
ABSTRACT
Because ADAM17 promotes colonic tumorigenesis, we investigated potential miRNAs regulating ADAM17; and examined effects of diet and tumorigenesis on these miRNAs. We also examined pre-miRNA processing and tumour suppressor roles of several of these miRNAs in experimental colon cancer. Using TargetScan, miR-145, miR-148a, and miR-152 were predicted to regulate ADAM17. miR-143 was also investigated as miR-143 and miR-145 are co-transcribed and associated with decreased tumour growth. HCT116 colon cancer cells (CCC) were co-transfected with predicted ADAM17-regulating miRNAs and luciferase reporters controlled by ADAM17-3’UTR. Separately, pre-miR-143 processing by colonic cells was measured. miRNAs were quantified by RT-PCR. Tumours were induced with AOM/DSS in WT and transgenic mice (Tg) expressing pre-miR-143/miR-145 under villin promoter. HCT116 transfection with miR-145, −148a or −152, but not scrambled miRNA inhibited ADAM17 expression and luciferase activity. The latter was suppressed by mutations in ADAM17-3’UTR. Lysates from colonocytes, but not CCC, processed pre-miR-143 and mixing experiments suggested CCC lacked a competency factor. Colonic miR-143, miR-145, miR-148a, and miR-152 were downregulated in tumours and more moderately by feeding mice a Western diet. Tg mice were resistant to DSS colitis and had significantly lower cancer incidence and tumour multiplicity. Tg expression blocked up-regulation of putative targets of miR-143 and miR-145, including ADAM17, K-Ras, XPO5, and SET. miR-145, miR-148a, and miR-152 directly suppress colonocyte ADAM17 and are down-regulated in colon cancer. This is the first direct demonstration of tumour suppressor roles for miR-143 and miR-145 in an in vivo model of colonic tumorigenesis.
Introduction
Colonic tumorigenesis involves genetic and epigenetic changes that lead to progressive loss of growth control. Recent advances in exome sequencing, DNA copy number analysis, and quantification of promoter methylation and messenger RNA and microRNA expression have uncovered an increasingly complex picture of deregulated genes driving colonic tumorigenesis [Citation1]. MicroRNAs (miRNAs) are short non-coding RNAs (~21 bp) that regulate gene expression by base pairing to 3’-untranslated regions (3’UTR) of target genes. miRNAs are transcribed as primary miRNA (pri-miRNA) by RNA polymerase II and processed in the nucleus to precursor miRNA (pre-miRNA) by Drosha, an RNase III enzyme, and then transported by exportin5 (XPO5) to the cytoplasm. In the cytoplasm Dicer, another RNase III enzyme, cleaves pre-miRNA, releasing mature miRNA. The mature miRNA is directed to the microRNA-induced silencing complex (miRISC complex) where the complementary base pairing of miRNA seed region (conserved sequence near the miRNA 5´-end) to the 3’UTR of mRNA target leads to mRNA destabilization or stalling of mRNA translation [Citation2]. Numerous studies have demonstrated that expression levels of miRNAs are deregulated in diverse tumours including colon cancer [Citation3–7]. The growth phenotypes induced by changes in miRNAs that mimic changes that occur in tumours suggest that some miRNAs, when upregulated, function as oncogenes, whereas others, when downregulated, function as tumour suppressors in cancer [Citation7]. Data supporting such conjectures were provided by tumour xenografts derived from colon cancer cells (CCC) bioengineered to overexpress or suppress specific miRNAs [Citation8,Citation9].
We previously reported that metalloproteinase ADAM17 is upregulated in colon cancer and exerts a tumour promoter role [Citation10]. We also showed that the Western diet increased ADAM17 [Citation10]. To uncover potential tumour suppressor miRNAs that inhibit ADAM17, here we report a bioinformatic search that identified miR-145, miR-148a, miR-152, and miR-338 as putative ADAM17 regulators using a publicly available miRNA dataset [Citation11,Citation12]. Because miR-145 is polycistronic and co-transcribed with miR-143, we also chose to include miR-143 in our analysis. Both miR-143 and miR-145 were identified as down-regulated in human colon cancer and subsequently in many other epithelial cancers suggesting their tumour suppressor function [Citation3,Citation13,Citation14]. In the current study, we hypothesized that the Western diet or tumorigenesis might suppress expression levels of miRNAs that are predicted inhibitors of ADAM17. We also examined stromal cells and colonocytes separately for these miRNAs to begin to understand their cell-specific roles. Since pre-miR-143 appeared to be maintained, whereas mature miR-143 was reduced in human colon cancers [Citation3], we asked if lysates from colon cancer cells were able to process pre-miR-143 to mature miR-143 and compared processing activity in CCC to normal colonocytes.
Finally, our laboratory and other investigators have demonstrated that miR-143 or miR-145 suppressed tumour xenograft growth when transfected into fully transformed colon cancer cells [Citation8,Citation15,Citation16]. No studies to date, however, have addressed whether maintaining these miRNAs in non-transformed colonocytes could suppress the development of endogenous colon tumours. To address this question, we expressed pre-miR-143 and pre-miR-145 as transgenes under a villin-promoter in colonocytes and examined their effects in a model of colitis-associated colon cancer. Results of these in vitro and in vivo studies and a discussion of their implications are the focus of this report.
Materials and methods
Materials
Defined standard diet (Std diet, 5% fat) and Western diet (WD, 20% fat) were obtained from Harlan Teklad (Madison, WI) [Citation17]. Azoxymethane was obtained from Midwest Research (Kansas City, MO), the NCI Chemical Carcinogen Reference Standard Repository. ADAM17 3’UTR vector was purchased from Switchgears Genomics (Menlo Park CA, 94,025). Antibodies included rabbit polyclonal ADAM17 (Abcam Catalogue: ab2051); rabbit polyclonal anti-Dicer antibodies (Abcam ab227518); rabbit polyclonal K-Ras (Santa Cruz Catalogue: SC-522); mouse monoclonal SET (Santa Cruz SC-133138); mouse monoclonal XPO5 (Santa Cruz Catalogue: SC-271036); and mouse monoclonal β-actin antibodies (#A-544, Sigma-Aldrich St. Louis, MO). Colitis grade dextran sulphate sodium salt (18,000–19,000) was purchased from MP Biomedicals.
