
ABSTRACT
Although the mechanism of DNA demethylating drugs has been understood for many years, the direct effect of these drugs on methylation of the complementary strands of DNA has not been formally demonstrated. By using hairpin-bisulphite sequencing, we describe the kinetics and pattern of DNA methylation following treatment of cells by the DNA methyltransferase 1 (DNMT1) inhibitor, decitabine. As expected, we demonstrate complete loss of methylation on the daughter strand following S-phase in selected densely methylated genes in synchronized Jurkat cells. Thereafter, cells showed a heterogeneous pattern of methylation reflecting replication of the unmethylated strand and restoration of methylation.
KEYWORDS:
Introduction
Decitabine and azacytidine are synthetic nucleoside analogues of cytosine used as demethylating anti-cancer drugs in the treatment of acute myeloid leukaemia and myelodysplastic syndrome [Citation1–5]. These demethylating drugs are incorporated into DNA during DNA replication or S-phase of the cell cycle, and irreversibly bind and inactivate DNA methyltransferase 1 (DNMT1) thereby preventing methylation of the newly synthesized strand [Citation6–8].
Although there is compelling evidence for this mechanism of decitabine and azacytidine-induced demethylation, it has not been directly observed through sequencing of the complementary DNA strands at the time of replication. Hairpin-bisulphite sequencing, in which complementary DNA strands are linked together prior to sodium bisulphite treatment, provides a method by which the demethylation and re-methylation of DNA can be observed and monitored following exposure of cells to demethylating agents [Citation9–11].
Our hairpin protocol incorporated a random ‘barcode’ composed of 14 G, A, or T nucleotides within the loop of the hairpin linker, giving 4.8 million possible combinations. By using high throughput barcoded hairpin-bisulphite sequencing we generated thousands of unique barcoded sequences to examine the pattern and kinetics of methylation at two densely methylated gene promoters.
Methods
Treatment of synchronized cells with decitabine
Jurkat and MOLT4 (T lymphoblastic leukaemia) and NALM6 (B lymphoblastic leukaemia) cell line cells were grown to 1 x 106/mL in RPMI-Glutamax media (Gibco, ThermoFisher) supplemented with 10% heat inactivated foetal calf serum, then incubated with 2 mM thymidine for 18 h to arrest the cell cycle at the G1/S boundary. Following the thymidine block, DNA synthesis was reinitiated (designated as time 0 h) by replacement with media containing 50 µM deoxycytidine [Citation12]. Decitabine was added to the media at time 0 h; Jurkat cells were incubated with 5 µM decitabine, whereas MOLT4 and NALM6 were incubated with 1 µM decitabine. Cells were collected at 0, 2, 4, 6, 8, 24, 48 and 72 h for DNA extraction (Qiagen QIAamp DNA Blood MinikitTM) and flow cytometric cell cycle analysis. To assess cell cycle progression, cells were fixed with 70% ethanol, stained with FxCycleTM PI/RNase (Thermo Fisher Scientific) kit, and ploidy was measured by flow cytometry. Flow cytometry data were analysed with FlowJo® 10.4.2 software (FlowJo, LLC). For the 120 h culture, media was replaced with non-decitabine-containing media at 48 h and at 96 h.
Low coverage bisulphite sequencing (post-bisulphite adapter tagging)
Global 5-mC levels were determined using post-bisulphite adapter tagging (PBAT) sequencing as previously described [Citation13](Ortega, Peat and Hore, 2020 in press). Briefly, 100 ng of purified DNA was bisulphite converted and first strand synthesis was performed with a biotin-labelled adapter sequence possessing seven random nucleotides at its 3ʹ end (BioP5N7, ) followed by purification using streptavidin-coated Dynabeads (Thermo Fisher Scientific, 11205D) and magnetic immobilization. Immobilized first strand was used as the template for second strand synthesis using an adapter with seven random nucleotides at the 3ʹ end (P7N7, ), to create double-stranded DNA. Finally, a unique molecular barcode and the Illumina adapter sequences were added by PCR (98°C for 5 min; 13 cycles of 98°C for 80 sec, 65°C 30 sec and 72°C 30 sec), using the KAPA HiFi Uracil Plus system. Libraries were sequenced on a MiSeq instrument (Illumina), the first 10 bp of each read was trimmed along with adapter and poor quality sequences using TrimGalore, and remaining reads were mapped to the human genome (NCBI build 37) using Bismark [Citation14]. The resulting CG methylation calls were extracted from the Bismark mapping report, and the margin of error calculated from this sample using a standard asymptotic estimator [Citation13] (Ortega, Peat and Hore, 2020 in press).
