
ABSTRACT
The epigenetic regulator Dot1, the only known histone H3K79 methyltransferase, has a conserved role in organismal development and homoeostasis. In yeast, Dot1 is required for telomeric silencing and genomic integrity. In Drosophila, Dot1 (Grappa) regulates homoeotic gene expression. Dysregulation of DOT1L (human homologue of Dot1) causes leukaemia and is implicated in dilated cardiomyopathy. In mice, germline disruption of Dot1L and loss of H3K79me2 disrupt vascular and haematopoietic development. Targeted inactivation of Dot1L in principal cells of the mature collecting duct affects terminal differentiation and cell type patterning. However, the role of H3K79 methylation in mammalian tissue development has been questioned, as it is dispensable in the intestinal epithelium, a rapidly proliferating tissue. Here, we used lineage-specific Cre recombinase to delineate the role of Dot1L methyltransferase activity in the mouse metanephric kidney, an organ that develops via interactions between ureteric epithelial (Hoxb7) and mesenchymal (Six2) cell lineages. The results demonstrate that Dot1LHoxb7 is dispensable for ureteric bud branching morphogenesis. In contrast, Dot1LSix2 is critical for the maintenance and differentiation of Six2+ progenitors into epithelial nephrons. Dot1LSix2 mutant kidneys exhibit congenital nephron deficit and cystic dysplastic kidney disease. Molecular analysis implicates defects in key renal developmental regulators, such as Lhx1, Pax2 and Notch. We conclude that the developmental functions of Dot1L-H3K79 methylation in the kidney are lineage-restricted. The link between H3K79me and renal developmental pathways reaffirms the importance of chromatin-based mechanisms in organogenesis.
Keywords:
Introduction
Post-translational modifications of histones regulate gene expression by acting as a platform for recruitment of transcriptional complexes and nucleosome remodellers thus controlling access to DNA-binding transcription factors [Citation1–3]. Chromatin profiling in stem cells and a variety of progenitor and differentiating cell types has revealed that, in general, active gene promoters harbour H3K4me3, active enhancers harbour H3K27ac/H3K4me1, and bivalent promoters (H3K4me3/H3K27me3) and enhancers (H3K4me1/H3K27me3) denote poised genes that are primed for later activation or repression during differentiation [Citation4,Citation5]. Indeed, loss of H3K27 methylation or disrupted histone deacetylation in nephron progenitors are associated with unscheduled activation of the epithelial differentiation programme and nephron deficit [Citation6,Citation7].
The histone mark, H3K79me2, decorates nucleosomes of active promoters and gene bodies and generally denotes an active transcriptional state [Citation8,Citation9]. H3K79 methylation is catalysed by Disruptor of Telomere Silencing (Dot1) in yeast and its Drosophila (Grappa) and mammalian homologues (Dot1L). Dot1 does not have a classical SET domain (as in other histone methyltransferase) but contains four conserved sequence motifs of class I SAM-dependent methyltransferases. Of note, Dot1 is the only known H3K79 methyltransferase, and a H3K79 demethylase has yet to be identified. Unlike most histone H3 modifications which occur on residues on the N-terminal tail of histone, H3K79 is located in the globular domain on the nucleosome surface [Citation10].
H3K79me mediates several important and conserved functions, such as telomere silencing, response to DNA damage, cell cycle and leukaemogenesis in association with MLL1 gene rearrangements [Citation11–13]. Gene targeting in mouse embryonic stem cells revealed that Dot1L is essential for vascular morphogenesis and haematopoiesis [Citation14,Citation15]. Global Dot1L−/- mutant embryos die at E9.5-E10.5, which precludes full assessment of H3K79me role in organogenesis. Published studies in mice with targeted tissue-specific deletion of Dot1L have given conflicting results regarding the physiological relevance of H3K79me in normal homoeostasis. For example, lack of H3K79me in the myocardium results in Dystrophin dysregulation and dilated cardiomyopathy [Citation16,Citation17], whereas elimination of H3K79me in the intestinal crypt epithelium causes an increase in apoptosis but has no significant effect on intestinal morphogenesis or Wnt-target gene expression [Citation18]. Loss of Dot1L-H3K79me in principal cells of the mature collecting duct of the kidney results in an increase in intercalated cell number [Citation19]. Collectively, these findings raise an important question of whether H3K79 methylation has an active role in epigenetic regulation of organ development and function or merely represents an epigenetic mark of the active transcriptional state.
Recent studies from our group and others have documented an important role for epigenetic mechanisms in renal development and disease [Citation6,Citation7,Citation20–28]. In this study, we investigated the role of Dot1L/H3K79me in kidney development, an organ that develops via interactions between the metanephric mesenchyme and ureteric bud lineages. Using lineage-specific Cre drivers to disrupt Dot1L, we show here that H3K79me is dispensable for the overall formation of the collecting system. By contrast, loss of H3K79me from the Six2-mesenchyme lineage results in dysplastic kidney disease. The results of this study provide new insights into the tissue- and lineage-specific roles of Dot1L-H3K79me and uncovers putative Dot1L developmental targets.
Materials and Methods
Animals
All experiments involving mice were conducted in compliance with guidelines outlined by the institutional IACUC. Mice harbouring the Six2-GFP::Cretg/+ (Six2GC) transgene [Citation29] were obtained from the laboratory of Dr A. P. McMahon (University of Southern California). The Hoxb7-eGFP-Cre mice [Citation30] were obtained from the laboratory of Dr. Carl Bates (University of Pittsburgh), while the Dot1Lfl/fl mice were obtained from Dr Taiping Chen (Novartis). The specifications for genotyping were furnished by the respective contributors of animals, as previously described [Citation15]. All animals in this study were maintained on a mixed background. The age of embryos in timed mating were approximated by designating noon of the day the vaginal plug was detected as embryonic (E) day 0.5.