Methods
Cell culture
HCT116 colon cancer cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). HCT116 cells were used between passages 26–29. HCT116 cells were authenticated by IDEXX (Westbrook, Maine) using short tandem repeat DNA fingerprinting and were found to be mycoplasma free. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2-95% air and cultured in McCoy’s 5A Modified Medium containing 10% foetal bovine serum and penicillin/streptomycin under conditions recommended by ATCC. For transfections, cells were incubated with the indicated concentrations of mature mimic miRNA alone [Citation20] [Western blotting studies], and with WT or mutant ADAM17 3’UTR fused to a luciferase reporter. After 48 h, cells were broken and lysates probed for proteins by Western blotting or assayed for luciferase as described [Citation8].
Western Blotting
Proteins were extracted in SDS-containing Laemmli buffer, quantified by RC-DC protein assay and subjected to Western blotting as previously described with primary antibodies in 5% Blotto blocking buffer [Citation17]. Antibody concentrations included SET (I2PP2A) 1:500 dilution; Exportin5 (XPO5) 1:500 dilution; ADAM17 1:250 dilution; K-Ras 1:200 dilution; and Dicer 1:1000 dilution. Protein expression levels in tumours and adjacent mucosa were expressed as fold of indicated protein in control (wt-mouse) mucosa (mean ± SD). Separate aliquots were probed for β-actin to confirm comparable protein loading.
Mutagenesis of ADAM17-3’UTR
ADAM17 is a putative target of miR-145, miR-148a miR-152, and miR-338 as ADAM17 mRNA 3’UTR (NM_003183.6) contains bases complementary to the seeds of these miRNAs [Citation21]. ADAM17-3’UTR was obtained from Switchgear Genomics (S807284). The sequences complementary to seeds of miR-145, miR-148a, and miR-152 were mutated using Lightning quick change mutagenesis kit (Agilent #210515). Primers for mutating ADAM17-3’UTR bases complementing miR-148a-miR152 were: forward primer: 5’ggattgggttatgaatatttacgtttttgtaaattaatcttttatattgataacacgtgactctagggaaatgatcagttttttttt-atacactgtaatg antisense-3’ reverse primer: 5’cattacagtgtataaaaaaaaactgatcatttccctaga- gtcacgtgttatcaatataaaagattaatttacaaaaacgtaaatattcataacccaatcc-3’. Primers for mutating ADAM17-3’UTR bases complementing miR-145 were: forward primer 5’-aatgcag-ctgttctgtttttcagcattctttag-3’ and reverse primer 5’ctaaagaatgctgaaaaacagaacagctgcatt-3’. The mutated constructs were confirmed by DNA sequencing.
Transfection of HCT116 cells with ADAM17 3’UTR and miRNA
HCT116 cells were transferred from a T75 flask into a 24-well plate. Wild type or mutant ADAM17-3’UTR (100 ng) was diluted in 192.7 µl of Opti-MEM reduced serum media (Invitrogen) in one reaction tube. Scrambled control oligonucleotide or mimics of mature miRNA (4 µL, 20 nmol final) were added to this solution [Citation20]. Lipofectamine 2000 transfection reagent (8 µL) was diluted in 192 µl of Opti-MEM media in a separate tube. After 5 min, the two reaction tubes were combined to form a single complex solution for transfection. Following washing of HCT116 cells with Opti-MEM, 400 µl of Opti-MEM was added to each well, along with 100 µl of transfection complex solution. The cells were incubated for 5 hours at 37°C. The transfection media was then removed and HCT116 media with 10% FBS was added to each well (1 ml/well). The cells, in replicates of 8, were incubated at 37°C for 48 h prior to luciferase assay and mean luciferase expression differences compared using a Student’s t-test.
Luciferase assay
The vector for ADAM17-3UTR codes for the Renilla luciferase gene. HCT116 cells were seeded in a 24-well plate and co-transfected with wild type or mutant ADAM17-3UTR vector together with a scrambled oligonucleotide or indicated mimic of mature miRNA (Ambion). Metalloprotease inhibitor, Marimastat (1 mM, Tocris), was added to prevent ADAM17 proteolysis. Renilla luciferase was assayed 48 h post-transfection following manufacturer’s recommendation (Switchgear Genomics, Menlo Park, CA).
Pre-miRNA-143 processing assay using cellular extracts
Primary mouse colonocytes were prepared as described [Citation10]. HCT116 cells were rinsed several times in cold PBS and then scraped into 10 ml PBS and pipetted into 15 conical tubes. Cells were collected by centrifugation for 5 min at 500 g and then resuspended in 2 ml hypotonic buffer and transferred to 2 ml tubes. Hypotonic buffer contained 30 mM HEPES-KOH pH 7.4, 2 mM Mg acetate, 5 mM DTT, and Roche protease inhibitor pellet. Cells were incubated on ice for 10 min and lysed by 5 passages through 27.5 gauge needle. Lysates were centrifuged for 20 min at 500 g. The soluble fraction was transferred to a fresh tube and potassium acetate added to final concentration of 100 mM. Lysate was then centrifuged 15,000 rpm for 15 min at 4°C. Protein in the soluble S500 fraction was adjusted to 5 µg protein/µL and used for subsequent miRNA processing assays.