Table 1. Primers and linkers
Generation of bisulphite converted DNA hairpin molecules
Two genes, RASSF1 and PCDHGA12, whose promoters were reported to be densely methylated in Jurkat cells [Citation15–17] were studied. The dense methylation of these promoters was confirmed by targeted bisulphite sequencing. The hairpin-bisulphite technique [Citation10] was modified to document methylation of the complementary strands simultaneously. A random 14-bp molecular barcode sequence (G, A, and T) was included in the hairpin loop. To ligate the hairpins, Jurkat cell DNA was digested with either SacI (for RASSF1) or BamHI (for PCDHGA12) and 400 ng was ligated to 5 μM hairpin linkers () using T4 DNA ligase (ThermoFisher Scientific). Hairpin-ligated DNA was bisulphite-converted using EZ DNA Methylation-GoldTM (Zymo Research). Following conversion, hairpins were PCR-amplified using primers complementary to the bisulphite-converted forward and reverse strands (). First round primers were tagged to enable addition of indexed Illumina adaptor sequences in a second round of PCR (). Samples were pooled, cleaned to remove primer-dimer and short non-specific amplification products using Ampure XP beads, diluted to 1 ng/µL and assessed using a Bioanalyzer (Agilent Technology, Germany).
Figure 1. Hairpin amplification, sequencing and processing. A. Following bisulphite conversion, the region of interest was amplified using tagged specific primers. A second round of PCR was used to label the samples with indexed Illumina primers. B. Summary of the analysis of methylation data. The methylation of each CpG is displayed and the status of each hairpin is interpreted after ‘folding’ the complimentary strands
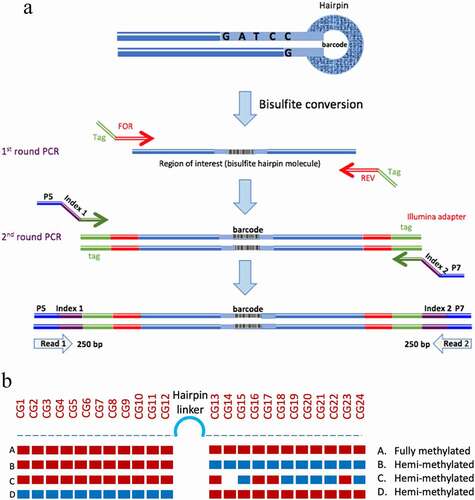
Sequencing of hairpin libraries
The pooled PCR products were sequenced on an Illumina MiSeq System using Nano kit V2 500 bp to generate 2 × 250 bp paired-end reads [Citation18]. Based on index sequences the paired-end sequence files were demultiplexed and saved as two sequence files (fastq.gz format) for each sample (read 1 and read 2). MiSeq sequence output files were uploaded to Galaxy online platform (usegalaxy.org). Paired-end sequence files were joined with PEAR and FASTQC was used to check the quality of the joined sequence, with poor-quality sequences (average Phred quality score <30) being removed. The ‘Barcode collapser’ program was used to identify and remove reads containing duplicate barcodes, so that only unique reads were retained. These unique reads were then imported to ‘BiQ Analyzer HT’ [Citation19] for analysis and visualization.
Processing and ‘folding’ hairpin sequences
To calculate the percentages of hemi-methylated hairpin reads, the binary methylation data (1, 0 or x) were extracted from the BiQ Analyzer HT output ‘methylation.tsv’ result files. Using R (www.r-project.org), the data were analysed after ‘folding’ the hairpin reads to calculate the methylation proportion of each hairpin read (). The hairpin reads were classified based on the methylation pattern of the complementary CpGs sites. Sequence reads were classified as ‘methylated’ when the proportion of methylated complementary CpGs ≥0.51; ‘hemi-methylated’ if the proportion of hemi-methylated CpGs ≥0.51 and ‘unmethylated’ if the proportion of unmethylated CpGs ≥0.51. A small proportion of reads (<1%) remained unclassified.