Gross morphology
Phase images of the kidneys were captured with a SMZ1000 stereomicroscope fitted with DS-Fi1 camera enabled by the NIS-Elements F2.20 software. To measure the surface area of the kidneys, a picture of a ruler was taken as a reference under the same 1.5x magnification through the dissecting microscope. The imaging software calculates 1 mm = 200 pixels. After taking kidney the photographs, the software calculates the kidney surface areas in pixels, and then the pixel areas were converted into sq. mm.
Histology
Routine Haematoxylin and Eosin and PAS staining (Richard Allan Scientific) was performed on age matched wild type and mutant kidney sections (4 microns thick). The protocol used was as per the manufacturer’s instructions.
Immunostaining
Tissues were fixed in 10% formalin at 4°C and dehydrated prior to paraffin embedding. Four-micron tissue sections were cleared in xylene and rehydrated in a graded series of ethanol prior to antigen retrieval in 10 mM Sodium Citrate (pH6.0). Endogenous peroxidase was quenched by incubating slides in 3% hydrogen peroxide at room temperature. Sections were blocked for 90 min at room temperature in 0.5% blocking reagent (Perkin-Elmer FP1012) in Tris-buffered saline supplemented with 10% normal donkey serum together with unconjugated, monovalent donkey anti-Rabbit Fab (711–007-003 Jackson Immunoresearch Laboratories, Inc., PA) and donkey anti-Mouse Fab (715–007-003) fragments at 15ul/ml each. The primary antibodies used are listed in . For secondary detection, we used donkey anti-rabbit, donkey anti-mouse, donkey anti-goat or goat anti-chicken as was required and conjugated to one of the following Alexafluor dyes_ AlexaFluor555, AlexaFluor488 or AlexaFluor 647 (Molecular Probes, Invitrogen). For IF using Tyramide Signal Amplification we used the TSA fluorescence kit (Perkin Elmer NEL760001KT) as per the manufacturer’s recommendations. For immunohistochemistry, we used the VECTASTAIN Elite ABC kit (cat# PK6001). Images were captured using the deconvolution Olympus (IX51) microscope using the Metamorph software version 7.0 (Molecular Devices Corporation, USA).
Table 1. Antibodies used in the immunostaining assays
In situ hybridization
The protocol used was described previously [Citation31]. The samples were fixed overnight in 4% PFA/PBS and then dehydrated through a graded alcohol series before paraffin embedding. Sections were cut to 10-micron thickness. The non-radioactive RNA probes carried a digoxigenin label which was detected using anti-digoxigenin Fab fragments coupled to alkaline phosphatase (Roche Diagnostics). BM purple was used as the chromogenic substrate for Alkaline Phosphatase.
Organ culture
Embryonic kidneys were aseptically micro-dissected from timed-pregnant mice and cultured on polycarbonate Transwell filters (0.4 μm pore size, Corning Co-Star) over medium [DMEM/F−12 containing 10% foetal bovine serum (FBS)] at 37°C and 5% CO2.
Genome-wide microarray analysis
Fluorescently labelled cRNA was generated from 0.5 μg total RNA in each reaction using the Agilent Fluorescent Direct Label Kit and 1.0 mM Cyanine 3’- or 5’- labelled dCTP (PerkinElmer). Hybridization was performed using the Oligonucleotide Microarray Hybridization and In Situ Hybridization Plus Kit (Agilent). The labelled cRNA was hybridized to Agilent 44 K whole mouse genome oligonucleotide microarray. The arrays were scanned using a dual-laser DNA microarray scanner (Agilent). The data were then extracted from images using Feature Extraction software 6.1 (Agilent). Microarray data are available at GEO under accession number GSE110920 and GSE110928.
Nephron progenitor isolation/counting
Pairs of dissected kidneys from P0 Six2CreGFP, Six2CreGFP:Dot1Lfl/+, Six2CreGFP:Dot1Lf/fl mice were digested in 1 mL ACCUTASE (Stemcell, Catalogue #07922) at 37°C for 1 h with rotation. Cells were completed dissociated by pipetting up and down 10–15 times and centrifuged at 350 rpm for 5 min followed by removal of supernatant without disrupting the cell pellet. The cell pellet was re-suspended in FACS Buffer (PBS with 10 mM EDTA and 2% FBS) and filtered with 30 µm pro- separation filter (Miltenyi Biotech, Catalogue #130-041-407). The filtered cells were then subjected to FACS to obtain the number of GFP+ and total cells.
Results
Expression of Dot1L in the developing kidney
The kidney develops via iterative interactions between the ureteric bud and metanephric mesenchyme [Citation32–35]. The epithelium of the ureter, renal pelvis and collecting ducts is derived from the Hoxb7+ lineage [Citation36]. Six2+ lineage cells form a crescent-shaped nephrogenic niche surrounding the ureteric branch tips (called cap mesenchyme) and undergo mesenchyme-to-epithelium transition to form the pretubular aggregates and nascent nephrons (renal vesicle, comma-shaped and S-shaped bodies). Six2-lineage cells give rise to the epithelium of glomerulus, proximal tubule, loop of Henle, and distal tubules [Citation29,Citation37]. We previously demonstrated that Dot1L gene expression is up-regulated during mouse nephrogenesis [Citation25]. Using section immunofluorescence, we show here that Dot1L is expressed in the cap mesenchyme and nascent nephrons such as S-shaped bodies and maturing glomerular epithelial cells (). Likewise, Dot1L is abundantly expressed in the ureteric bud branches and maturing collecting ducts (). The cortical stromal (interstitial) cells located in between individual nephrons also express Dot1L, albeit at lower levels ().
