
ABSTRACT
Early developmental environment can influence long-term health through reprogramming of the epigenome. Human environmental epigenetics studies rely on surrogate tissues, such as blood, to assess the effects of environment on disease-relevant but inaccessible target tissues. However, the extent to which environment-induced epigenetic changes are conserved between these tissues is unclear. A better understanding of this conservation is imperative for effective design and interpretation of human environmental epigenetics studies. The Toxicant Exposures and Responses by Genomic and Epigenomic Regulators of Transcription (TaRGET II) consortium was established by the National Institute of Environmental Health Sciences to address the utility of surrogate tissues as proxies for toxicant-induced epigenetic changes in target tissues. We and others have recently reported that perinatal exposure to lead (Pb) is associated with adverse metabolic outcomes. Here, we investigated the sex-specific effects of perinatal exposure to a human environmentally relevant level of Pb on DNA methylation in paired liver and blood samples from adult mice using enhanced reduced-representation bisulphite sequencing. Although Pb exposure ceased at 3 weeks of age, we observed thousands of sex-specific differentially methylated cytosines in the blood and liver of Pb-exposed animals at 5 months of age, including 44 genomically imprinted loci. We observed significant tissue overlap in the genes mapping to differentially methylated cytosines. A small but significant subset of Pb-altered genes exhibit basal sex differences in gene expression in the mouse liver. Collectively, these data identify potential molecular targets for Pb-induced metabolic diseases, and inform the design of more robust human environmental epigenomics studies.
Introduction
It is increasingly evident that environmental exposures and nutritional perturbations during critical windows of development can influence the long-term risk of disease. One important mechanism by which early-life environment can reprogram disease risk is via modification of the epigenome. Normal human development is governed by precise spatio-temporal regulation of gene expression, and this is driven in large part by epigenetic mechanisms. Methylation of DNA at the 5th-position of cytosines (5-methylcytosine, 5mC) occurring in cytosine-phosphate-guanine (CpG) dinucleotides is critical for establishment of tissue-specific gene expression patterns, regulation of imprinted genes, maintenance of genome stability, and silencing of transposable elements[Citation1]. 5mC undergoes distinct waves of reprogramming during early embryonic development and in primordial germ cells, and is stable and mitotically heritable [Citation2]. Epigenetic reprogramming during these periods of rapid growth and differentiation can be altered by environmental or nutritional perturbation. 5mC can undergo further oxidation to 5-hydroxymethylcytosine (5hmC) [Citation3]. Although 5hmC is an intermediate in the DNA demethylation pathway, recent studies suggest that the genomic distribution, downstream targets, and functional roles of 5hmC are distinct from that of 5mC [Citation3]. Environmental exposures may also lead to reprogramming of 5hmC, and these changes are associated with altered transcription and adverse health outcomes [Citation4–6].
Exposure to heavy metals, including lead (Pb), remains a significant public health concern, particularly in poor urban communities due to various sources of contamination (e.g., leaded paint and pipes, mining, lead-glazed ceramics). Although the neurological effects of early-life Pb exposure have been well-documented, substantially less is known about how Pb exposure impacts the liver and other metabolic organs. Notably, recent human studies suggest that blood Pb levels are associated with liver disease, metabolic syndrome, and diabetes [Citation7–10]. Likewise, maternal blood Pb levels are associated with increased DNA methylation of the imprinted MEG3 locus in offspring blood, concomitant with rapid gains in body fat [Citation11]. In animal models, we and others have recently reported that perinatal [Citation12–15] and post-natal [Citation16–18] exposure to Pb leads to sex-specific changes in body weight, metabolic function, and gut microbiota. Post-natal Pb exposure leads to hypermethylation of genes associated with glucose and lipid metabolism in the rat liver [Citation16]. However, the effects of Pb exposure during gestation and lactation on hepatic DNA methylation, and the implications this may have for long-term disease risk, are unclear.
In human population-based environmental epigenetics studies, easily obtainable sources of DNA (e.g. hair, blood, stool, saliva) are used as surrogates for the tissue(s) targeted by the environmental exposure. It is unknown, however, to what extent exposure-induced epigenetic changes in surrogate tissues reflect those occurring in target tissues, and whether this overlap differs by sex. Likewise, whether toxicant-induced epigenomic changes persist in both target and surrogate tissues over time within individuals is unclear. To address these important questions, the National Institute of Environmental Health Sciences (NIEHS) established the TaRGET II consortium [Citation19]. Using mouse models of human-relevant environmental exposures, the objective of TaRGET II is to assess the conservation of environment-induced epigenomic perturbations between easily obtainable surrogate and disease-relevant, but inaccessible, target tissues. As part of this effort, we used an established mouse model of perinatal Pb exposure to determine the effects of Pb on genome-wide DNA methylation in the liver and blood of 5-month old, adult animals. We hypothesized that perinatal Pb exposure would lead to sex-specific alterations in DNA methylation in liver at genes important for metabolic homoeostasis. We further hypothesized that a subset of differentially methylated sites would overlap between liver and blood.
Materials and methods
Animal exposure paradigm
Mice utilized for breeding and exposure were obtained from a colony maintained for over 230 generations with the Avy allele passed through the male line, resulting in forced heterozygosity on a genetically invariant background with 93% identity to C57BL/6 J [Citation20,Citation21]. Virgin a/a females (6–8 wks old) were mated with virgin a/a males (7–9 wks old), and randomly assigned to control or Pb exposure via drinking water. For Pb exposure, Pb-acetate was mixed with water to result in a Pb concentration of 32 ppm in drinking water, to model human-relevant maternal exposure in the 16–60 μg/dL range [Citation12]. Exposure water was made by dissolving Pb (II) acetate trihydrate (Sigma‐Aldrich) in a single batch of distilled water, and Pb concentrations were verified using inductively coupled plasma mass spectrometry with a limit of detection of 1.0 µg/L (ICPMS; NSF International). Animals were maintained on a phytoestrogen‐free modified AIN‐93 G diet (TD.95092, 7% Corn Oil Diet, Harlan Teklad). The a/a dams in the Pb group were exposed to Pb‐supplemented drinking water for 2 weeks prior to mating a/a males. Exposure was continued during gestation and lactation. After weaning, the resulting pups were weighed and switched to Pb‐free drinking water (). A subset of pups from each exposure group, representing approximately 1–2 male and 1–2 female offspring per litter, was maintained to 5 months of age (n = 6 males and 6 females per treatment). All animals had access to food and drinking water ad libitum throughout the experiment while housed in polycarbonate‐free cages. The study protocol was approved by the University of Michigan Institutional Animal Care and Use Committee (IACUC).
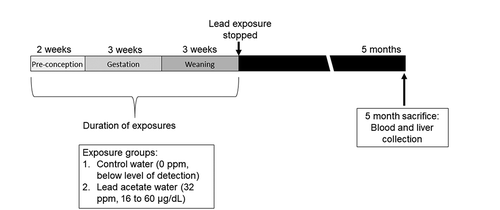
Figure 1. Mouse treatment paradigm showing timing of Pb exposure and animal sacrifice/tissue collection. Dams were exposed to normal chow and either control or Pb‐acetate water, starting two weeks prior to mating, through gestation, until offspring were weaned on postnatal day 21. At this point, Pb exposure ceased and offspring were administered control water and chow. At 5 months of age, matched liver and blood tissues were collected for DNA methylation analysis. Pb doses and average blood lead levels are shown
Body composition and tissue collection
Body weights were measured for each mouse on a weekly basis (Mettler Toledo). Total body fat and lean mass were measured via EchoMRI (Houston, TX USA). Body fat percentage and lean mass percentage were calculated as a percent of total body weight. Upon euthanasia at 5 months of age, mesenteric fat, blood, and liver samples were collected in accordance with protocols established by the TaRGET II Consortium ( and [Citation19]). Briefly, mice were fasted for 6 h prior to euthanasia. Euthanasia was carried out via CO2 asphyxiation and bilateral pneumothorax. Blood was collected by cardiac puncture, followed by whole body perfusion with cell culture grade 0.9% saline solution (Sigma Life Sciences). Next, liver and mesenteric fat were collected and weighed. Mesenteric fat was collected by dissecting white adipose tissue away from the stomach, spleen, pancreas, and intestines. Relative liver and mesenteric fat weights were expressed as a percent of total body weight. Liver was separated into individual lobes, and the left lobe was used for the analyses in this study. Liver and fat samples were immediately snap-frozen in liquid nitrogen and stored at −80° C. Blood was collected with EDTA anticoagulant, centrifuged, and plasma was removed. Red blood cells were lysed using Erythrocyte Lysis Solution (Qiagen, 79,217). The remaining white blood cell fraction was washed with PBS, pelleted by centrifugation, and resuspended in Buffer RLT (Qiagen, cat # 1,053,393). Crude cell lysate was stored at −80 C until DNA extraction.