32P-labelled pre-miR-143
pre-miR-143 was labelled with α 32P-UTP in an in vitro transcription assay using the Promega Riboprobe kit under conditions described by the manufacturer (Promega Chicago, IL). Briefly, genomic pre-miR-143 DNA was amplified from HCT116 genomic DNA with forward primer 5’-TGAGGTGCAGTGCTGCATC-3’ and reverse primer 5’-GCTACAGTGCTTCATCT-CAGACTC-3’ with Taq polymerase to generate A-overhangs on the 3’-end. The pGEM-T vector was linearized with SmaI and terminal T’s added to the 3’ end with ddTTP and terminal transferase. The pre-miR-143 amplicon was then subcloned into the linearized pGEM-T vector downstream of the T7 polymerase promoter. In vitro transcription was performed in the presence of T7 polymerase, dNTP and α32P-UTP. 32P-labelled pre-miR-143 was isolated (~100 bp) and specific activity determined as recommended by the manufacturer (Promega Technical Bulletin product L5610).
32P-pre-miR-143 processing assay
The 5x processing buffer contained 150 mM HEPES-KOH pH7.4, 500 mM potassium acetate, 10 mM magnesium acetate, 25 mM DTT. Final processing reactions (10 µL) contained 10 mM creatine phosphate, 0.3 µg creatine kinase (prepared fresh), 0.5 mM ATP, 4 U RNasin, 0.1 µg yeast RNA, 25,000–50,000 cpm 32P-pre-miR-143 and 5 µL S500 enzyme fraction. Reactions were incubated at 37°C for 6 h. The reaction was stopped, and RNA extracted in 100 µL phenol: chloroform (5:1) pH 4.7 followed by extraction in 100 µl chloroform: isoamyl (24:1). RNA was precipitated at −20°C for 1 h by addition of 250 µL ethyl alcohol, 25 µL sodium acetate pH 5.5, 2.5 µL glycogen and 0.5 µL yeast tRNA (1 mg/ml). Samples were centrifuged for 5 min at 14,000 rpm and pellets washed with 800 µL 70% ethanol, centrifuging 5 min at 14,000 rpm. After a second wash, the pellet was dissolved in 5–10 µL RT-PCR grade H2O. Samples were mixed 1:1 in 2x sample buffer, centrifuged and heated to 70°C for 3 min followed by cooling on ice. Samples were loaded on a 5% denaturing polyacrylamide gel and separated at 150 V for 1 h using 1x BE running buffer. The gel was transferred to nylon membranes at 200 mAmps for 30 min and membranes exposed to film for an autoradiogram.
Human tissue
Fresh human colonic tissue was obtained under an approved IRB (10–209-A). Colonic tumours and adjacent normal-appearing colonic mucosa were isolated from surgical specimens. Colonocytes and stromal fractions were prepared and assayed for purity by Western blotting for cytokeratin 20 (CK20, colonocyte marker) and vimentin (VIM, stromal cell marker) as described [Citation22].
Mouse tissues
All animal studies were approved by the Animal Care and Use Committee at the University of Chicago and are fully compliant with the NIH guidelines for the humane use of animals. Archived samples were available from prior studies taken from control mice and AOM-treated mice fed Std diet (5% fat) or WD (20% fat) for 40 wks [Citation8]. Samples included distal colonic segments fixed in 10% formalin or frozen in OCT, as well as RNA and proteins from scrape-isolated colonic mucosa. For studies to determine stromal cell vs. colonocyte distribution of specific miRNAs, archived tissue was used from a prior study with controls and Apc+/LoxP-cdx2P-Cre mice [Citation22]. Colonocytes and stromal cells were isolated as previously described [Citation22]. Briefly, colons were removed and mucosa scrape-isolated and minced with a razor blade into 2 mm pieces. Fragments were collected in tubes containing 6 ml sterile ice-cold transport media, 50 IU/ml penicillin, 50 µg/ml streptomycin, and 0.5 mg/ml gentamycin. Tissue was washed 3 times by gentle inversion and collected by gravity sedimentation and re-suspended in 10 ml chelating buffer (media containing 1 mM EDTA and 1 mM EGTA). Tissue was incubated on a shaker overnight at 4°C to release colonocytes into the supernatant. The pellet was washed three times with 3 ml ice-cold PBS, releasing residual colonocytes, and colonocyte-containing supernatants were combined. Epithelial cell fractions (supernatants) and stromal cell fractions (pellets) were centrifuged at 400 x g for 5 min at 4°C, yielding loosely packed pellets from which proteins and RNA were extracted as described [Citation22]. Western blotting fractions were assayed for CK20 (colonocyte marker) and vimentin (stromal cell marker) to assess purity.
Pre-miR-143-pre-miR-145 transgenic mice
We cloned 2.1kb genomic fragment containing pre-miR-143-145 cluster into XhoI and KpnI sites of villin promoter construct obtained from Addgene. Based on the map, 12.4 kb Villin promoter sequence is located between EcoR1 (249 bp) and SmaI (13,278 bp) restriction sites. The pre-miR-143-145 cluster was amplified from genomic DNA from human HCT116 cells. Immediately 3’ to the villin promoter sequence we inserted the pre-miR-143-145 cluster between the Xho site (13,282 bp) and KpnI site (13,317 bp). The PCR primers for amplification were forward F1: CCGCTCGAGCGGTTTGGTCCT and reverse R1: TCTTTCCATCGGGGTACCCCG. The construct also contained SV40 poly A tail from 13,399 to 14,174. The villin promoter-pre-miRNA plasmid was amplified using Copy Cutter Competent bacteria (Epicenter). The transgene was excised by digesting with PmeI. The 14.5 kb PmeI-PmeI DNA construct was confirmed by DNA sequencing and purified on a caesium chloride gradient and used for pronuclear microinjection into pseudo-pregnant CD1 female mice. The injections were performed by the Transgenic Mouse Core Facility at the University of Chicago. Pups born from microinjected females were screened by PCR-based genotyping using pre-miR-143; pre-miR-145 specific primers. Transgene-positive mice were crossed with CD1 mice to obtain germ line transmission. Three founder lines #2, #30, and #34 were identified and all were shown to be resistant to DSS colitis compared to wild type CD1 control mice. Founder line #2, which showed the highest expression levels of pre-miR-143 and pre-miR-145, was expanded for subsequent experiments. We also obtained miR-143 KO and miR-145 KO mouse from our collaborator, Dr Eric Olson (UT Southwestern TX), as negative controls for these miRNAs.