Figure 1. (a) Dot1L protein expression in the developing kidney. Dot1L is expressed in the cap mesenchyme and its nephron derivatives, as well as in the ureteric bud, and stroma. (b) Strategy for Cre-mediated deletion of Dot1L methyltransferase domain. (c-j) Targeted disruption of Dot1L in the ureteric bud lineage (Hoxb7-Cre). (c,e) In E14.5 control kidneys, H3K79me2 is abundantly expressed in both cap mesenchyme and ureteric bud. (d,f) Dot1LHoxb7 kidneys show selective reduction of H3K79me2 in the cytokeratin-positive ureteric bud. (g,i) In E17.5 control kidneys, H3K79me2 is expressed in AQP2-positive collecting ducts. (h,j) E17.5 Dot1LHoxb7 kidneys show selective reduction of H3K79me2 in AQP2+ collecting ducts (closed arrowheads). (k-n) Targeted disruption of Dot1L in the Six2 lineage (Six2-Cre). (k,m) H3K79me2 is expressed abundantly in Six2+ progenitors in control kidneys. (l,n) In contrast, there is reduction of H3K79me2 in the Six2+ cap mesenchyme but not the ureteric bud branch of Dot1LSix2 kidneys. cm: cap mesenchyme; ub: ureteric bud; ssb: S-shaped body; cd: collecting duct; glom: maturing glomerulus. N = 6–7 animal samples per group
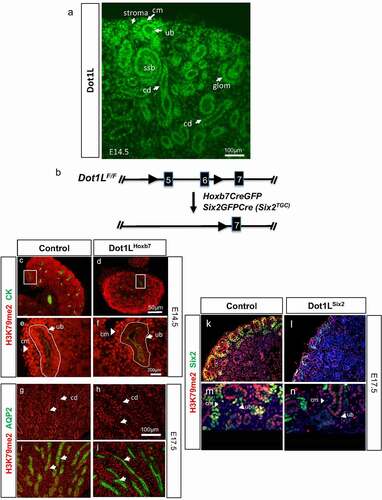
Lineage-specific deletion of Dot1L/H3K79me2
To inactivate Dot1L from the ureteric bud or nephron progenitor lineages, we crossed Dot1Lfl/fl mice [Citation15] to transgenic Hoxb7CreGFP [Citation30] or Six2GFPCre (Six2TGC) [Citation29] mice. The LoxP sites flank exons 5 and 6 encompassing the catalytic histone methyltransferase domain of Dot1L (). To examine the efficacy of the targeting strategy, we performed immunostaining for H3K79me2 on kidney sections from littermate Controls (Dot1Lfl/fl) and Dot1LHoxb7 or Dot1LSix2 mice. C, E demonstrate that H3K79me2 is expressed in the ureteric bud branches and surrounding mesenchyme in control E14.5 kidneys. In Dot1LHoxb7 kidneys, there is significant reduction of H3K79me2 in the UB tip/stalk but not in the surrounding cap mesenchyme ( D, F). At E17.5, selective reduction of H3K79me2 is seen in AQP2+ collecting ducts of Dot1LHoxb7 as compared to control kidneys ( G-J), white arrowheads and Fig. S1 A,B.
The transcription factor Six2 is expressed in the metanephric mesenchyme as early as embryonic day E10.5 [Citation29]. Later in development, Six2 is expressed in the cap mesenchyme surrounding the ureteric branch tips. Six2 marks both the self-renewing and the induced nephron progenitors committed to differentiation. To determine the role of Dot1L in Six2+ nephron progenitors, we crossed Six2-CreTGC and Dot1Lfl/fl mice. Da-d and Fig. S1 C-H show significant reduction of H3K79me2 from the Six2+ cells in E17.5 Dot1LSix2 kidneys confirming successful inactivation of Dot1L H3K79 methyltransferase activity in nephron progenitors. In summary, these findings confirm that conditional deletion of exons 5 and 6, which eliminates the methyltransferase domain of Dot1L, results in significant reduction of H3K79me2. Because we continue to see the protein by section IF using the polyclonal Dot1L antibody (Abcam ab64077), we examined the original targeting strategy [Citation15] and found that lack of exons 5 and 6 indeed results in a 108-aa in-frame deletion in the catalytic domain of Dot1L. This may explain why we continue to see Dot1L immunoreactivity following cre-mediated recombination because the polyclonal Dot1L antibody does not distinguish between the wild-type and shorter mutant protein.
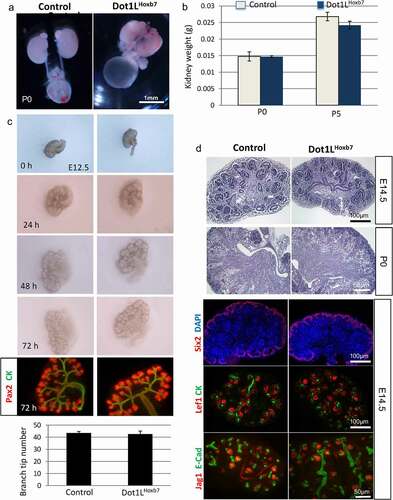
H3K79 methylation is dispensable for ureteric bud branching. Dot1LHoxb7 mice were born at the expected Mendelian ratio. Postnatal survival (up to 6 months of age) was comparable between control and Dot1LHoxb7 mice (not shown). Gross morphological development of the ureters and kidneys at P0 () and kidney weights at P0 and P5 () were unaffected. Culture of E12.5 metanephroi for up to 72 h revealed similar overall growth and branching patterns, including the number of ureteric bud tips, between mutant and control kidneys (). Moreover, histological examination and marker analysis of nephron progenitors (Six2), nascent nephrons (Lef1, Jag1), and collecting ducts (cytokeratin and E-cadherin) did not reveal any differences in the structure and patterning of the nephrogenic zone or medulla between Control and Dot1LHoxb7 kidneys (). In addition, in situ hybridization revealed that expression of the ureteric bud tip markers c-Ret and Wnt11 was unaltered in Dot1LHoxb7 compared to control kidneys (Fig. S2 A-D). Similarly, expression of the ureteric bud stalk makers, Wnt7b and Wnt9b was not unaffected (Fig. S2 E-H). Thus, Dot1L/H3K79me are dispensable for ureteric bud branching morphogenesis. Of note, our study confirmed the previously identified and reported terminal differentiation function of Dot1L/H3K79me in maintenance of principal and intercalated cell identity since Hoxb7Cre-mediated deletion of Dot1L resulted in increased number of carbonic anhydrase-2-positive intercalated cells (Fig. S3).