DNA extraction and enhanced reduced representation bisulphite sequencing
The left lobe of the liver was cryo-pulverized and suspended in Buffer RLT (Qiagen, cat # 1,053,393). Blood and liver DNA extraction were performed using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen #80,224). Enhanced reduced representation bisulphite (ERRBS) was performed at the University of Michigan Epigenomics and Advanced Genomics Cores as described previously [Citation22,Citation23]. Briefly, 50 ng of genomic DNA was digested using MspI, a restriction enzyme that preferentially cuts CG-rich sites. The digested DNA was then purified using phenol:chloroform extraction and ethanol precipitation in the presence of glycogen, before blunt-ending and phosphorylation. A single adenine nucleotide was next added to the 3′ end of the fragments in preparation for the ligation of the adapter duplex with a thymine overhang. The ligated fragments were cleaned, then processed for size selection on agarose gel. Selected fragments were treated with sodium bisulphite to convert unmethylated cytosines to uracils, which are then replaced with thymines during PCR amplification. Both methylated and hydroxymethylated CpGs are protected from bisulphite conversion. Therefore, ERRBS detects a combination of 5mC and 5hmC. Final libraries were next cleaned up with AMPure XP beads (Product #A63880; Beckman Coulter), quantified using the Agilent TapeStation genomic DNA kit (Catalogue #G2991AA; Agilent) and Qubit High Sensitivity dsDNA (Catalogue #Q32850; Invitrogen). Single-end, 50 bp reads were obtained for each library by sequencing on the HiSeq 4000 system (Illumina). ERRBS samples were multiplexed, with three samples per sequencing lane.
Bioinformatics pipeline, quality control, and differential methylation analysis
Snakemake [Citation24] was used to manage the bioinformatics workflow, and Anaconda (Anaconda Software Distribution) was used to manage software dependencies and create reproducible software environments. For quality control, FastQC (v0.11.3) was employed to assess the overall quality of each sequenced sample and identify specific reads and regions for trimming. TrimGalore (v0.4.5) was used to trim low-quality bases (quality score lower than 20), adapter sequences (required overlap of 6bp), and end-repair bases from the 3ʹ end of reads. Reads shorter than 20bp after trimming were discarded. For alignment and methylation calling we used Bismark [Citation25] (v0.19.0), an integrated alignment and methylation call program that performs unbiased alignment (by converting residual cytosines to thymines prior to alignment in both reads and reference). Within Bismark, reads were aligned to the reference genome (mm10) using Bowtie2 [Citation26] (v2.3.4) with default parameter settings (multi-seed length of 20bp with 0 mismatches). Methylation calls were reported for all nucleotides with a read depth of at least 5. We used the methylSig R package [Citation27] (v0.5.0) to identify differentially methylated CpG positions and/or regions. Differential methylation for each comparison was tested using methylSigDSS(), which tests for differential methylation under general experimental design using a beta-binomial approach with the ‘arcsine’ link function [Citation28]. To control for batch effects, run was included as a covariate in the model. After obtaining p-values, we adjusted for multiple testing using the FDR approach. The results of the differential methylation tests were then annotated using the annotatr R Bioconductor package [Citation29] (v1.5.9). The annotate_regions function was used to generate genomic annotations, which include CpG annotations [CpG islands (CGI), shores, shelves, open sea (InterCGI)], genic annotations (exon, intron, promoter, 5ʹUTR, 3ʹUTR), and gene IDs. CpGs with read coverage > 1000 were removed because they were likely the result of PCR amplification, and CpGs with read coverage < 10 were removed due to decreased power to detect differential methylation. Opposite strand CpGs at the same position were combined via destranding. We performed differential methylation testing on individual CpG sites (DMCs), requiring sufficient sequencing coverage for a minimum of 4 samples from the Pb group, and 4 samples from the control group for a site to be tested. Differentially methylated regions (DMRs) were identified in 50 bp tiles using the same requirements. For both DMCs and DMRs, sites with FDR <0.05 and an absolute difference in methylation of > 10% were considered significant.
Identification of DMRs within sex-specific mouse liver enhancers
ChromHMM tracks from a 2013 study by Sugathan et al [Citation30]. were kindly provided by Dr. David Waxman. Bedtools version 2.28.0 was used to identify the intersections between male or female DMRs and their respective sex-specific mouse liver enhancer elements. Specifically, intersections were generated between DMRs and 5 chromatin states that were identified as enhancers in this study based on a combination of DNAse hypersensitivity and the presence of enhancer-specific chromatin marks (states 5, 6, 9, 10, 11) [Citation30].
Pathway analysis of DMRs
Poly-Enrich [Citation31] was used to assess the biological pathways enriched among the DMRs. Separate pathway analyses were conducted for each sex, tissue, and direction of differential methylation, and were restricted to proximal promoter regions (within 1 kb of transcription start sites). The following Gene Ontology gene sets used for this analysis: Biological Process, Cellular Component, and Molecular Function. Pathways with FDR <0.05 were considered statistically significant.
Analysis of overlap in differentially methylated sites between blood and liver
Annotated lists of DMCs and DMRs for males and females were compared between blood and liver to identify chromosomal locations that directly overlapped between the two tissues. Overlapping sites with changes in methylation in the same direction in both tissues were considered biologically relevant. In a further analysis, the full lists of DMR-associated genes in blood were compared to those in liver to identify a list of genes in common between the two tissues. Separate lists were generated for males and females. These gene lists were analysed using DAVID version 6.8, Laboratory of Human Retrovirology and Immunoinformatics (LHRI),
(https://david.ncifcrf.gov) to identify pathways that were significantly enriched (FDR <0.05).
qRT-PCR
Quantitative real time reverse transcription PCR (qRT-PCR) was carried out using RNA samples from male and female livers. RNA was converted to cDNA using iScript cDNA Synthesis Kits (Bio-Rad cat# 1,708,891) following manufacturers’ instructions. Quantitative PCR reactions were set up for Gnas and 3 housekeeping genes (Actb, Hmbs, Psmc4) using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) via the manufacturer’s instructions. Gnas primer sequences are shown in Supplemental Table S1. Primers for Actb, Hmbs, and Psmc4 were PrimePCR primers purchased from Bio-Rad. Samples were run in triplicate, and controls included no template and genomic DNA conditions. Quantitative real-time PCR was conducted using the CFX384 Real-Time PCR Detection System (Bio-Rad). The cycling protocol was as follows: 1) 2 mins at 95 C x 1 cycle, 2) 5 seconds at 95 C and 30 seconds at 60 C x 40 cycles, and 3) melt curve with 0.5 C 5 second increments from 65 C – 95 C. Gnas expression was calculated relative to the geometric mean of the Ct values for the housekeeping genes using the delta Ct method.