For the DSS colitis studies, mice were treated with 3% DSS supplemented in the drinking water for 7 d followed by unsupplemented water for 14 d (21-d DSS cycle). The DSS cycles were repeated twice (3-cycles total) and mice then sacrificed at week 10. Disease activity index was scored as described [Citation23]. For the AOM/DSS studies, AOM 12.5 mg i.p./kg body weight or vehicle (saline) was administered weekly x 2 weeks and mice then given 3% DSS in the drinking water or water alone (control) for 7 d followed by water alone for 14 d. The DSS cycle was repeated twice. Mice were sacrificed 12 weeks after initial AOM treatment. Colons were resected and tumours enumerated. Tissue was either fixed in formalin (IHC), flash frozen in OCT (IF), stored in RNAlater (RNA) or frozen in liquid nitrogen (protein). Adjacent normal-appearing mucosa from AOM/DSS-treated mice and normal colons from control mice (no AOM/DSS treatment) were also collected and stored under the same conditions.
Real-time PCR
RNA was extracted from snap-frozen tissue using RNeasy Lipid Tissue Mini Kit (Qiagen, Germantown, MD). Samples were homogenized in a Bullet blender and loaded onto an RNA-binding spin column, washed, digested with DNase I and eluted in 30 µl elution buffer. Samples were examined by Agilent chip for RNA purity and quantified by Ribogreen. For messenger RNA, 100 ng total RNA was reverse transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit (# 4387406, Applied Biosystems, Foster City, CA) in 20 µL total volume. The resulting first-strand complementary DNA (cDNA) was used as a template for quantitative PCR in triplicate using Fast SYBR Green QPCR Master Mix kit (# 4,385,612, Applied Biosystems). Primers were designed using primer3 [Citation24]. Where possible one of the primers was designed to span intron-exon junctions (see Supplemental Table S1). Reverse transcribed cDNA (1 µL of 1:10 dilution) and primers were mixed with Fast SYBR Green master mixture in final volume of 25 µl. Negative controls (reactions lacking either reverse transcriptase or template) yielded no PCR products. Primers and TaqMan probes for mature miRNAs were obtained from ABI and conditions for reverse transcription and PCR amplification followed the manufacturer’s recommendations. Reverse transcriptase reactions were run in duplicate and PCR reactions in triplicate. Data were analysed using the comparative 2^(-∆∆Ct) method [Citation25,Citation26]. mRNA levels were normalized to β-actin and miRNA levels normalized to RNU48 (human tissue) or snoRNA202 (murine tissue) and expressed as fold-control [Citation26]. We also measured precursor and mature forms of miRNAs by SYBR green real-time PCR. See supplemental methods and miRNA primer sequences (Supplemental Table S2).
Immunohistochemistry
Five-micron sections of formalin-fixed paraffin-embedded tissue were cut and mounted on Vectabond-coated Superfrost Plus slides. Slides were heated to 60°C for 1 h, deparaffinized by 5 min washes x3 in xylene, hydrated in a graded series of ethanol washes, and rinsed with distilled water. Epitope retrieval was achieved by heating with a pressure cooker for 20 min in Tris-EDTA buffer for Ki67 staining (10 mM Tris-Base, 1 mM EDTA, 0.05% Tween 20, pH 9.0); or in 0.01 M citrate buffer, pH 6 for other antibodies. Sections were incubated overnight at 4°C. For Ki67 antibodies (clone SP6 Cat. #RM-9106-S0, Fisher Scientific, 1:500 final dilution), staining was quantified using the Fiji image software (National Institutes of Health) with ImmunoRatio plug-in [Citation27]. Antibody dilutions for other proteins included anti-ADAM17 antibodies 1:200 dilution; anti-K-Ras antibodies 1:250 dilution; anti-SET (I2PP2A antibodies) 1:500 dilution; anti-Exportin5 (XPO5) antibodies 1:500 dilution.
Putative targets for miR-143 and miR-145 miRNA
We used publicly available databases, including Target scan, to identify potential targets of miR-143 and miR-145 [Citation21]. We also used The Human Protein Atlas to screen predicted miRNA targets for up-regulation of RNA and protein in human colon cancers. For protein studiers, we narrowed the search for relevant targets by looking for those with nuclear expression to facilitate image quantification of DAB stained sections [Citation28]. The lists of Target scan predicted miR-143 and miR-145 targets were then combined and duplicate genes were removed. Ingenuity Pathway Analysis (Qiagen) was used to perform a Core Analysis query using the list of genes and an assumed overexpression level of the targets (assigned 10 in fold-change gene expression) to model the scenario in which miRNAs did not repress the transcripts of these targets because miR-143 and miR-145 were down-regulated in tumours. Following the analysis, a summary of the Diseases and Functions Analysis of IPA was downloaded and a list of pathways with predicted increased or decreased signalling was culled.
Statistical methods
Continuous data (animal weights, tumour sizes, and Western blotting densitometry units) were normally distributed as assessed by the Shapiro-Wilk normality test and summarized as means ± SD [Citation29]. All analyses of normally distributed variables involving more than two groups were calculated by one-way ANOVA and subsequent two group comparisons made using two-sided unpaired Student’s t-test. The Tukey’s HSD correction was applied to control for multiple comparisons for independent univariable tests and a nominal p-value <0.05 was used to determine statistical significance.
Cancer incidence was defined as the percentage of mice with at least one cancer and significance was calculated by Chi-square test. Tumour multiplicity (TM) was defined as the average number of tumours and includes both cancers and adenomas per tumour bearing mouse. Tumour multiplicities were compared among groups using the non-parametric Kruskal–Wallis (KW) test and significance determined by p-values with p < 0.05 considered significant.