Figure 2. Dot1L-H3K79me is dispensable for ureteric bud branching and formation of the collecting system. (a) Gross morphology of the urinary tracts. Dot1LHoxb7 pups have normal appearing kidneys, ureters and bladder. (b) Kidney weights are similar in control and Dot1LHoxb7 newborns at P0 and P5 (n = 6–8 per group). (c) In vitro culture of E12.5 metanephroi reveals that inactivation of Dot1L in the ureteric bud lineage has no discernible effect on normal growth and branching (n = 5/group). (d) Histological examination and immunostaining of nephron progenitors (Six2), nascent nephrons (Lef1, Jagged1), and collecting ducts (CK, E-Cadherin) show normal nephron differentiation (n = 4/group)
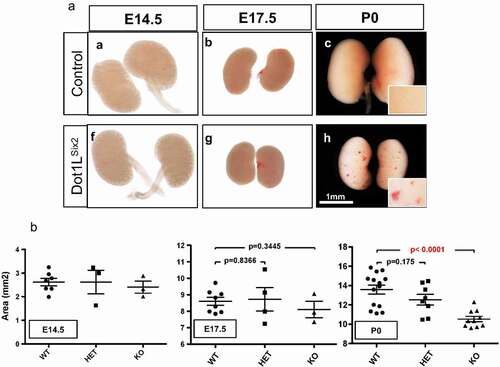
Inactivation of Dot1L methyltransferase activity in the Six2 lineage causes congenital renal dysplasia. Dot1LSix2 mice are born at the expected Mendelian ratio. However, renal size is significantly compromised in homozygous Dot1LSix2 kidneys at P0 ( A, B). Dot1LSix2 kidneys also exhibit surface ‘petechial’-like lesions” ( Ah).
Figure 3. Targeted disruption of Dot1L H3K79 methyltransferase activity in Six2+ nephron progenitors impairs renal growth. (a) Gross morphology views showing that homozygous disruption of Dot1L (Dot1LSix2) causes small kidneys with petechial surface lesions. (b) Surface area analysis (see methods) of control and Dot1LSix2 kidneys during development. Targeted disruption of one allele of Dot1L in nephron progenitors did not affect renal size during maturation; however, disruption of both Dot1L alleles caused a marked reduction in renal size
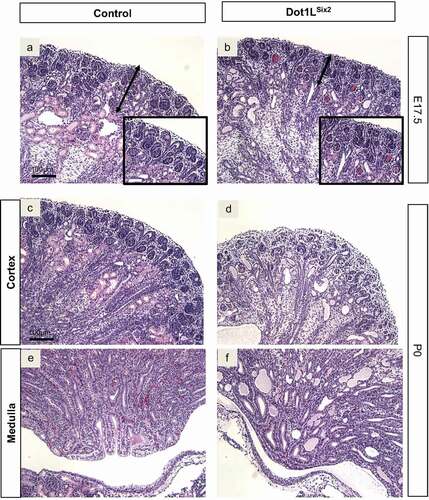
Histological examination of PAS-stained sections at E17.5 revealed that the nephrogenic zone of Dot1LSix2 kidneys was thinner than control kidneys and contained ‘blood-filled’ glomeruli (hence the surface petechial lesions), and dilated/cyst-like tubules ( A,B and insets). P0 Dot1LSix2 kidneys showed depletion of the nephrogenic zone with multiple-dilated tubules in the medulla filled with proteinaceous material ( C-F).
Figure 4. Targeted disruption of Dot1L-H3K79 methyltransferase activity in Six2+ nephron progenitors causes congenital renal dysgenesis. (a, b) PAS-stained sections. E17.5 Dot1LSix2 kidneys display loss of cortical architecture, thinning of the nephrogenic zone, and blood-filled glomerular tufts. (c-f) PAS-stained sections. P0 Dot1LSix2 kidneys show progressive loss of corticomedullary differentiation, glomerular blood extravasation, depletion of the nephrogenic zone, cortical and medullary tubular dilation/cyst-like formation. Consistent with disrupted glomerular architecture, many cortical and medullary dilated tubules contain proteinaceous material. N = 5–7/group
Section immunofluorescence using markers of proximal tubules, thick ascending loop of Henle and collecting ducts was performed to determine the segmental origin of the dilated tubules/cysts. The results revealed that the majority of the cysts originated from LTL-positive (proximal) tubules ( A-D). In addition, the cystic LTL-positive tubules were de-differentiated as suggested by loss of angiotensinogen ( A, B). Only few cysts originated from NKCC2-positive (thick ascending limb of Henle) tubules ( C, D). AQP2-positive collecting ducts did not exhibit cystic changes ( E, F). Thus, it appears that the cystic-dysplastic changes in Dot1LSix2 mutant kidneys are cell autonomous and restricted to the nephron-lineage and not the ureteric bud lineage.