RNA-seq gene expression
RNA was isolated from the left lobe of PND21 or 5 month mouse liver using the Qiagen AllPrep DNA/RNA/miRNA Universal Kit. Library preparation and sequencing were conducted on postnatal day 21 and 5- month liver and blood at the University of Michigan Advanced Genomics Core. Liver and blood samples from mice that had gross liver tumours (1 female Control and 1 female Pb) were excluded. RNA Integrity Numbers (RIN) were assessed by Agilent 2200 TapeStation analysis. Electropherograms indicated that the RINs were greater than 6 for most liver RNA samples greater than 8 for most blood samples. Library preparation was conducted using the Illumina TruSeq stranded mRNA Library Prep Kit (5- month samples) or the KAPA mRNA Hyper Prep Kit with Dual Indexing Adapters (PND21 samples) according to manufacturer instructions. Quantity and quality of the prepared libraries were confirmed with the Agilent 2200 TapeStation. Sequencing (paired-end 50 base pair reads) was carried out on the Illumina HiSeq 4000 (5 month samples) and the NovaSeq S2 flow cell (PND21). Data quality control and analysis were conducted by the TaRGET II Consortium Data Coordinating Centre at Washington University in St. Louis, MO. Data were normalized using the RUVr method of the RUVseq R package [Citation32]. Genes were first selected with more than 1 cpm in more than 10 RNA-seq samples. The resulting gene signal matrix was divided into adult liver, PND21 liver, adult blood and PND21 blood. RUVr with k = 3 was run separately for these four groups and samples were stratified base on sex and treatment. Additional methods for data processing can be found at https://targetepigenomics.org/documents/. Read count data for individual mice were plotted, and gene expression differences were analysed for statistical significance using linear mixed effects regression with the lme4 package and R version 3.5.1.
Results
Litter parameters and phenotype
Perinatal lead exposure did not significantly alter the offspring sex ratio, litter size, or mortality rates (p = 1.0, 0.9, and 0.2, respectively, Fisher’s Exact Test) (). We have previously reported that perinatal Pb exposure leads to sex-specific changes in food intake, body weight, and body fat later in life (n = 12–18 mice per sex) [Citation13]. In this study, we observed no significant differences in body weight, total body fat, or body fat percentages in either males or females, an observation that may have been due to a smaller sample size (n = 6 mice per sex) (Supplemental Figure 1 A-F) than our previous Pb studies observing significant metabolic effects [Citation12,Citation13]. Nevertheless, consistent with our previous observations, we observed a trend towards increased total and relative mesenteric fat weights in males at 5 months, although this did not reach statistical significance (p = 0.06, Supplemental Figure 1 G-J). Total and relative liver weights did not differ significantly between exposed and control groups in either males or females (Supplemental Figure 1 K-N).
Table 1. Litter parameters
Epigenome-wide differential DNA methylation with Pb exposure
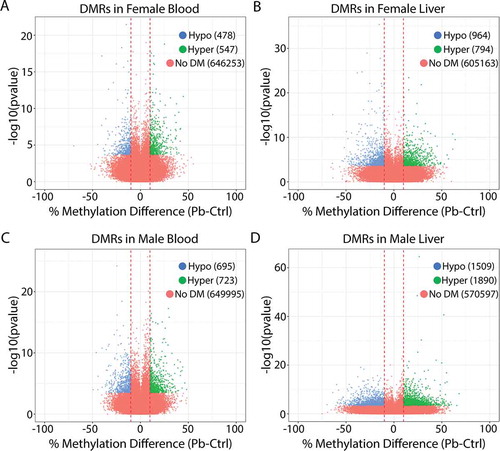
To assess the effects of perinatal Pb exposure on DNA methylation across target and surrogate tissues, ERRBS was conducted on liver and blood samples from 5-month-old Pb and control-exposed offspring. illustrate the total number of regions tested and the number of differentially methylated cytosines (DMCs) and DMRs identified in each exposure group. Although Pb exposure ceased at 3 weeks of age, this analysis revealed thousands of sex-specific DMCs and DMRs in the adult liver and blood ( and Supplemental Tables 2–9). The relative proportion of hypo- and hyper-methylated regions/cytosines was similar for each condition (). Although observed methylation changes were between 10 and 20% at most sites with Pb exposure, numerous sites exhibited changes in methylation that were much greater, up to 70% in females and 74% in males () and Supplemental Figure 2). We annotated the DMCs and DMRs to the mouse mm10 genome to determine the genomic locations of DMCs and DMRs relative to all sites tested, stratified by sex, tissue, and direction of differential DNA methylation. Compared to all regions tested, DMCs/DMRs were significantly depleted from CpG islands, promoters, 5ʹUTRs, and exons, and were significantly enriched for regions greater than 4000 bp from a CpG island (interCGI) and intergenic regions (chi squared test, p <.05) (Supplemental Figure 3 A-H). This pattern was similar across sex and tissue (Supplemental Figure 3 A-H).
Table 2. Differentially methylated cytosines (DMCs) in female and male blood and liver identified using ERRBS
Table 3. Differentially methylated regions (DMRs) in female and male blood and liver identified using ERRBS
Figure 2. Volcano plots showing differentially methylated regions (DMRs) for lead vs. control in female blood (a), female liver (b), male blood (c), and male liver (d). Blue: regions significantly hypomethylated with Pb exposure. Green: regions significantly hypermethylated with Pb exposure
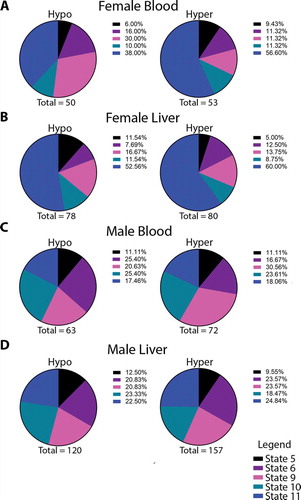
The liver exhibits marked sexual dimorphism, with distinct patterns of chromatin accessibility and enhancer modifications driving sex-biased gene expression [Citation30]. Given the sex specificity of differential methylation, we investigated whether DMRs occurred at sex-specific enhancers in the mouse liver. To this end, we stratified DMRs by sex, tissue, and direction of DNA methylation, and determined the overlap with previously published, sex-specific mouse liver enhancers [Citation30]. Specifically, we determined whether DMRs in females fell within female-specific enhancers, and whether DMRs in males occurred within male-specific enhancers. Five chromatin states were identified as enhancers using ChromHMM, based on a combination of DNAse hypersensitivity and specific chromatin marks. These include the presence of enhancer chromatin marks H3K27 acetylation and/or H3K4 mono-methylation, and a lack of the promoter-enriched H3K4 trimethylation mark. (see and reference [Citation30]). Notably, in both sexes and tissues we observed a subset of DMRs falling within their respective sex-specific enhancer regions. Chromatin states 5 and 6 are characterized by robust DNAse hypersensitivity [Citation30], suggesting an open enhancer state [Citation33]. In contrast, states 9, 10 and 11 exhibit low DNAse hypersensitivity [Citation30], suggesting closed chromatin and an inactive enhancer [Citation33]. In all conditions, the majority of DMRs were found in closed enhancer regions ()). The relative proportions of different enhancer states overlapping DMRs were similar for both liver and blood, but differed substantially between sexes ()). Compared to females, in male blood and liver a greater proportion of DMRs were found in open enhancer regions ()).
Figure 3. Pie charts depicting the overlap between DMRs in each sex and their respective sex-specific enhancer states in female blood (a), female liver (b), male blood (c) and male liver (d). Totals are the number of DMR-enhancer overlaps for each condition. Published enhancers are from Sugathan and Waxman, Mol. Cell. Biol. 2013, and chromatin states are defined according to the degree of enrichment with H3K4me1, H3K27Ac, and DNAse hypersensitivity (DHS): State 5 – low K4me1, medium K27ac, high DHS; State 6 – high K4me1, high K27ac, high DHS; State 9 – high K4me1, high K27ac, low DHS; State 10 – low K4me1, medium K27ac, low DHS; State 11 – high K4me1, low K27ac, low DHS
Pathway analysis of DMRs
Having established that early life Pb exposure leads to sex-specific changes in DNA methylation in adulthood, we next determined the cellular pathways enriched among the DMRs (Supplemental Tables S10-S13). We stratified our analyses by sex, tissue, and direction of differential methylation, and focused on proximal promoter regions (within1 kb of transcription start sites). Pathways with a FDR<0.05 were considered significant. This analysis revealed that the pathways enriched among hypo-methylated sites were distinct from those enriched among hyper-methylated sites. In addition, the enriched pathways differed across sex and tissue. In spite of these differences, overall, we observed that Pb-altered methylation of CpG sites in key organ developmental pathways including mammary gland, bone, nervous system, and kidney. Interestingly, we observed enrichment of pathways involved in regulation of neurotransmitter levels and activity in female and male liver (Supplemental Tables S10-S13).