Results
Regulation of ADAM17 by miRNAs in HCT116 colon cancer cells
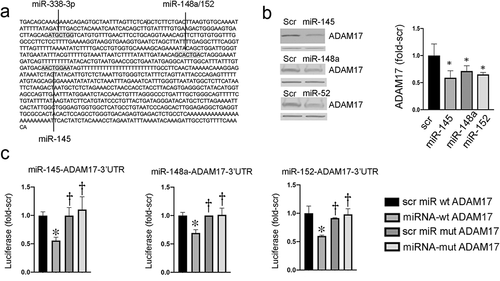
In prior studies, we showed that ADAM17 promotes tumour development in both genetic and carcinogen models of colon cancer [Citation10]. ADAM17 was reported to be regulated by miRNAs in other cancers, and numerous studies have reported that miRNAs are implicated in colonic tumorigenesis [Citation30–32]. Since we previously found that Western diet and tumorigenesis increased ADAM17 [Citation10], we asked whether changes in miRNAs might contribute to the effects of diet and malignant transformation on ADAM17 expression. Using Targetscan, a predictive miRNA target tool, we searched for miRNAs with seed sequences complementing bases in the 3’UTR of ADAM17. The search identified four putative ADAM17-regulating miRNAs: miR-145, miR-148a, miR-152, and miR-338 [Citation11]. Shown in ) are ADAM17 3’UTR bases complementing the seed sequences of miR-145, miR-148a/-152, and miR-338. To interrogate the inhibitory activity of these four miRNAs, we next transfected HCT116 colon cancer cells (CCC) with mature mimics of these miRNAs as described [Citation20]. As shown in ), miR-145, miR-148a, and miR-152 reduced ADAM17 expression, whereas miR-338 did not change ADAM17 expression. To directly test whether these miRNAs regulate ADAM17, we prepared luciferase reporters regulated by wild type (WT) or mutant ADAM17-3’UTR with mutations deleting bases complementing miRNA seed bases and confirmed mutations by sequencing. As shown in ), exogenous transfected mature mimics of miR-145, miR-148a, and miR-152 significantly reduced luciferase expression in cells transfected with luciferase regulated by wild type, compared to mutant ADAM17-3’UTR. In contrast, miR-338 did not reduce luciferase regulated by WT ADAM17-3’UTR (see supplemental data Fig S1). The combination of miR-145, −148a, and −152 also significantly suppressed ADAM17 compared to scrambled miRNA controls (supplemental Fig S3). Together, these data indicate that miR-145, miR-148a, and miR-152 can directly regulate ADAM17 via direct interaction with ADAM17- 3’UTR in colon cancer cells. Whether these miRNA effects on ADAM17 expression involve decreases in ADAM17 mRNA stability or protein translation will require further study [Citation33,Citation34]. We chose to focus on these ADAM17-regulating miRNAs for subsequent studies. We also examined ADAM17 and Dicer expression in primary human colonocytes and HCT116 cells. As shown in supplemental Fig S2 and supplemental Fig S6, ADAM17 and Dicer expression levels were higher in colon cancer cells compared to primary colonocytes, in agreement with prior studies [Citation35,Citation36].
Figure 1. miRNA regulation of ADAM17. (a) Predicted miRNA binding sites for ADAM17. (b) Exogenous miRNAs suppress ADAM17 in HCT116 cells. Left panel representative blots; β-actin band below ADAM17 is shown as a loading control. right panel densitometry of two independent experiments, *p < 0.05 compared to scrambled (Scr) miRNA. (c) Expression of luciferase regulated by WT or mutant ADAM17-3’UTR in cells transfected with indicated miRNAs. *p < 0.05 comparing targeted miRNA to control Scr assayed with WT ADAM17-3’UTR; †p < 0.05 comparing targeted or Scr miRNA + mutant ADAM17-3’UTR to targeted miRNA +WT ADAM17-3’UTR (n = 3 independent platings in replicates of 8)
Cell distribution of miRNAs regulating ADAM17 in human colon and colonic tumours
In prior studies, miR-143 and miR-145 were shown to be predominantly expressed in stromal cells, including fibroblasts in normal colon [Citation37]. To extend these studies, we examined expression levels of mature miR-143 and mature forms of putative ADAM17-regulating miRNAs (miR-145, miR-148a, and miR-152) in colonocytes and stromal cells from normal human colon and colonic tumours using Taqman assays. As shown in ), miR-143 and miR-145 were predominantly expressed in stromal cells in agreement with prior studies [Citation37], whereas miR-148a was predominantly expressed in colonocytes and miR-152 was abundant in both. Furthermore, when examined in isolated colonocytes or stromal cells, we observed these miRNAs were down-regulated (miR-143, miR-148a, miR-152) or trending towards down-regulation (miR-145) in purified cell types from human colonic tumours. These results are consistent with our observation that both miR-143 and miR-145 were down-regulated when measured in intact tissue from human or experimental colon tumours [Citation8].