Figure 5. Dot1LSix2 renal cysts are predominantly of proximal tubule origin. Section immunofluorescence of P0 Control and mutant Dot1LSix2 using markers of proximal tubule (angiotensinogen [AGT], LTL), thick ascending limb of Henle (NKCC2), and collecting duct (AQP2). (a, b) In (a) AGT and LTL decorate the proximal tubules in control kidney. In (b) some less affected LTL-positive tubules retain AGT staining, whereas de-differentiated cystic-dysplastic tubules lost AGT staining. (c, d) In control kidney (c), LTL-positive proximal tubules and NKCC2-positive thick ascending limb of Henle-positive tubules are seen in the inner cortex. In mutant kidney (d), the majority of cystic-dysplastic tubules are LTL-positive; only one cystic structure is NKCC2-positive. Note the reduction in number of NKCC2-positive segments in mutant kidneys. (e, f) Staining of AQP2 reveals no evidence of cystic changes in the collecting ducts of mutant kidneys. PT: proximal tubules, TAL: thick ascending limb of Henle, CD: collecting duct. N = 5/group
![Figure 5. Dot1LSix2 renal cysts are predominantly of proximal tubule origin. Section immunofluorescence of P0 Control and mutant Dot1LSix2 using markers of proximal tubule (angiotensinogen [AGT], LTL), thick ascending limb of Henle (NKCC2), and collecting duct (AQP2). (a, b) In (a) AGT and LTL decorate the proximal tubules in control kidney. In (b) some less affected LTL-positive tubules retain AGT staining, whereas de-differentiated cystic-dysplastic tubules lost AGT staining. (c, d) In control kidney (c), LTL-positive proximal tubules and NKCC2-positive thick ascending limb of Henle-positive tubules are seen in the inner cortex. In mutant kidney (d), the majority of cystic-dysplastic tubules are LTL-positive; only one cystic structure is NKCC2-positive. Note the reduction in number of NKCC2-positive segments in mutant kidneys. (e, f) Staining of AQP2 reveals no evidence of cystic changes in the collecting ducts of mutant kidneys. PT: proximal tubules, TAL: thick ascending limb of Henle, CD: collecting duct. N = 5/group](/cms/asset/5ad3acdf-9fc8-498c-95b0-c2f8da0dfe83/kepi_a_1861168_f0005_oc.jpg)
Dot1LSix2 kidneys exhibit depletion of nephron progenitors and nascent nephrons.By section immunofluorescence, we found that the transcription factors, Pax2, and Six2, which are required for maintenance and differentiation of the cap mesenchyme, were both suppressed in P0 mutant kidneys ( A-D). Quantitative analysis of FACS isolated Six2GFP progenitors revealed a marked reduction in the number of nephron progenitors in homozygous Dot1LSix2 as compared to control or heterozygous kidneys (). Moreover, expression of Lhx1, a transcription factor required for differentiation of renal vesicles and nascent nephrons, is markedly decreased at E17.5 (not shown) and P0 ( A, B). Consistent with the downregulation of Lhx1, the mutant kidneys manifested a reduction in LTA+ proximal tubules ( E, F) and in the number of comma-, S-shaped bodies, and maturing glomeruli ( H). Given the importance of Notch signalling in maintenance and differentiation of Six2 progenitors, we noted that Notch2 but not NICD1, showed reduced abundance in nascent nephrons ( A-F).
Figure 6. Dot1LSix2 kidneys exhibit depletion of nephron progenitors and nascent nephrons. Section immunofluorescence using markers of nephron progenitors (Six2, Pax2), nascent nephrons (Pax2, Lhx1), maturing proximal tubules (LTL) and collecting ducts (AQP2). Dot1LSix2 kidneys show marked downregulation of Six2, Lhx1 (a, b) and Pax2 (c,d) expression. There is a concomitant reduction in LTL-positive proximal tubules and secondary diminution of collecting ducts (e, f). (g) Quantitation of Six2 progenitors was performed using FACS of Six2GFP cells (see methods) and expressed as the percent of GFP+/total cells. (h) Quantitative analysis of nascent nephrons in Dot1LSix2 kidneys at E17.5 (N = 7–8/group). Counts were performed on 3 alternating sections per kidney (n = 4–5 mice per group). RV: renal vesicles; CSB: Comma-shaped nephrons; SSB: S-shaped nephrons; Glom: maturing glomeruli. Means SEM
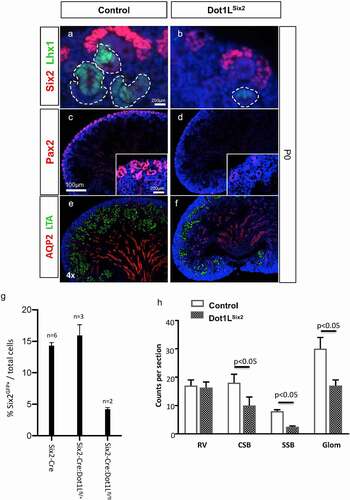
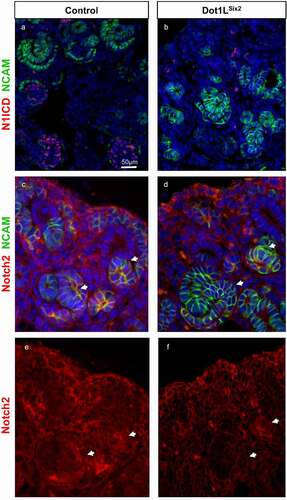
Figure 7. Notch2 is downregulated in nascent nephrons of Dot1LSix2 kidneys. Section immunofluorescence for Notch 1 intracellular domain, Notch2, and NCAM (a marker of induced nephron progenitors and nascent nephrons). Arrows denote nascent nephrons. N = 3–4/group
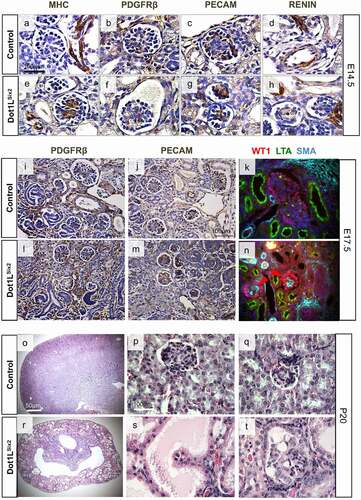
Dot1lSix2 kidneys have abnormal glomerular morphology. Examination of renal microvascular and glomerular morphology revealed abnormal expression of vascular smooth muscle and mesangial cell markers in the mutant Dot1LSix2 kidneys. At E14.5, Myosin Heavy Chain (MHC) is ectopically expressed in the mesangium of mutant glomeruli ( A, E). The spatial expression of PDGFRβ (mesangial cells), PECAM (endothelium) and renin (juxtaglomerular cells) was not altered at E14.5 and E17.5 ( B-H, I-M). At E17.5, small amount of smooth muscle-actin (SMA) is normally expressed in the mesangium of control mice but is substantially induced in the mesangium of Dot1LSix2 glomeruli ( K, N). The podocyte transcription factor WT1 was maintained, although its immunoreactivity appeared more intense in mutant glomeruli ( K, N). At P20, examination of the kidneys of two homozygous mutant survivors showed widespread cystic tubules and glomeruli with microaneurysm formation ( O-T). Since the mesangium is derived embryologically from stromal progenitors (which retain Dot1L/H3K79me2), the observed changes in glomerular/vascular development in mutant kidneys are non-cell autonomous effects.