Perinatal Pb exposure and regulation of imprinted loci
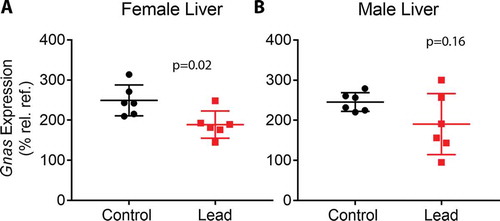
Previous findings from our lab and others demonstrated that developmental exposure to BPA and Pb led to altered CpG methylation at imprinted loci [Citation4,Citation11,Citation34]. Therefore, we investigated whether perinatal Pb exposure disrupted methylation at imprinted loci in liver and blood, and whether these changes overlapped between the two tissues. To do this, we compared the lists of Pb-induced DMCs/DMRs for each condition to a database of mouse-imprinted genes [Citation35]. Of 139 imprinted genes interrogated, 112 were covered in this study using ERRBS (Supplemental Table S14). We observed Pb-induced DMRs/DMCs at 44 imprinted genes combined across tissue and sex. Specifically, we identified 3 in female blood, 18 in female liver, 22 in male blood, and 21 in male liver. The enrichment of imprinted genes among the full set of DMC-associated genes (both tissues and sexes) was statistically significant (p < 0.01, hypergeometric test). See for DMCs mapping to imprinted genes in females and males, respectively, and Supplemental Tables S15-S18 for DMRs. In liver samples, we discovered 5 imprinted genes with DMCs in both males and females, including Commd1, Gnas, Nespas, Pde10a, and Pde4d (). Methylation at CpG sites in Gnas/Nespas was in the same direction in both males and females ()). In blood, CpG sites in Meg3 were hypermethylated in both males and females ()). We next compared DMC-associated imprinted genes between blood and liver within each sex. In females, no imprinted genes overlapped between blood and liver. In males, we observed DMCs at Begain, Cdkn1c, Commd1, Peg12, Rasgrf1, and Snrpn in both blood and liver, and found that CpGs in Begain, Peg12, Rasgrf1, and Snrpn exhibited changes in methylation in the same direction in both blood and liver ( and )). We previously observed that BPA exposure led to changes in methylation and hydroxymethylation at the Gnas locus, concomitant with increased Gnas transcript expression [Citation4]. Therefore, we investigated whether Pb-induced changes in methylation at the Gnas locus were accompanied by changes in Gnas transcript expression using qRT-PCR. We observed a significant reduction in Gnas expression with Pb exposure in female mice, and a trend towards reduced expression in male mice (). Collectively, these data suggest that Pb exposure alters methylation at imprinted loci in a manner that is largely tissue and sex-dependent.
Table 4. Differentially methylated cytosines (DMCs) identified at imprinted loci in female blood and liver
Table 5. Differentially methylated cytosines (DMCs) identified at imprinted loci in male blood and liver
Figure 4. Lollipop diagrams showing differentially methylated cytosines (DMCs) with Pb exposure in: (a) Gnas in male and female liver, (b) Meg3 in male and female blood, (c) Begain in male blood and liver, (d) Peg12 in male blood and liver, (e) Rasgrf1 in male blood and liver, and (f) Snrpn in male blood and liver
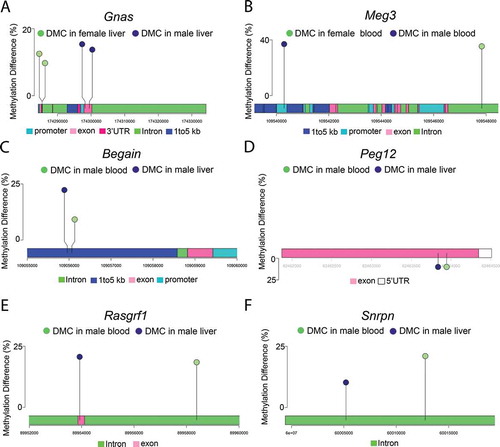
Figure 5. qRT-PCR data depicting Gnas expression in livers from female (a) and male (b) mice. Gnas expression was determined relative to the geometric mean of the Ct values for housekeeping genes Actb, Hmbs, and Psmc4. Since some of the male and female mice were littermates, data were analysed using linear mixed effects regression with litter-specific random effects to account for within-litter correlation
Tissue and sex specificity of DMCs and DMRs
An important objective of the TaRGET II Consortium, and a need for the epigenetic research community at large, is to identify signatures of environmental exposures in surrogate tissues that mirror those in target tissues. To this end, we first compared the chromosomal locations for DMCs and DMRs between liver and blood to determine whether there was direct overlap in differentially methylated sites between the two tissues. In females, 4 DMCs and 3 DMRs overlapped between blood and liver; however, the Pb-induced change in methylation at each of the overlapping sites was in the opposite direction for each tissue (). In males, 4 DMCs and 5 DMRs each overlapped between blood and liver (). In contrast with females, several sites in males exhibited changes in methylation that were in the same direction in both blood and liver (). However, the overlaps of DMCs/DMRs between tissues were not statistically significant, as assessed by hypergeometric test (). Next, we evaluated the overlap in genes with at least one DMC between blood and liver in males and females. We identified 188 DMC-associated genes in females and 325 genes in males that overlapped between blood and liver () and Supplemental Tables S19-S20). In both sexes, this overlap was statistically significant (p < 2.7x10−[Citation36] and p < 1.1x10−[Citation37] for females and males, respectively, hypergeometric test). Gene ontology analysis of the overlapping genes in females revealed enrichment for cell junction, membrane, and synapse pathways. In males, we observed enrichment for cell adhesion, protein binding, synapse, and neuronal process pathways ()).
Table 6. Overlap between differentially methylated cytosines/regions (DMCs/DMRs) in female blood and liver
Table 7. Overlap between differentially methylated cytosines/regions (DMCs/DMRs) in male blood and liver
Figure 6. Analysis of overlapping DMC-associated genes in females (a) and males (b). Panels C-D denote genes in blood (c) and liver (d) that are in common across sexes. Panel E depicts the number of genes in common across sex and tissue. Numbers in circles indicate the total number of DMC-associated genes in each tissue. All overlaps were statistically significant, as indicated by hypergeometric test (panel A: p < 2.7x10−46, panel B: p < 1.1x10−73, panel C: p < 9.459x10−59, panel D: p < 2.656x10−90, panel E: p < 3.227x10−30). In the tables, # Genes indicates the number of genes in the specified GO pathway that appear in the experimental dataset. Pathway analysis was conducted using DAVID, version 6.8 (Laboratory of Human Retrovirology and Immunoinformatics (LHRI))
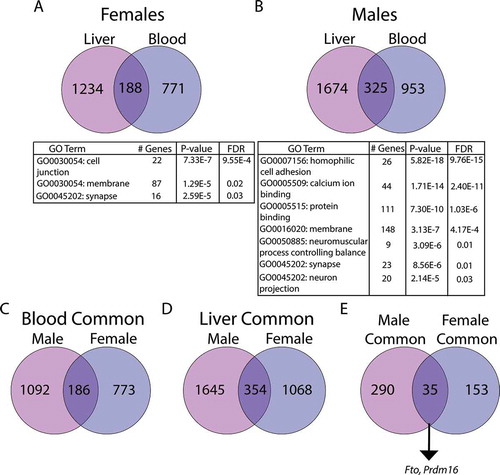
We next compared the sex-specific overlap between genes in both tissues. In blood and liver, respectively, we observed 186 and 354 genes in common () and Supplemental Tables S21-S22). Although the intersecting genes were a minority of the total number of DMC-associated genes, the overlaps were statistically significant (p < 9.46x10−59 and 2.66 × 10−90 for blood and liver, respectively, hypergeometric test). We then investigated the intersection of DMC-associated genes across tissues and sexes, and found a small number of genes (35 genes) were common across tissues and sexes () and Supplemental Table S23). This was significantly greater than what would be expected by chance p < 3.23x10−30, hypergeometric test). These included several genes associated with obesity, metabolic syndrome and hepatocellular carcinoma, including Prdm16 [Citation38–40], Fto [Citation41,Citation42], and Wwox [Citation43].