Results
Effect of decitabine on cell cycle progression
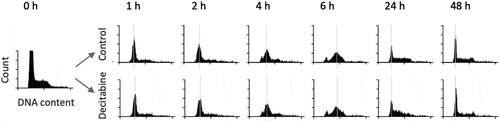
To study the effect of decitabine on DNA methylation, Jurkat cells were first synchronized using a single thymidine block. Asynchronous Jurkat cells showed a predominance of cells in G1/G0 phase, along with some S-phase and G2 phase cells (). Thymidine-blocked Jurkat cells showed a larger proportion of cells in G1, with a significant reduction in G2 cells consistent with cell cycle block at G1/S (, 0 h), as expected. Following release, the majority of synchronized cells progressed through S-phase (4 h) and into S/G2 phase (6 h).
To determine the effect of decitabine on cell growth and cell cycle progression, synchronized Jurkat cells were treated with a single dose of decitabine (5 μM) at release and incubated for 72 h. These results demonstrate that decitabine had little to no effect on cell cycle kinetics, although we did observe a small difference in cell distribution in S phase at 24 h suggesting mild retardation of cell division in decitabine-treated cells, but this difference was no longer apparent at 48 h ().
Figure 2. DNA ploidy analysis of Jurkat cells following release from thymidine block. The upper series shows untreated Jurkat cells whereas the lower series shows cells treated with 5 μM decitabine. DNA content of cells was measured by propidium iodine staining
Impact of decitabine on global genomic DNA methylation in Jurkat cells
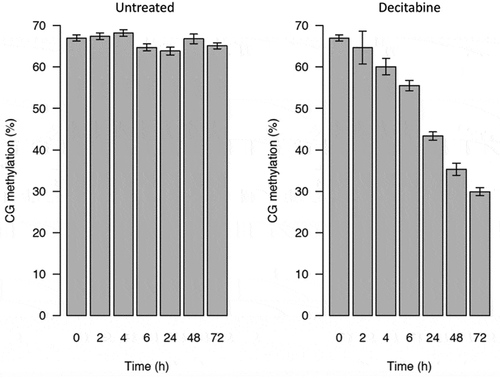
The genome-wide DNA methylation of the Jurkat cells treated with 5 µM decitabine was determined by using low-coverage bisulphite sequencing [Citation13](Ortega, Peat and Hore, 2020 in press). Samples had mapping efficiencies of 53–67%, and the number of analysable CpGs ranged from 4000 to 31 000. Apparent methylation at non-CpG sites was less than 1%, indicating that the bisulphite conversion was at least 99% efficient.
The levels of CpG methylation were the same in non-blocked (67%) and synchronized Jurkat cells (68%). As expected, there was a gradual loss of CpG methylation in Jurkat cells treated with decitabine in a time-dependent manner following release from cell cycle arrest (from 68% in arrested Jurkat cells to 30% in decitabine-treated cells at 72 h) ().
Figure 3. Global CpG methylation levels of decitabine-treated and untreated Jurkat cells measured by low-coverage bisulphite sequencing. The error bars indicate the theoretical 99.5% confidence intervals of the methylation value (see methods)
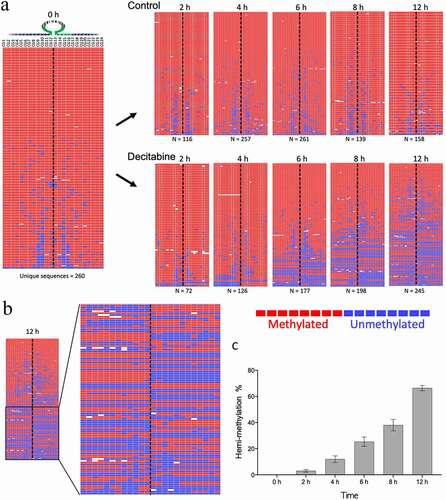
Decitabine-induced hemi-methylation in the PCDHGA12 promoter.