Figure 2. (a) Cell-specific distribution of miRNAs in normal human colon and colon cancers. Stromal cells and colonocytes were prepared from surgical specimens of human tumours and adjacent normal appearing tissue as described in the Methods. Indicated miRNAs were quantified by real-time PCR with values normalized to 1 in whole (non-fractionated) tissue. Note the relative enrichment of miR-143, miR-145, and miR-152 in stromal cells and the relative enrichment of miR-148a in the colonocyte fraction. miR-143 ap<0.05 whole tissue vs stroma; bp<0.005 whole tissue versus colonocytes; cp<0.001 stroma vs colonocytes; dp<0.005 stroma vs tumour stroma. miR-145: ep<0.05 stroma vs colonocytes; miR-148a: fp<0.005 stroma vs colonocytes; gp<0.05 colonocytes vs tumour colonocytes, miR-152: hp<0.05 stroma vs tumour stroma. (b) Cell-specific distribution of miRNAs in normal mouse colon and mouse colon tumours. Cellular fractions were prepared from fresh colonic mucosa from control and conditional Apc+/LoxP-cdx2-P-Cre mice as described in the Methods. RNA was prepared and indicated miRNAs quantified by real-time PCR. Expression levels were normalized to 1 in whole (non-fractionated) tissue. Note the relative enrichment of miR-143, miR-145a and miR-152 in stromal cells and relative enrichment of miR-148a in the colonocyte fraction. miR-143: ap<0.05, whole tissue vs stroma, bp<0.05, stroma vs colonocytes. cp<0.05, stroma vs tumour stroma, dp<0.05, stroma vs tumour colonocytes, ep<0.05 colonocytes vs tumour stroma; miR-145: fp<0.005 whole tissue vs colonocytes, gp<0.05 whole tissue vs tumour stroma, hp<0.005 stroma vs colonocytes, ip<0.05 stroma vs tumour stroma; miR-148a: jp<0.05 whole tissue vs tumour stroma, kp<0.05 stroma vs colonocytes, lp<0.05 colonocytes vs tumour stroma, mp<0.005 colonocytes vs tumour colonocytes; miR-152: np<0.005 whole tissue vs colonocytes, op<0.05 whole tissue vs tumour colonocytes. (c) Western diet and tumour development down-regulate miR-143 and miR-145a with a similar trend for miR-148a and miR-152. Mice were fed standard diet (Std diet, 5% fat) or Western diet (20% fat) and treated with AOM or saline as described [Citation8]. After 40 wks mice were sacrificed and colonic mucosa from left colon scrape-isolated and RNA extracted. miRNAs were measured by real-time PCR. Diet significantly reduced miR-143 and miR-145a with even greater reductions in tumours. miR-143: ap<0.005; bp<0.01 cp<0.0005 compared to Std diet; miR-145a: dp<0.01; ep<0.02; fp<0.005; compared to Std diet. miR-148a: gp<0.05, hp<0.005 compared to Std diet; miR-152: ip<0.001, compared to standard diet
![Figure 2. (a) Cell-specific distribution of miRNAs in normal human colon and colon cancers. Stromal cells and colonocytes were prepared from surgical specimens of human tumours and adjacent normal appearing tissue as described in the Methods. Indicated miRNAs were quantified by real-time PCR with values normalized to 1 in whole (non-fractionated) tissue. Note the relative enrichment of miR-143, miR-145, and miR-152 in stromal cells and the relative enrichment of miR-148a in the colonocyte fraction. miR-143 ap<0.05 whole tissue vs stroma; bp<0.005 whole tissue versus colonocytes; cp<0.001 stroma vs colonocytes; dp<0.005 stroma vs tumour stroma. miR-145: ep<0.05 stroma vs colonocytes; miR-148a: fp<0.005 stroma vs colonocytes; gp<0.05 colonocytes vs tumour colonocytes, miR-152: hp<0.05 stroma vs tumour stroma. (b) Cell-specific distribution of miRNAs in normal mouse colon and mouse colon tumours. Cellular fractions were prepared from fresh colonic mucosa from control and conditional Apc+/LoxP-cdx2-P-Cre mice as described in the Methods. RNA was prepared and indicated miRNAs quantified by real-time PCR. Expression levels were normalized to 1 in whole (non-fractionated) tissue. Note the relative enrichment of miR-143, miR-145a and miR-152 in stromal cells and relative enrichment of miR-148a in the colonocyte fraction. miR-143: ap<0.05, whole tissue vs stroma, bp<0.05, stroma vs colonocytes. cp<0.05, stroma vs tumour stroma, dp<0.05, stroma vs tumour colonocytes, ep<0.05 colonocytes vs tumour stroma; miR-145: fp<0.005 whole tissue vs colonocytes, gp<0.05 whole tissue vs tumour stroma, hp<0.005 stroma vs colonocytes, ip<0.05 stroma vs tumour stroma; miR-148a: jp<0.05 whole tissue vs tumour stroma, kp<0.05 stroma vs colonocytes, lp<0.05 colonocytes vs tumour stroma, mp<0.005 colonocytes vs tumour colonocytes; miR-152: np<0.005 whole tissue vs colonocytes, op<0.05 whole tissue vs tumour colonocytes. (c) Western diet and tumour development down-regulate miR-143 and miR-145a with a similar trend for miR-148a and miR-152. Mice were fed standard diet (Std diet, 5% fat) or Western diet (20% fat) and treated with AOM or saline as described [Citation8]. After 40 wks mice were sacrificed and colonic mucosa from left colon scrape-isolated and RNA extracted. miRNAs were measured by real-time PCR. Diet significantly reduced miR-143 and miR-145a with even greater reductions in tumours. miR-143: ap<0.005; bp<0.01 cp<0.0005 compared to Std diet; miR-145a: dp<0.01; ep<0.02; fp<0.005; compared to Std diet. miR-148a: gp<0.05, hp<0.005 compared to Std diet; miR-152: ip<0.001, compared to standard diet](/cms/asset/a561a1f0-d15d-4461-afb8-74341e47c388/kepi_a_1863117_f0002_b.gif)
To assess whether the cell-type distributions of mature forms of these miRNAs in mouse colon mimic their distributions in human colon and assess effects of tumorigenesis on their expression levels, we examined their distributions in a genetically engineered mouse model expressing a conditional Apc gene in mouse colonocytes [Citation22]. As shown in ), distributions of these miRNAs were similar to those in human colonic cells. Furthermore, miR-143, miR-145a, and miR-148a were downregulated in murine tumour-associated stromal cells (miR-143, miR-145a) or tumour colonocytes (miR-148a), with a similar trend for miR-152.
In subsequent experiments, we examined both precursor and mature forms of these miRNAs in SYBR green qPCR assays in mouse and human colonic tissue and in HCT116 colon cancer cells and CCD-18Co colonic fibroblasts. The precursor and mature miRNA levels in mouse tissue are shown in supplemental Fig S4A and the precursor and mature miRNA levels in human tissue shown in supplemental Fig S4B. In general, the relative stromal cell to colonocyte distributions of the mature forms of the miRNAs agree broadly with ratios determined by our Taqman assays shown in ). The exception was miR-148a that appeared to be more enriched in the stromal cells in the SYBR green assay. Since the TaqMan assay adds a greater degree of specificity, we are inclined to give it more weight. In supplemental Fig S5, we noted that mature miR-143, miR-145, miR-148a, and miR-152 were down-regulated in HCT116 cells compared to levels in primary human colonocytes (supplemental Fig S5). The precursors for these miRNAs were also lower in HCT116 cells compared to primary colonocytes, though pre-miR-145 and pre-miR-152 were detectable in the cancer cells. Further studies will be needed to assess whether reductions of these precursors reflect impaired transcription and/or reduced stability of these pre-miRNAs in cancer.