Pb-induced changes in gene expression
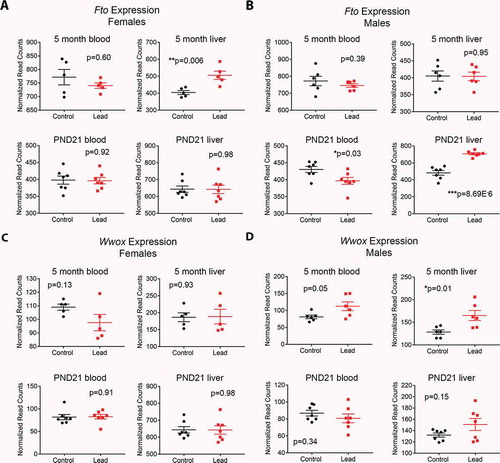
In order to determine whether altered DNA methylation was accompanied by changes in expression of Fto, Wwox and Prdm16 in both tissues, we interrogated publicly available RNA-seq read count data from the same animals that we generated as part of the TaRGET II consortium (see Methods). Notably, Fto expression was significantly increased in female liver, but not blood at 5 months of age (), top). Males showed no change in Fto expression in either tissue at this time point (), top). In contrast, females showed no change in Wwox expression at 5 months of age, but expression in males was increased with Pb exposure in both tissues in adulthood (), top). Cessation of Pb exposure occurred at postnatal day (PND) 21, and we wondered whether changes in gene expression were present at this early time point. Interestingly, Fto expression was significantly increased in male, but not female liver at PND21 (), bottom). Wwox expression was not significantly changed at PND21 in either sex or tissue (), bottom).
Figure 7. Effects of Pb exposure on gene expression in mouse blood and liver at two time points. Normalized RNA-seq read count data for females (a,c) and males (b,d) for Fto and Wwox. Top graphs in each panel depict gene expression in a cohort of animals sacrificed at 5 months of age (the same animals used for ERRBS), and bottom graphs show gene expression at postnatal day (PND) 21, when animals were weaned and Pb exposure stopped. A cohort of animals were sacrificed for gene expression analysis at this time point. Statistical significance was determined using linear mixed effects regression
Differential methylation at sexually dimorphic genes
Given the established sex-biased gene expression the liver [Citation44,Citation45], we next investigated whether genes exhibiting sex-specific methylation with Pb exposure were among those that show basal sex differences in expression in the adult mouse liver. To this end, we compared our findings with those of a previously published study that examined sex differences in mouse hepatic gene expression at multiple time points [Citation44]. Of the 1422 genes containing DMCs with Pb exposure in the female liver, 46 were significantly differentially expressed between males and females at 8 weeks of age () and Supplemental Table S24). Likewise, 78 of the 1999 genes showing differential methylation with Pb exposure in males were also differentially expressed in the male vs. female adult mouse liver () and Supplementary Table S25). These overlaps were statistically significant (p = 0.001 and 0.02 for females and males, respectively). A subset of these genes was also differentially methylated in the blood, including 2 in common across tissue and sex ()). Together, these data suggest that there exist sex-specific changes in DNA methylation with perinatal Pb exposure that are consistent across blood and liver in adulthood, with significant gene overlap between tissues and sexes. Furthermore, several Pb-induced changes in methylation in both males and females occur at sexually dimorphic genes.
Figure 8. Overlap between genes with differential methylation (DM genes Pb) and genes that are differentially expressed in male vs. female liver at 8 weeks of age (DE genes 8 weeks). Separate analyses were conducted in females (a) and males (b). Genes listed below each diagram are those that also exhibit differential methylation in the blood of either females (A) or males (B). Bold genes are differentially methylated with Pb exposure in both tissues and sexes, and are differentially expressed between sexes in 8 week mouse liver. P values (hypergeometric test) represent the significance of overlap between gene lists

Discussion
In this work, we have investigated the effects of perinatal Pb exposure on DNA methylation in the liver and blood of adult mice. The findings from this study are novel for several reasons. First, although increasing evidence links Pb exposure to adverse metabolic outcomes [Citation46], the mechanistic basis for these associations is unclear. Indeed, little is known about the effects of Pb exposure on the liver. To our knowledge, this is the first report demonstrating that Pb exposure during pregnancy and lactation leads to sex-specific alterations in hepatic DNA methylation in adulthood. Second, we discovered that perinatal Pb exposure leads to changes in methylation at several imprinted loci that are also reprogrammed by perinatal exposure to another common toxicant, BPA [Citation4]. Finally, we mapped DNA methylation genome-wide in paired blood and liver samples from the same mice after perinatal Pb exposure. Few differentially methylated sites directly overlapped between blood and liver; however, we discovered a small subset of DMC-associated genes with significant overlap between tissues in both males and females, including several that play critical roles in obesity and metabolic syndrome. This finding has important implications for the design and interpretation of human population-based epigenomics studies.
Tissue- and sex-specific changes in DNA methylation with Pb exposure
Although Pb exposure ceased at 3 weeks of age, we discovered thousands of tissue- and sex-specific DMCs/DMRs in both male and female mice at 5 months of age. The sex-specific effects of Pb on DNA methylation have been reported by our lab and others [Citation12,Citation13,Citation47]. Indeed, a subset of differentially methylated regions were found in published sex-specific mouse liver enhancers [Citation30]. As sex-specific chromatin structure drives sex differential gene expression, it is plausible that Pb exposure may reprogram the sexually dimorphic patterns of gene expression in the liver. Consistent with this, we observed altered DNA methylation at a subset of genes that are differentially expressed between male and female mice at 8 weeks of age [Citation44]. Likewise, when comparing males and females, pathway analyses revealed enrichment of distinct biological pathways. These data suggest that sex will be a critical factor in the design, analysis, and interpretation of human- and animal-based environmental epigenetics studies.
Although we investigated the effects of perinatal Pb exposure on DNA methylation in liver and blood, pathway analysis of DMRs in promoter regions revealed enrichment for pathways involved in neurological development and function in male and female liver. Moreover, among overlapping genes between blood and liver, gene pathways associated with synapse function were significantly enriched in both males and females. The nervous system is a well-known target of Pb, and although we did not directly measure nervous system tissues in this study, it is nonetheless striking that neurological pathways were enriched in genes annotated to DMCs/DMRs in both liver and blood. However, it is also important to note that the liver receives significant innervation from the autonomic nervous system, which functions in the sensing of glucose and lipid concentrations and maintenance of metabolic homoeostasis[Citation48]. Intriguingly, several nervous system-related genes identified in the pathway analysis are also expressed in the liver in normal or disease states. For example, Slc18a3, Rab3b, and Nos1, are expressed in normal liver, although their hepatic functions are not well-characterized [Citation36,Citation49,Citation50]. Likewise, Slc6a8, Tenm4, Celsr1, and Dvl1 have all been reported to be mutated, hyper-methylated or over-expressed in hepatocellular carcinoma or colon cancer liver metastases [Citation51–54]. Analysis of other tissues, including brain, from Pb-exposed mice is currently underway, and will provide important insight into whether there is a common subset of tissue-independent, environmentally labile genes or CpG sites.