Methylation of the PCDHGA12 promoter was investigated in synchronized Jurkat cells treated with decitabine for 12 h. Decitabine induced a gradual increase in hemi-methylated DNA in the PCDHG12 promoter as early as 2 h after release from synchronization (). Hemi-methylation of reads, where one side of the hairpin is methylated and the other side is unmethylated, was clearly observed ( shows a magnification of the 12 h timepoint from ). The percentage of fully methylated, hemi-methylated and unmethylated CpGs sites for each sample was determined. By 12 h the percentage of hemi-methylated reads was 65% ().
Figure 4. Example of decitabine-induced hemi-methylation in Jurkat cells. A. The left panel shows the almost fully methylated PCDHGA12 promoter at time 0 h. The right panels show the development of hemi-methylation of the PCDHGA12 promoter in decitabine-treated cells 2 to 12 h after initiation of DNA replication. Each line is a unique sequence read; the number of unique sequences is shown at the bottom of each heatmap. B. Enlargement of the lower portion of the heatmap of the decitabine-treated cells at 12 h to show the hemi-methylated reads. The dotted line indicates the location of the hairpin barcode. The heatmaps in A and B are outputs from BiQ Analyzer HT. Red = methylated CpG, blue = unmethylated CpG, white = non-aligned sequence. C. Percentage of hemi-methylated sequences in the PCDHGA12 promoter in decitabine-treated Jurkat cells following initiation of DNA replication. The data presented are means of three replicates ± SEM. The level of hemi-methylation in the untreated controls was <1% at all time points
Kinetics of methylation up to 120 h after decitabine
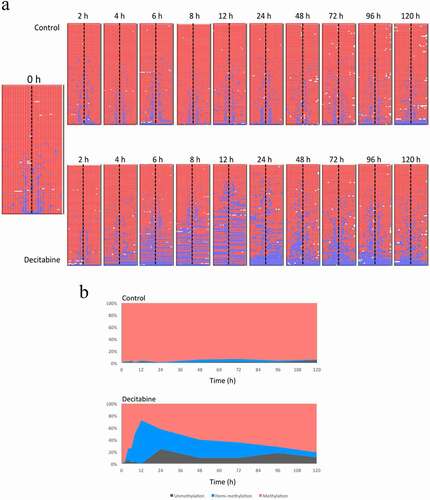
To investigate the effects of a single decitabine treatment (at time 0 h) on long-term methylation, we repeated the experiment and collected cells at 0, 2, 4, 6, 8 and 12, 24, 48, 72, 96 and 120 h post-release from the thymidine block. Media was replaced with non-decitabine containing media at 48 h and at 96 h.
As before, we observed a decrease in methylation of the PCDHGA12 promoter in decitabine-treated cells and this was associated with an increase in the proportion of hemi-methylated reads (). The amount of hemi-methylation reached 65% at 12 h. From 24 h, we observed increasing levels of methylation and loss of hemi-methylation, consistent with re-methylation of the PCDHGA12 promoter.
Figure 5. Demethylation and re-methylation of the PCDHGA12 promoter in Jurkat cells. A. Methylation pattern of hairpin sequence reads from untreated and decitabine-treated cells for up to 120 h after initiation of DNA replication. The heatmaps are outputs from BiQ Analyzer HT. Red = methylated CpG, blue = unmethylated CpG, white = missing sequence. B. Distribution of methylation, hemi-methylation and unmethylation in untreated and decitabine-treated cells as a function of time
Decitabine-induced hemi-methylation in the RASSF1 promoter
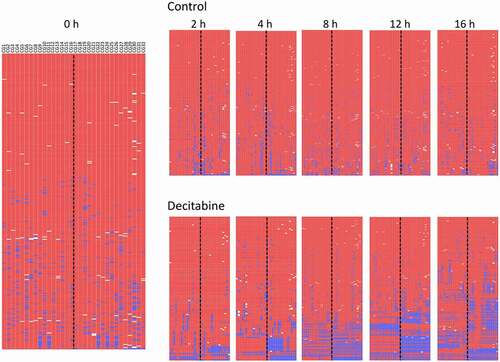
To confirm the changes seen in PCDHGA12, the methylation of the RASSF1 promoter was similarly examined in Jurkat cells following decitabine treatment. Similar to PCDHGA12, the RASSF1 promoter was shown to be densely methylated in untreated cells. Jurkat cells showed increasing decitabine-induced hemi-methylation of the RASSF1 promoter with time, whereas controls remained fully methylated (). The extent of hemi-methylation was usually less than that seen for the PCDHGA12.