Western diet inhibits expression of miR-143 and miRNAs that regulate ADAM17
Since Western diets promote colon cancer and increase ADAM17 expression and activity [Citation10], we assessed the effects of the Western diet on these miRNAs. We used archived RNA isolated from scraped colonic mucosa from mice fed standard diet (5% fat) or Western diet (20% fat) [Citation8] and treated with azoxymethane (AOM) or vehicle. As shown in ), colonic mucosa from mice fed a Western diet had significantly lower levels of miR-143 and miR-145a compared to mice fed standard diet (5% fat). There were similar trends for miR-148a and miR-152. In prior studies, we showed that colonocyte proliferation in mice on WD was higher than those on Std diet, consistent with observations that ADAM17-EGFR signals were increased by WD [Citation8,Citation10]. In AOM-induced tumours from mice fed WD, miR-143, miR-145a, miR-148a, and miR-152 were significantly downregulated compared to control mice fed a Std diet.
Colon cancer cells cannot process pre-miR-143 to mature miR-143
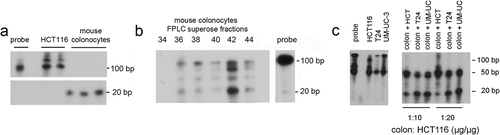
In the current study in agreement with prior investigations [Citation8], we demonstrated reduced levels of mature miR-143 and miR-145 in mouse and human colon tumours. Studies by Michael et al. suggested that pre-miR-143 was not altered in human colon cancers [Citation3]. We, therefore, examined the ability of mouse colonocytes and human CCCs to process pre-miRNA to mature miRNA. We radio-labelled pre-miR-143 transcripts with α32P-UTP. Mouse colonocytes or HCT116 colon cancer cell lysates were incubated with the 32P-pre-miR-143 and reactants separated on 8% denaturing polyacrylamide gels. As shown in ), the mature form of miR-143 was detected in lysates from mouse colonocytes, but not in lysates from HCT116 colon cancer cells. As a first step in isolating this processing activity, we separated a soluble fraction of mouse colonocyte lysates on a superpose column by HPLC and fractions (0.5 ml) were assayed for the ability to process 32P-pre-miR-143 to mature miR-143. As shown in ), fraction 42 contained the greatest Dicer-like activity. In separate experiments, adding HCT116 colon cancer cell or bladder cancer cell lysate to mouse colonocyte lysate (in a cancer cell/mouse colonocyte protein ratio of 10:1 or 20:1 µg/µg) did not reduce processing. This suggested that loss of pre-miR-143 processing in the HCT116 cells was not caused by the presence of an inhibitor of processing, but rather the absence of an activating factor ()). To provide more understanding of this difference in processing, we compared precursor levels in primary colonocytes compared to HCT116 cells. As shown in supplemental Fig S5 the precursors are abundant in colonocytes, but low to undetectable in HCT116 cells, whereas Dicer1 was more abundant in HCT116 cells compared to human colonocytes (supplemental Fig S6).
Figure 3. Lysates from mouse colonocytes, but not human colon cancer cells, can process pre-miR-143 to mature miR-143. Pre-miR-143 was transcribed in vitro with α32P-UTP. Lysates from primary mouse colonocytes and HCT116 colon cancer cells were prepared. 32P-pre-miR-143 processing was assayed as described in the Methods. (a) Processing by soluble extracts from human HCT116 cells and mouse colonocytes. (b) Processing by mouse colonocyte fractions separated on a superpose HPLC column. Fractions (0.5 ml) were assayed for ability to process 32P-pre-miR143 to mature 32P-miR-143. C. Pre-miR-143 processing by mouse colonocytes is not inhibited by addition of human colon cancer (HCT116) or bladder cancer cell (T24, UM-UC-3) extracts. Comparable results were obtained in two independent experiments
Generation of villin promoter regulated pre-miR-143, pre-miR-145 expressing mice
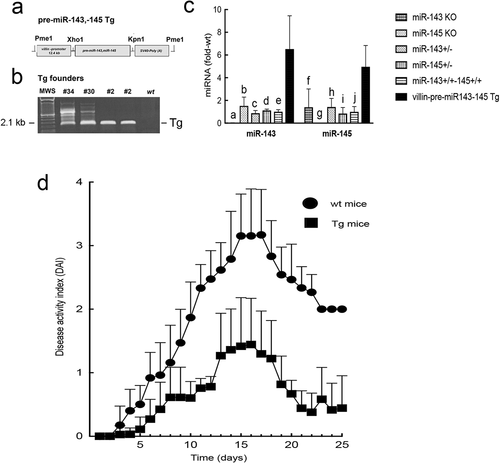
We chose to focus on miR-143 and miR-145 for the remainder of our studies because these polycistronic miRNAs are coordinately transcribed, and we observed that the Western diet alone induced significant reductions in their levels. The available miR-143 and miR-145 KO mice were not further investigated as these mice were on a mixed background that could confound the effects of miRNA KO on tumour development. To directly test the tumour suppressor activity of miR-143 and miR-145 in a mouse model of colon cancer, we generated transgenic mice expressing pre-miR-143 and pre-miR-145 under a mouse villin promoter as described in the Methods. While miR-143 and miR-145 are predominantly expressed in stromal cells, miR-145 is also detectable in colonocytes. Furthermore, there are no suitable promoters to drive transgene expression in colonic stromal cells.