Figure 8. (a-n) Immunohistochemical analysis of selected glomerular and vascular smooth muscle markers in Dot1LSix2 kidneys. Dot1LSix2 glomeruli show ectopic/enhanced Myosin Heavy Chain (MHC) expression and Smooth Muscle Actin (SMA) in the mesangium at E14.5 and E17.5, respectively. Endothelial and juxtaglomerular cell markers are preserved. (o-t) P20 Dot1LSix2 kidneys exhibit diffuse glomerulocystic disease with microaneurysm formation. (N = 4/group)
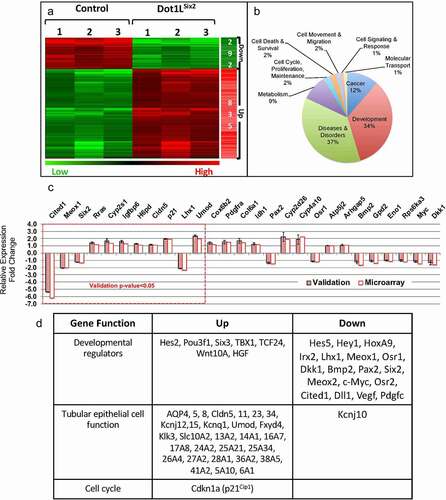
To gain insights into the pathogenesis of the Dot1LSix2 phenotype, we compared the transcriptomes of E16.5 control and Dot1LSix2 kidneys using Agilent 44 K microarray analysis of whole kidney RNA. Using p < 0.05 and 1.5-fold difference cut-offs, we identified 292 downregulated and 835 upregulated transcripts (). The differentially expressed genes clustered in the development, cancer, and diseases and disorders categories (). Validation using the NanoString Platform (n = 3 per genotype) confirmed the directional expression changes (up or down) in 27 developmental and metabolic genes; of these, 11 genes met the statistical significance in both the NanoString and Microarray (). In the upregulated categories, tubular epithelial transporters/channels and the cell cycle inhibitor Cdkn1a (p21Cip1) predominated, whereas in the downregulated categories developmental regulators of nephron progenitor renewal and differentiation and segmental patterning (e.g., Six2, c-Myc, Pax2, Meox1/2, Cited1, Osr1, Osr2, Lhx1, Irx2) were most prominent ().
Figure 9. Transcriptional profiling of whole kidney RNA identifies disrupted developmental and differentiation programmes in E16.5 Dot1LSix2 kidneys. (a,b) Microarray analysis (n = 3 per group). Heatmap and GO analysis of differentially expressed transcripts. Dot1LSix2 mutant kidneys have 2.8-fold increase in upregulated transcripts that cluster in development, cancer and metabolism. (c) NanoString Platform (n = 3 per group) was used to validate differentially expressed genes observed in the microarray. The dotted-red line demarcates those genes that reached statistical significance in both NanoString and microarray. (d) A selected list of transcripts that changed >1.5-fold (p < 0.05) in Dot1LSix2 compared to control kidneys. In general, nephron progenitor genes are downregulated, whereas tubular transport genes are upregulated in Dot1LSix2 kidneys
DISCUSSION
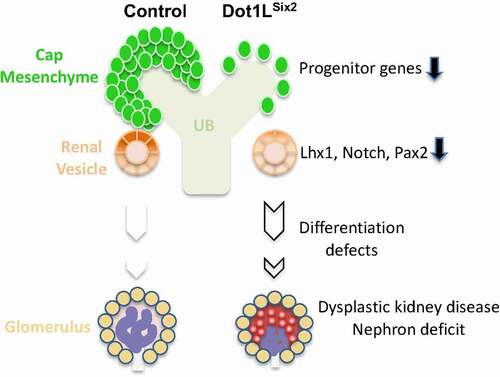
The present study demonstrates that conditional inactivation of the gene encoding the histone H3K79 methyltransferase, Dot1L, impairs kidney development in mice. Interestingly, the full developmental effects of Dot1L inactivation are much more striking upon disruption in the Six2+ lineage than in the Hoxb7+ lineage. Furthermore, this study identifies progenitor genes such as Six2, and differentiation genes such as Pax2, Notch, and Lhx1 as putative Dot1L-H3K79me regulated genes (). Our findings link Dot1L dysfunction to renal dysplasia, a developmental phenotype encountered in syndromic and isolated CAKUT in humans.