Imprinted genes
Genomic imprinting, which results in mono-allelic, parent-of-origin expression of a subset of genes, is mediated in large part by DNA methylation, and establishment of 5mC at imprinted loci occurs during early embryonic development [Citation55,Citation56]. Importantly, we and others have reported that environmental exposures during this critical developmental window lead to altered 5mC and 5hmC at imprinted loci [Citation4,Citation11,Citation34,Citation57,Citation58] and alterations in imprinted gene expression [Citation4,Citation58]. Imprinted genes play a critical role in growth and metabolism, and abnormal DNA methylation and expression of imprinted genes are associated with several diseases, including growth, metabolic and developmental disorders, and cancer [Citation59]. Notably, we discovered that Pb exposure during gestation and lactation leads to changes in DNA methylation at 44 imprinted genes, and this enrichment is statistically significant. The Gnas locus encodes for three distinct protein products, and disruptions in the regulation of this locus have been implicated in a number of disorders, including early-onset obesity [Citation60]. We recently reported that developmental BPA exposure leads to changes in DNA methylation and hydroxymethylation at the Gnas locus, concomitant with increased Gnas expression [Citation4]. In this study, hypermethylation at the Gnas locus in both males and females was accompanied by reduced Gnas transcript expression. Thus, Gnas may be an important target of multiple environmental exposures, in both males and females. When comparing male liver and blood, we observed DMCs at several imprinted genes. For example, Snrpn exhibited changes in methylation in the same direction in both blood and liver. Snrpn encodes for an RNA binding protein that is critical for neurological function, and disruption of Snrpn expression is implicated in Prader-Willi syndrome, a neuroendocrine disorder associated with intellectual disability and obesity [Citation38]. Work from us and others suggests that environmental exposures are associated with changes in methylation of the Snrpn locus [Citation4,Citation58,Citation61,Citation62]. Given that the Srnpn locus is hypermethylated in both blood and liver with gestational Pb exposure, it is plausible that it may represent an important biomarker of early life Pb exposure. We recently reported that perinatal BPA exposure leads to reprogramming of DNA methylation and hydroxymethylation at 12 imprinted genes [Citation4]. Interestingly, 9 of the imprinted loci altered by Pb exposure were among those reprogrammed by BPA exposure. These data suggest that a core set of imprinted genes may be vulnerable to epigenetic reprogramming by multiple environmental exposures. Recent work suggests that the late stages of oocyte development and early embryonic development are particularly vulnerable to altered genomic imprinting by environmental exposures [Citation58]. As both Pb and BPA exposure have been implicated in adverse metabolic health outcomes [Citation46,Citation57,Citation63], it is plausible that reprogramming of genomic imprinting may represent a common mechanism linking both toxicants to these outcomes. The mechanistic consequences of environment-mediated dysregulation of imprinted genes, and the implications this may have for human health, should be investigated in future studies.
Potential mechanisms for altered DNA methylation
The mechanism(s) by which developmental Pb exposure alter DNA methylation are currently unknown. DNA methyltransferases (DNMTs) methylate the 5-position of cytosine bases in DNA using S-adenosylmethionine (SAM), which is generated via one-carbon metabolism [Citation64]. Pb may disrupt DNA methylation via alteration in DNMT expression/activity or through perturbation of SAM levels. Consistent with the first hypothesis, Pb exposure in vitro and in vivo leads to inhibition and altered expression of DNMTs [Citation65]. In support of the second possibility, administration of exogenous SAM mitigates the deleterious effects of Pb exposure, suggesting that Pb may deplete endogenous levels of SAM [Citation66,Citation67]. DNA methylation can be erased through an active process that includes 5hmC as an intermediate, and altered 5hmC has also been reported with Pb exposure [Citation68]. 5hmC is generated by the activity of TET methylcytosine dioxygenases (TETs), using the cofactor alpha-ketoglutarate (α-KG), which is generated by the TCA cycle [Citation69]. Interestingly, Pb exposure has been shown to reduce activity of TCA cycle enzymes, including isocitrate dehydrogenase, which catalyzes the formation of α-KG [Citation70,Citation71]. In spite of the substantial differences between Pb and BPA, similar mechanisms have been reported for BPA. Indeed, the effects of developmental BPA exposure on DNA methylation can be countered by methyl donor supplementation [Citation72]. Likewise, BPA has been reported to alter expression of DNMT1 in other tissues [Citation37,Citation73]. These mechanistic similarities, combined with the similar developmental window of exposure, may explain why BPA and Pb exhibit effects on DNA methylation at some common gene loci.
It is additionally notable that Pb-induced changes in DNA methylation are sex-specific. Although DNA methylation undergoes reprogramming during fertilization and primordial germ cell development, the kinetics of, and mechanisms underlying, this reprogramming differ between male and female genomes [Citation2,Citation74]. In the preimplantation embryo, demethyation of the paternal genome occurs via an active, TET-mediated process [Citation74]. In contrast, demethylation of the maternal genome occurs through a slower, passive loss of DNA methylation over the course of cell divisions [Citation74]. In the primordial germ cells, similar sex differences occur during the restoration of sex-specific DNA methylation. In sperm, this process is completed prior to birth; however, in oocytes, sex-specific remethylation continues until puberty [Citation2,Citation74]. Thus, sex differences in Pb-mediated alterations in DNA methylation may be due to these mechanistic and temporal differences. The precise mechanisms underlying the observed Pb-induced changes in DNA methylation are currently under investigation.
Utility of blood as a surrogate tissue for Pb effects on liver
When comparing the chromosomal locations of DMCs/DMRs between blood and liver, only a nonsignificant number of sites directly overlapped between the two tissues. However, we observed a significant number of DMC-associated genes in common between blood and liver, and 35 genes showed differential methylation across tissues and sexes. These overlaps were statistically significant. Importantly, altered methylation at Fto and Wwox was accompanied by changes in gene expression which were time, tissue and sex-dependent, highlighting the complex nature of sex- and tissue-specific gene regulation. Fto expression was elevated in the female liver at 5 months, and in the male liver at PND21. Moreover, Wwox expression was elevated in blood and liver of males at 5 months of age, suggesting that it may serve as a biomarker for Pb exposure. SNPs in the FTO gene are associated with increased energy intake and obesity in human and rodent studies [Citation42]. WWOX is a tumour suppressor that is lost in hepatocellular carcinoma and is also de-regulated in metabolic syndrome [Citation43,Citation75]. Whether altered regulation of these genes plays a mechanistic role in Pb-induced adverse metabolic outcomes requires further investigation. The genes identified herein, particularly those that are commonly differentially methylated and expressed in blood and liver, may represent candidates for analysis in human environmental epigenetics studies. Future studies are needed to validate the plausibility of these loci as potential biomarkers of Pb exposure, as well as to investigate the functional and health-related consequences of altered DNA methylation at these sites.
Limitations
This study has several important limitations. First, we utilized ERRBS, a method that, while providing resolution at the level of individual CpG sites, does not cover the entire genome. The use of whole-genome bisulphite sequencing techniques may yield additional insight into the effects of Pb exposure on the liver, and identify additional Pb-induced signatures that overlap between blood and liver. Second, ERRBS does not differentiate between 5mC and 5hmC. Thus, the differentially methylated loci identified in this study may be a combination of both 5mC and 5hmC. Third, changes of interest in DNA methylation may only occur in specific subpopulations of cells [Citation76]. Thus, our approach using unfractionated tissues may have missed other potential markers of interest for Pb exposure. Additionally, blood cells undergo rapid turnover, as well as exposure-, age-, and disease-dependent changes in cellular composition [Citation77–79]. Future studies using single-cell transcriptomics and epigenomics approaches will help to address issues of tissue-specific cellular heterogeneity and composition [Citation76,Citation80]. It is also important to note that DAVID gene ontology analysis does not consider the differing number of CpG sites per gene, and therefore may exhibit bias. Although we found that altered methylation was accompanied by changes in expression of a few genes, we observed few significant changes in gene expression with RNA-seq (reference https://targetepigenomics.org/). Thus, the functional significance of altered methylation is unclear based on this study. However, recent work suggests that developmental exposures may induce epigenetic changes that are silent until a further physiologic stimulus or environmental insult is encountered [Citation81,Citation82]. Finally, as we did not measure Pb levels in the blood and liver of offspring mice, we cannot exclude the possibility that some Pb remains in these tissues at 5 months of age. Thus, some changes in methylation and gene expression observed at this time point may be due to ongoing exposure to residual Pb and not to a programming event that occurred during early development. However, the majority of Pb is absorbed in bone, with a small fraction found in blood and soft tissues [Citation83,Citation84]. While Pb is stably stored in bone, children are less likely to retain Pb in bone compared to adults, and turnover is much more dynamic in soft tissues [Citation83,Citation84]. Ongoing release of Pb from bone is of concern; however, this occurs primarily during pregnancy, stress, and age-related bone loss [Citation85,Citation86]. Thus, given the early exposure window, the relatively young age of the adult mice and the dynamic turnover of Pb in soft tissues, we would expect little Pb to be present in blood and liver at 5 months of age. Nevertheless, whether Pb remains in the body after early developmental exposure or not, our data suggest that Pb exposure during early life causes epigenetic and gene expression changes that may have important implications for health across the life course.
