ultra-deep sequencing method to uncover DNA methylation at imprinted regions. Nucleic Acids Res. 2020;48:e92-e92.
- Zhang P, Boisson B, Stenson PD, et al. SeqTailor: a user-friendly webserver for the extraction of DNA or protein sequences from next-generation sequencing data. Nucleic Acids Res. 2019;47:W623–W631.
- Wickham H. ggplot2. New York, NY: Springer New York; 2009.