Figure 10. A working model depicting defective nephrogenesis programme in Dot1LSix2 mice. Deletion of Dot1L/H3K79me in Six2-positive nephron progenitors results in depletion of Six2 progenitors, reduced expression of genes mediating progenitor epithelial differentiation into nascent nephrons. This results in nephron deficit as well as secondary abnormalities in glomerular maturation
Several lines of evidence support an important and conserved role for Dot1L in embryogenesis. During Drosophila development, H3K79me2 deposition on chromatin is first observed after gastrulation and increases later in epidermal cells during dorsal closure where it is confined to proliferating cells [Citation38]. Mutagenesis of the fly ortholog of Dot1, Grappa, causes phenotypes reminiscent of both Polycomb Group (PcG) and Trithorax Group (TrxG) mutants [Citation38]. PcG and TrxG mediate the deposition of repressive (H3K27me3) and activating (H3K4me3) histone marks, respectively. Another function of Dot1 in the fly is regulation of the Wingless target genes, senseless and frizzled 3 [Citation38]. Knockdown of Dot1L in Xenopus Tropicalis induces a growth retardation phenotype at the tadpole stage [Citation39]. Similar to observations in the fly, during mouse embryogenesis high levels of H3K79me2 are not detectable until the blastocyst stage [Citation40]. Germline disruption of Dot1L in mice causes a severe vascular phenotype, anaemia, and embryonic lethality [Citation14,Citation15]. A dilated cardiomyopathy also occurs following cardiac myocyte-specific inactivation of Dot1L [Citation17]. In these studies, Dot1L was found to directly methylate the Dystrophin gene. Dystrophin is required of cardiac muscle contraction/relaxation and integrity, and mutations in Dystrophin cause Duchenne Muscular Dystrophy (DMD), also known to cause a dilated cardiomyopathy. Interestingly, patients with DMD expresses lower levels of Dot1L than normal [Citation17]. In contrast to the importance of Dot1L in mammalian cardiac development, Dot1L disruption and elimination of H3K79me2 in Lgr5+ intestinal stem cells or in whole intestinal epithelium had little if any effect on animal survival, intestinal functions or structure (except for an increase in apoptosis) [Citation18]. Moreover, while genomic H3K79me2 occupancy correlated with gene transcription in general, it did not correlate with expression of Wnt-responsive genes [Citation18]. These findings questioned the role of Dot1L in regulation of mammalian tissue homoeostasis.
The present study demonstrates that targeted inactivation of Dot1L-H3K79 methyltransferase activity in the ureteric bud lineage is dispensable for branching morphogenesis and formation of the renal collecting system. This finding was somewhat surprising given the strong phenotype we observed as a result of Dot1L disruption in the Six2 lineage. It is conceivable that lack of branching defects in Dot1LHoxb7 mutants may be compensated for by activation of other pathways, and does not rule out a role for Dot1L in collecting duct cell regeneration following injury in the mature kidney, as recently shown [Citation41]. To our knowledge, the role of Dot1L in other branching organs such as the lungs, salivary gland, and pancreas is unknown.
The present study demonstrates that unlike the ureteric bud lineage, reduced H3K79 methylation in the Six2 nephron progenitor lineage results in a congenital dysplastic kidney disease. Developing Dot1LSix2 kidneys have thinning of the cap mesenchyme, fewer nascent nephrons, and dilated tubules predominantly of proximal tubular origin. Dot1LSix2 nephrons also manifest glomerular defects, such as disrupted glomerular tuft, aneurysms and aberrant mesangial expression of MHC and SMA. Transcriptional profiling showed reduced expression of Notch2 which is important for tubular epithelial differentiation [Citation42]. Although Notch signaling is dispensable during terminal differentiation of podocytes [Citation43], other studies suggest that Notch signaling is required for development of podocytes prior to the S-shaped stage [Citation44]. Thus, defective Notch signaling may be partly responsible for the glomerular abnormalities in the Dot1LSix2 mutants. However, it is not possible at this time to determine whether the changes in Notch2 are primary or secondary to the deficit in nephron progenitors.
As mentioned earlier in the Results, the gene targeting strategy employed in this study may result in generation of a shorter Dot1L protein which can have neomorphic effects in addition to loss of histone methyltransferase activity and this should be kept in mind in the interpretation of the phenotype.
Dot1L mutations have not been described in humans with congenital anomalies of the kidney and urinary tract (CAKUT). However, in this study, many of the CAKUT genes are dysregulated in the Dot1LSix2 mutants (e.g., Six2, Pax2, and Notch). Only 20–30% of human CAKUT can be attributed to monogenic mutations. The remaining cases are presumably secondary to undiscovered mutations and gene-environment interactions, in which epigenetic influences play a greater role. The functional link between Dot1L-H3K79me and human CAKUT is worthy of further investigations.
Supplemental Material
Download Zip (49.3 MB)Acknowledgments
We thank the Tulane Renal and Hypertension of Excellence for use of Core facilities. This work is supported by National Institutes of Health Grant RO1 DK114050 and P50 DK096373. These studies constituted the dissertation thesis for Fenglin Wang in the Biomedical Sciences Graduate as well as in Program at Tulane School of Medicine.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Guo C, Morris SA. Engineering cell identity: establishing new gene regulatory and chromatin landscapes. Curr Opin Genet Dev. 2017;46:50–57.
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080.
- Paksa A, Rajagopal J. The epigenetic basis of cellular plasticity. Curr Opin Cell Biol. 2018;49:116–122.
- Harikumar A, Meshorer E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015;16:1609–1619.
- Tchasovnikarova IA, Kingston RE. Beyond the Histone code: A physical map of chromatin states. Mol Cell. 2018;69:5–7.