Figure 6. Methylation of RASSF1 promoter in Jurkat cells treated with decitabine compared to untreated cells. Heatmap output of BiQ Analyzer HT
Impact of decitabine on methylation in MOLT4 and NALM6 cell lines
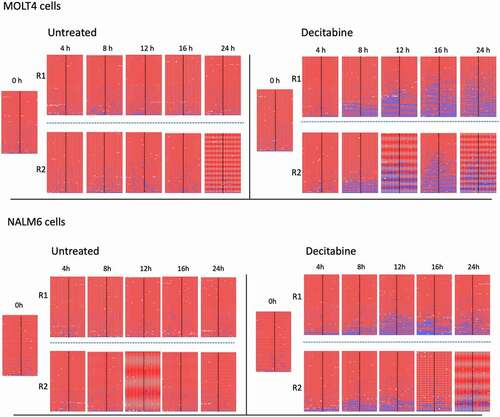
To confirm that the observations from Jurkat cells occur similarly in other cell lines we examined the effect of decitabine on MOLT4 and NALM6 cells. The PCDHGA12 promoter was densely methylated in untreated MOLT4 and NALM6 cell lines, with overall >95% mean methylation (). As seen with Jurkat cells, synchronized MOLT4 and NALM6 cells treated with 1 µM decitabine also showed an increase in the proportion of hemi-methylated sequences at increasing timepoints whereas untreated cells did not show hemi-methylation ().
Figure 7. Methylation of PCDHGA12 promoter in decitabine-treated MOLT4 and NALM6 cells. Biological replicates (R1 and R2) are shown for each condition
Discussion
The mechanism of the demethylating drugs, as specific inhibitors of DNMT1, was demonstrated many years ago [Citation6–8,Citation20,Citation21]. Here, with the use of hairpin-bisulphite sequencing, we have directly observed the pattern and kinetics of methylation following DNMT1 inhibitor treatment. Using two genes that are highly methylated in Jurkat cells, we demonstrate decitabine-induced DNA hemi-methylation in densely methylated loci within 2 h of release from G1-arrest, with increasing levels of hemi-methylation as DNA replication proceeds. In most hemi-methylated sequences the newly synthesized DNA strand was completely devoid of methylation. Even though these results were predicted by previous studies, decitabine-induced hemi-methylation has not been documented in such detail.
Since low coverage global bisulphite sequencing demonstrated a similar degree of demethylation on a global scale, we conclude that hemi-methylation demonstrated at these two genes is recapitulated throughout the genome.
We predicted that nearly all cells would show hemi-methylated DNA sequences 6–12 h after exposure to decitabine, as nearly all cells were initiated into the S phase by then. We observed, however, 15 ~ 20% fully methylated hairpin reads at 12 h, representing cells that either did not undergo demethylation, or did not divide. Incomplete demethylation might be due to replication of DNA prior to depletion of available DNMT1 activity, immediate repair of demethylated strands by de novo methylases, or the presence of dead or non-replicating cells. It is unclear how long the effect of a single addition of decitabine will last since this reflects the effects of degradation of decitabine and cellular uptake. In RPMI 1640 media (pH 7.2) containing calf serum, the half-life of decitabine at 37°C was reported to be 3.5 h [Citation22]. However, intracellular decitabine levels continued to rise for at least 9 hours and incorporation into DNA reached a maximum at about 72 h [Citation23]. We observed maximal demethylation 12–16 h after a single addition of decitabine to the media. By 24 h many cells will have entered S phase for the second time giving rise to the heterogeneous pattern of DNA methylation observed; some cells had lost methylation of the remaining strand leading to unmethylated hairpins, whereas others appeared to have acquired methylation, presumably through re-methylation of the previously demethylated strand.