Conclusions
In summary, we have investigated the effects of perinatal Pb exposure on DNA methylation in paired samples of mouse blood and liver. There are several key strengths of this study, including the side-by-side comparison of blood and liver to identify signatures of Pb exposure present in the blood that reflect changes in the liver. Future studies should investigate the reproducibility of these findings, the mechanistic and functional consequences of persistent, Pb-induced changes in hepatic methylation, and the implications these changes have for human health.
Author Contributions
All authors provided critical feedback and helped shape the research, analysis and manuscript. KN, CAR, TRJ, CL performed experiments. LKS, RC, CAR, ZT, SL, JMG, JAC, MAS, DCD analyzed data. LKS wrote manuscript in consultation with DCD, JAC, MAS, and JMG.
Supplemental Material
Download Zip (14.1 MB)Disclosure statement
The authors declare no conflict of interest.
Data Availability
ERRBS sequencing data have been uploaded to GEO (accession number GSE150670). RNA-seq data are available from the TaRGET II Consortium: https://targetepigenomics.org/.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492.
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293(5532):1089–1093.
- Lopez V, Fernandez AF, Fraga MF. The role of 5-hydroxymethylcytosine in development, aging and age-related diseases. Ageing Res Rev. 2017;37:28–38.
- Kochmanski JJ, Marchlewicz EH, Cavalcante RG, et al. Longitudinal effects of developmental bisphenol a exposure on epigenome-wide DNA hydroxymethylation at imprinted loci in mouse blood. Environ Health Perspect. 2018;126(7):077006.
- Ringh MV, Hagemann-Jensen M, Needhamsen M, et al. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine. 2019;46:290–304.
- Huang Y, Lin S, Jin L, et al. Decreased global DNA hydroxymethylation in neural tube defects: association with polycyclic aromatic hydrocarbons. Epigenetics. 2019;1–11.
- Zhai H, Chen C, Wang N, et al. Blood lead level is associated with non-alcoholic fatty liver disease in the Yangtze river delta region of China in the context of rapid urbanization. Environ Health. 2017;16(1):93. .
- Cave M, Appana S, Patel M, et al. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003-2004. Environ Health Perspect. 2010;118(12):1735–1742.
- Bener A, Obineche E, Gillett M, et al. Association between blood levels of lead, blood pressure and risk of diabetes and heart disease in workers. Int Arch Occup Environ Health. 2001;74(5):375–378.
- Park YJ, Jung Y, Oh CU. Relations between the blood lead level and metabolic syndrome risk factors. Public Health Nurs. 2019;36(2):118–125.
- Nye MD, King KE, Darrah TH, et al. Maternal blood lead concentrations, DNA methylation of MEG3 DMR regulating the DLK1/MEG3 imprinted domain and early growth in a multiethnic cohort. Environ Epigenet. 2016;2(1):1. .
- Faulk C, Barks A, Liu K, et al. Early-life lead exposure results in dose- and sex-specific effects on weight and epigenetic gene regulation in weanling mice. Epigenomics. 2013;5(5):487–500.
- Faulk C, Barks A, Sanchez BN, et al. Perinatal lead (Pb) exposure results in sex-specific effects on food intake, fat, weight, and insulin response across the murine life-course. PLoS One. 2014;9(8):e104273. .
- Wu J, Wen XW, Faulk C, et al. Perinatal lead exposure alters gut microbiota composition and results in sex-specific bodyweight increases in adult mice. Toxicol Sci. 2016;151(2):324–333. .
- Leasure JL, Giddabasappa A, Chaney S, et al. Low-level human equivalent gestational lead exposure produces sex-specific motor and coordination abnormalities and late-onset obesity in year-old mice. Environ Health Perspect. 2008;116(3):355–361. .
- Sun H, Wang N, Nie X, et al. Lead Exposure Induces Weight Gain in Adult Rats, accompanied by DNA hypermethylation. PLoS One. 2017;12(1):e0169958. .
- Xia J, Lu L, Jin C, et al. Effects of short term lead exposure on gut microbiota and hepatic metabolism in adult zebrafish. Comp Biochem Physiol C Toxicol Pharmacol. 2018;209:1–8.
- Xia J, Jin C, Pan Z, et al. Chronic exposure to low concentrations of lead induces metabolic disorder and dysbiosis of the gut microbiota in mice. Sci Total Environ. 2018;631-632:439–448.
- Wang T, Pehrsson EC, Purushotham D, et al. The NIEHS TaRGET II Consortium and environmental epigenomics. Nat Biotechnol. 2018;36(3):225–227. .
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23(15):5293–5300.
- Weinhouse C, Anderson OS, Bergin IL, et al. Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ Health Perspect. 2014;122(5):485–491. .
- Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87. .
- Garrett-Bakelman FE, Sheridan CK, Kacmarczyk TJ, et al. Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J Vis Exp. 2015;96:e52246.
- Koster J, Rahmann S. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics. 2012;28(19):2520–2522.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359.
- Park Y, Figueroa ME, Rozek LS, et al. MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics. 2014;30(17):2414–2422.
- Park Y, Wu H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics. 2016;32(10):1446–1453.
- Cavalcante RG, Sartor MA. annotatr: genomic regions in context. Bioinformatics. 2017;33(15):2381–2383.
- Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol. 2013;33(18):3594–3610.
- Lee CT, Cavalcante RG, Lee C, et al. Poly-Enrich: count-based methods for gene set enrichment testing with genomic regions. NAR Genom Bioinform. 2020;2(1):lqaa006.
- Risso D, Ngai J, Speed TP, et al. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2014;32(9):896–902.
- Chen A, Chen D, Chen Y. Advances of DNase-seq for mapping active gene regulatory elements across the genome in animals. Gene. 2018;667:83–94.
- Faulk C, Liu K, Barks A, et al. Longitudinal epigenetic drift in mice perinatally exposed to lead. Epigenetics. 2014;9(7):934–941.
- Williamson CMBA, Thomas S, Beechey CV, et al. World wide web site - mouse imprinting data and references. 2013. http://www.har.mrc.ac.uk/research/genomic_imprinting/
- Banerjee S, Melnyk SB, Krager KJ, et al. Trifluoperazine inhibits acetaminophen-induced hepatotoxicity and hepatic reactive nitrogen formation in mice and in freshly isolated hepatocytes. Toxicol Rep. 2017;4:134–142.
- Li Y, Duan F, Zhou X, et al. Differential responses of GC1 spermatogonia cells to high and low doses of bisphenol A. Mol Med Rep. 2018;18(3):3034–3040.
- Reed ML, Leff SE. Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi syndrome. Nat Genet. 1994;6(2):163–167.
- Nilsson E, Matte A, Perfilyev A, et al. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J Clin Endocrinol Metab. 2015;100(11):E1491–1501. .
- Seale P. Transcriptional regulatory circuits controlling brown fat development and activation. Diabetes. 2015;64(7):2369–2375.