- Liu H, Chen S, Yao X, et al. Histone deacetylases 1 and 2 regulate the transcriptional programs of nephron progenitors and renal vesicles. Development. 2018;145. DOI:https://doi.org/10.1242/dev.153619
- Zhang L, Ettou S, Khalid M, et al. EED, a member of the polycomb group, is required for nephron differentiation and the maintenance of nephron progenitor cells. Development. 2018;145. DOI:https://doi.org/10.1242/dev.157149
- Vlaming H, van Leeuwen F. The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma. 2016;125:593–605.
- Wong M, Polly P, Liu T. The histone methyltransferase DOT1L: regulatory functions and a cancer therapy target. Am J Cancer Res. 2015;5:2823–2837.
- Steger DJ, Lefterova MI, Ying L, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839.
- Bernt KM, Armstrong SA. A role for DOT1L in MLL-rearranged leukemias. Epigenomics. 2011;3:667–670.
- FitzGerald J, Moureau S, Drogaris P, et al. Regulation of the DNA damage response and gene expression by the Dot1L histone methyltransferase and the 53Bp1 tumour suppressor. PLoS One. 2011;6:e14714.
- Kim W, Choi M, Kim JE. The histone methyltransferase Dot1/DOT1L as a critical regulator of the cell cycle. Cell Cycle. 2014;13:726–738.
- Feng Y, Yang Y, Ortega MM, et al. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood. 2010;116:4483–4491.
- Jones B, Su H, Bhat A, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4:e1000190.
- Cattaneo P, Kunderfranco P, Greco C, et al. DOT1L-mediated H3K79me2 modification critically regulates gene expression during cardiomyocyte differentiation. Cell Death Differ. 2016;23:555–564.
- Nguyen AT, Xiao B, Neppl RL, et al. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011;25:263–274.
- Ho LL, Sinha A, Verzi M, et al. DOT1L- mediated H3K79 methylation in chromatin is dispensable for Wnt pathway-specific and other intestinal epithelial functions. Mol Cell Biol. 2013;33:1735–1745.
- Xiao Z, Chen L, Zhou Q, et al. Dot1l deficiency leads to increased intercalated cells and upregulation of V-ATPase B1 in mice. Exp Cell Res. 2016;344:167–175.
- Adli M, Parlak M, Li Y, et al. Epigenetic States of nephron progenitors and epithelial differentiation. J Cell Biochem. 2015;116(6):893–902.
- Beckerman P, Ko YA, Susztak K. Epigenetics: a new way to look at kidney diseases. Nephrol Dial Transplant. 2014;29:1821–1827.
- Chen S, Yao X, Li Y, et al. Histone deacetylase 1 and 2 regulate Wnt and p53 pathways in the ureteric bud epithelium. Development. 2015;142:1180–1192.
- Hilliard S, Song R, Liu H, et al. Defining the dynamic chromatin landscape of mouse nephron progenitors. Biol Open. 2019;8. DOI:https://doi.org/10.1242/bio.042754
- Majumder S, Thieme K, Batchu SN, et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J Clin Invest. 2018;128:483–499.
- McLaughlin N, Wang F, Saifudeen Z, et al. In situ histone landscape of nephrogenesis. Epigenetics. 2014;9:222–235.
- McLaughlin N, Yao X, Li Y, et al. Histone signature of metanephric mesenchyme cell lines. Epigenetics. 2013;8:970–978.
- Thomas MC. Epigenetic Mechanisms in Diabetic Kidney Disease. Curr Diab Rep. 2016;16:31.
- Wanner N, Bechtel-Walz W. Epigenetics of kidney disease. Cell Tissue Res. 2017;369:75–92.
- Kobayashi A, Valerius MT, Mugford JW, et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181.
- Zhao H, Kegg H, Grady S, et al. Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol. 2004;276:403–415.
- Hilliard S, Aboudehen K, Yao X, et al. Tight regulation of p53 activity by Mdm2 is required for ureteric bud growth and branching. Dev Biol. 2011;353:354–366.
- Costantini F, Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell. 2010;18:698–712.
- Kopan R, Chen S, Little M. Nephron progenitor cells: shifting the balance of self- renewal and differentiation. Curr Top Dev Biol. 2014;107:293–331.
- Little MH, Combes AN, Takasato M. Understanding kidney morphogenesis to guide renal tissue regeneration. Nat Rev Nephrol. 2016;12:624–635.
- Patel SR, Dressler GR. The genetics and epigenetics of kidney development. Semin Nephrol. 2013;33:314–326.
- Srinivas S, Goldberg MR, Watanabe T, et al. Expression of green fluorescent protein in the ureteric bud of transgenic mice: a new tool for the analysis of ureteric bud morphogenesis. Dev Genet. 1999;24:241–251.
- McMahon AP. Development of the Mammalian kidney. Curr Top Dev Biol. 2016;117:31–64.
- Shanower GA, Muller M, Blanton JL, et al. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics. 2005;169:173–184.
- Wen L, Fu L, Guo X, et al. Histone methyltransferase Dot1L plays a role in postembryonic development in Xenopus tropicalis. Faseb J. 2015;29:385–393.
- Ooga M, Inoue A, Kageyama S, et al. Changes in H3K79 methylation during preimplantation development in mice. Biol Reprod. 2008;78:413–424.
- Zhang L, Chen L, Gao C, et al. Loss of Histone H3 K79 methyltransferase Dot1l facilitates kidney fibrosis by upregulating endothelin 1 through Histone deacetylase 2. J Am Soc Nephrol. 2020;31:337–349.
- Chung E, Deacon P, Park JS. Notch is required for the formation of all nephron segments and primes nephron progenitors for differentiation. Development. 2017;144:4530–4539.
- Waters AM, Wu MY, Onay T, et al. Ectopic notch activation in developing podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2008;19:1139–1157.
- Cheng HT, Kopan R. The role of Notch signaling in specification of podocyte and proximal tubules within the developing mouse kidney. Kidney Int. 2005;68:1951–1952.