With the power of our hairpin-bisulphite sequencing technique, our results describe in detail the kinetics and pattern of DNA demethylation induced by a single exposure of decitabine in leukaemia cell lines. These results show that decitabine induced DNA hemi-methylation in PCDHGA12 and RASSF1 promoters as early as 2 h after treatment, that DNA hemi-methylation levels increased as DNA replication proceeded, but that by 24 h methylation levels begin to return to pre-exposure levels.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Crawford DJ, Liu MY, Nabel CS, et al. Tet2 catalyzes stepwise 5-methylcytosine oxidation by an iterative and de novo mechanism. J Am Chem Soc. 2016;138(3):730–733.
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–569.
- Silverman LR, Holland JF, Weinberg RS, et al. Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia. 1993;7(Suppl 1):21–29.
- Vogler WR, Miller DS, Keller JW. 5-Azacytidine (NSC 102816): a new drug for the treatment of myeloblastic leukemia. Blood. 1976;48(3):331–337.
- Zagonel V, Lo Re G, Marotta G, et al. 5-Aza-2ʹ-deoxycytidine (decitabine) induces trilineage response in unfavourable myelodysplastic syndromes. Leukemia. 1993;7(Suppl 1):30–35.
- Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2ʹ-deoxycytidine. J Biol Chem. 1982;257(4):2041–2048.
- Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93.
- Jones PA, Taylor SM. Hemimethylated duplex DNAs prepared from 5-azacytidine-treated cells. Nucleic Acids Res. 1981;9(12):2933–2947.
- Arand J, Wossidlo M, Lepikhov K, et al. Selective impairment of methylation maintenance is the major cause of DNA methylation reprogramming in the early embryo. Epigenetics Chromatin. 2015;8(1):1.
- Laird CD, Pleasant ND, Clark AD, et al. Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc Nat Acad Sci. 2004;101(1):204–209.
- Miner BE, Stoger RJ, Burden AF, et al. Molecular barcodes detect redundancy and contamination in hairpin-bisulfite PCR. Nucleic Acids Res. 2004;32(17):e135.
- Chen G, Deng X. Cell synchronization by double thymidine block. Biol Protoc. 2018;8:17.
- Peat JR, Ortega-Recalde O, Kardailsky O, et al. The elephant shark methylome reveals conservation of epigenetic regulation across jawed vertebrates. F1000Res. 2017;6:526.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27(11):1571–1572.
- Gordon M, El-Kalla M, Baksh S. RASSF1 polymorphisms in cancer. Mol Biol Int. 2012;(2012:365213.
- Milani L, Lundmark A, Kiialainen A, et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood. 2010;115(6):1214–1225.
- Taylor KH, Pena-Hernandez KE, Davis JW, et al. Large-scale CpG methylation analysis identifies novel candidate genes and reveals methylation hotspots in acute lymphoblastic leukemia. Cancer Res. 2007;67(6):2617–2625.
- Quail MA, Swerdlow H, Turner DJ. Improved protocols for the illumina genome analyzer sequencing system. Curr Protoc Hum Genet, Chapter. 2009;18:Unit 18.12.
- Lutsik P, Feuerbach L, Arand J, et al. BiQ analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res. 2011;39:W551–556. (Web Server issue)
- Friedman S. The inhibition of DNA(cytosine-5)methylases by 5-azacytidine. the effect of azacytosine-containing DNA. Mol Pharmacol. 1981;19(2):314–320.
- Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A. 1984;81(22):6993–6997.
- Hajek M, Votruba I, Holy A, et al. Alpha anomer of 5-aza-2ʹ-deoxycytidine down-regulates hTERT mRNA expression in human leukemia HL-60 cells. Biochem Pharmacol. 2008;75(4):965–972.
- Öz S, Raddatz G, Rius M, et al. Quantitative determination of decitabine incorporation into DNA and its effect on mutation rates in human cancer cells. Nucleic Acids Res. 2014;42(19):e152.