- Abbas S, Raza ST, Ahmed F, et al. Association of genetic polymorphism of PPARgamma-2, ACE, MTHFR, FABP-2 and FTO genes in risk prediction of type 2 diabetes mellitus. J Biomed Sci. 2013;20(1):80.
- Speakman JR. The ‘fat mass and obesity related’ (FTO) gene: mechanisms of impact on obesity and energy balance. Curr Obes Rep. 2015;4(1):73–91.
- Hua HW, Jiang F, Huang Q, et al. MicroRNA-153 promotes Wnt/beta-catenin activation in hepatocellular carcinoma through suppression of WWOX. Oncotarget. 2015;6(6):3840–3847.
- Conforto TL, Waxman DJ. Sex-specific mouse liver gene expression: genome-wide analysis of developmental changes from pre-pubertal period to young adulthood. Biol Sex Differ. 2012;3(1):9.
- Lau-Corona D, Bae WK, Hennighausen L, et al. Sex-biased genetic programs in liver metabolism and liver fibrosis are controlled by EZH1 and EZH2. PLoS Genet. 2020;16(5):e1008796.
- Niessen LW, Mohan D, Akuoku JK, et al. Tackling socioeconomic inequalities and non-communicable diseases in low-income and middle-income countries under the Sustainable Development agenda. Lancet. 2018;391(10134):2036–2046. .
- Sen A, Heredia N, Senut MC, et al. Early life lead exposure causes gender-specific changes in the DNA methylation profile of DNA extracted from dried blood spots. Epigenomics. 2015;7(3):379–393. .
- Mizuno K, Ueno Y. Autonomic Nervous System and the Liver. Hepatol Res. 2017;47(2):160–165.
- Field RB, Kruse DH, Redman RS. Immunohistochemical localization and mRNA detection of Rab3D and/or Rab3B in rat von Ebner’s glands, parotid gland, pancreas, and liver. Histochem J. 2001;33(2):71–77.
- Delbro DS, Hallsberg L, Wallin M, et al. Expression of the non-neuronal cholinergic system in rat liver. APMIS. 2011;119(3):227–228.
- Loo JM, Scherl A, Nguyen A, et al. Extracellular metabolic energetics can promote cancer progression. Cell. 2015;160(3):393–406. .
- Zhan H, Jiang J, Sun Q, et al. Whole-exome sequencing-based mutational profiling of hepatitis B virus-related early-stage hepatocellular carcinoma. Gastroenterol Res Pract. 2017;2017:2029315.
- Ammerpohl O, Pratschke J, Schafmayer C, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer. 2012;130(6):1319–1328. .
- Song J, Xie C, Jiang L, et al. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/beta-catenin pathway in hepatocellular carcinoma. Theranostics. 2018;8(13):3571–3583. .
- Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014;6(2):2.
- Okae H, Chiba H, Hiura H, et al. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet. 2014;10(12):e1004868. .
- Bansal A, Rashid C, Xin F, et al. Sex- and Dose-Specific Effects of Maternal Bisphenol A Exposure on Pancreatic Islets of First- and Second-Generation Adult Mice Offspring. Environ Health Perspect. 2017;125(9):097022. .
- Susiarjo M, Sasson I, Mesaros C, et al. Bisphenol a exposure disrupts genomic imprinting in the mouse. PLoS Genet. 2013;9(4):e1003401.
- Skaar DA, Li Y, Bernal AJ, et al. The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. Ilar J. 2012;53(3–4):341–358.
- Hanna P, Grybek V, Perez de Nanclares G, et al. Genetic and epigenetic defects at the GNAS locus lead to distinct patterns of skeletal growth but similar early-onset obesity. J Bone Miner Res. 2018;33(8):1480–1488.
- Soubry A, Hoyo C, Butt CM, et al. Human exposure to flame-retardants is associated with aberrant DNA methylation at imprinted genes in sperm. Environ Epigenet. 2017;3(1):dvx003. .
- Faulk C, Kim JH, Jones TR, et al. Bisphenol A-associated alterations in genome-wide DNA methylation and gene expression patterns reveal sequence-dependent and non-monotonic effects in human fetal liver. Environ Epigenet. 2015;1(1):1. .
- Nadal A, Quesada I, Tuduri E, et al. Endocrine-disrupting chemicals and the regulation of energy balance. Nat Rev Endocrinol. 2017;13(9):536–546.
- Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25(1):27–42.
- Sanchez OF, Lee J, Yu King Hing N, et al. Lead (Pb) exposure reduces global DNA methylation level by non-competitive inhibition and alteration of dnmt expression. Metallomics. 2017;9(2):149–160.
- Paredes SR, Fukuda H, Kozicki PA, et al. S-adenosyl-L-methionine and lead intoxication: its therapeutic effect varying the route of administration. Ecotoxicol Environ Saf. 1986;12(3):252–260.
- Cao XJ, Huang SH, Wang M, et al. S-adenosyl-L-methionine improves impaired hippocampal long-term potentiation and water maze performance induced by developmental lead exposure in rats. Eur J Pharmacol. 2008;595(1–3):30–34.
- Sen A, Cingolani P, Senut MC, et al. Lead exposure induces changes in 5-hydroxymethylcytosine clusters in CpG islands in human embryonic stem cells and umbilical cord blood. Epigenetics. 2015;10(7):607–621. .
- Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene. 2017;36(24):3359–3374.
- Basha DC, Basha SS, Reddy GR. Lead-induced cardiac and hematological alterations in aging Wistar male rats: alleviating effects of nutrient metal mixture. Biogerontology. 2012;13(4):359–368.
- Seddik L, Bah TM, Aoues A, et al. Elucidation of mechanisms underlying the protective effects of olive leaf extract against lead-induced neurotoxicity in Wistar rats. J Toxicol Sci. 2011;36(6):797–809.
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104(32):13056–13061.
- Kundakovic M, Gudsnuk K, Franks B, et al. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci U S A. 2013;110(24):9956–9961. .
- Zeng Y, Chen T. DNA methylation reprogramming during mammalian development. Genes (Basel). 2019;10:4.
- Abu-Remaileh M, Abu-Remaileh M, Akkawi R, et al. WWOX somatic ablation in skeletal muscles alters glucose metabolism. Mol Metab. 2019;22:132–140.
- Hui T, Cao Q, Wegrzyn-Woltosz J, et al. High-resolution single-cell DNA methylation measurements reveal epigenetically distinct hematopoietic stem cell subpopulations. Stem Cell Reports. 2018;11(2):578–592. .
- Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31.
- Su D, Wang X, Campbell MR, et al. Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PLoS One. 2016;11(12):e0166486. .
- Babio N, Ibarrola-Jurado N, Bullo M, et al. White blood cell counts as risk markers of developing metabolic syndrome and its components in the PREDIMED study. PLoS One. 2013;8(3):e58354. .
- Kelsey G, Stegle O, Reik W. Single-cell epigenomics: recording the past and predicting the future. Science. 2017;358(6359):69–75.
- Wang Q, Trevino LS, Wong RL, et al. Reprogramming of the epigenome by MLL1 links early-life environmental exposures to prostate cancer risk. Mol Endocrinol. 2016;30(8):856–871. .
- Trevino LS, Dong J, Kaushal A, et al. Epigenome environment interactions accelerate epigenomic aging and unlock metabolically restricted epigenetic reprogramming in adulthood. Nat Commun. 2020;11(1):2316. .
- Barry PS. A comparison of concentrations of lead in human tissues. Br J Ind Med. 1975;32(2):119–139.
- Schroeder HA, Tipton IH. The human body burden of lead. Arch Environ Health. 1968;17(6):965–978.
- Popovic M, McNeill FE, Chettle DR, et al. Impact of occupational exposure on lead levels in women. Environ Health Perspect. 2005;113(4):478–484.
- Berkowitz GS, Wolff MS, Lapinski RH, et al. Prospective study of blood and tibia lead in women undergoing surgical menopause. Environ Health Perspect. 2004;112(17):1673–1678.