A schema of the transgene, with restriction sites, is shown in ). Three founder mouse lines, #2, #30, and #34 were identified by PCR ()), and expression levels of colonocyte miR-143 and miR-145 were measured. All transgene-positive founder lines expressed comparable levels of up-regulated colonocyte miR-143 and miR-145. Shown in ) are comparisons of colonocyte miR-143 and miR-145 in wild type CD1 mice and transgenic founder line #2. We also included levels of colonocyte miR-143 and miR-145a in mice deleted of miR-143 or miR-145a as negative controls.
Figure 4. Villin-pre-miR-143, pre-miR-145 transgenic mouse. (a) Transgene (Tg) structure. (b) Genotyping of founder lines from tail snip DNA. (c) Expression levels of colonocyte miR-143 and miR-145 in WT and Tg mice. Levels of miR-143 and miR-145 in miR-143 KO and miR-145 KO mice are also shown. a, c, d, e p<0.0001, bp<0.0005, compared to miR-143 in Tg mice; f,hp<0.05, g p<0.0005, i, j p<0.005, compared to miR-145 in Tg mice. Note the first column for miR-143 group and the second column for miR-145 group were undetectable as these were KO mice for the respective miRNAs. (d) DSS colitis. *Disease activity index for all time points > 6 d were significantly lower in Tg mice compared to WT mice (p<0.005, Tukey’s HSD correction)
Up-regulation of pre-miR-143-pre-miR-145 in colonocytes inhibits DSS-colitis
We compared the severity of dextran sulphate sodium (DSS)-induced colitis in villin-regulated pre-miR-143, pre-miR-145 expressing mice to littermate-matched CD1 wildtype (WT) mice. Disease activity index (DAI) was calculated as previously described [Citation23]. In preliminary experiments, founder lines #2, #30, and #34 were comparable with respect to resistance to DSS-induced colitis. As shown in ), the expression of these microRNAs significantly delayed the onset and inhibited the severity of DSS-induced colitis.
Up-regulated colonocyte pre-miR-143, pre-miR145 inhibits tumour development
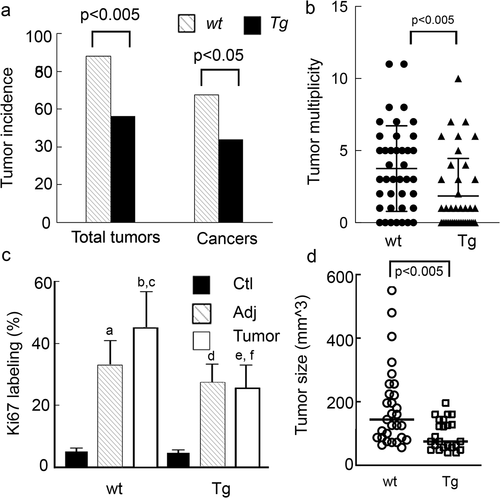
Since the CD1 strain that was used to make the Tg mouse is relatively resistant to AOM alone, we chose the AOM/DSS model to induce tumours as CD1 strain is relatively sensitive to AOM/DSS [Citation18]. We examined the effects of up-regulating these miRNAs in colonocytes on colon tumour development. As shown in ), total tumour incidence (adenomas + cancers) and cancer incidence, as well as tumour multiplicity, were significantly lower in Tg mice compared to littermate-matched wild type controls. Ki67 labelling and tumour size were also lower in Tg mice (), Supplemental Fig S7). These data indicate that upregulation of pre-miR-143 and pre-miR-145 in colonocytes inhibits tumour development and delays tumour progression, consistent with our conjecture that these miRNAs inhibit tumorigenesis.
Figure 5. AOM-DSS treated Tg mice have significantly lower tumour incidence and tumour burden and reduced colonocyte proliferation and tumour size. Tg mice and CD1 littermate controls were treated with AOM weekly for 2 wks followed by 3 cycles of DSS as described in the Methods. (a). Tumour and cancer incidence. (b) Tumour multiplicity. Kruskal Walis statistics. (c) Ki67 staining a, b p < 0.0001 compared to WT-ctl; cp<0.001 compared WT -adj, dp<0.0001 Tg-ctl vs. Tg-adj ep<0.0001 Tg-ctl vs Tg-Tumour, fp<0.0001 Tg Tumour vs WT Tumour. (d) Tumour size
Potential targets of miR-143 and miR-145 in colon cancer
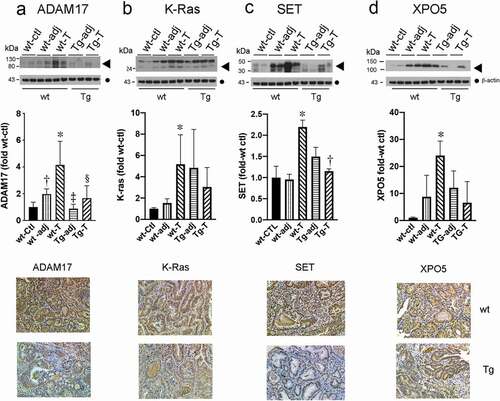
A number of genes have been validated as miR-143 and/or miR-145 targets in colon cancer cells by other strategies, including over-expression studies and mass spectrometry approaches involving transfected polycistronic pre-miR-143, -145 [Citation38,Citation39]. Since K-Ras [Citation40] and ADAM17 (this report) were identified as potential targets of miR-143 and miR-145 in colon cancer cells, respectively, we examined their expression levels in tumours from WT and Tg mice by Western blotting and immunostaining. Shown in are representative blots and immunostains of the indicated tissue. Compared to WT-ctl mucosa, K-Ras and ADAM17 were increased in tumours from WT mice, but not in tumours from Tg mice that express up-regulated colonocyte miR-143 and miR-145. These in vivo results are consistent with our predictions in cell culture ()) and in vitro studies by others [Citation40]. The IHC results, as shown in lower panels, were also consistent with the Western blotting data. Moreover, the Tg-induced changes in proto-oncogenes K-Ras and ADAM17 would be predicted to inhibit tumour development in the transgenic mice.
Figure 6. Putative targets of miR-143 and miR-145 are up-regulated in tumours from wild type (WT) but not Tg mice compared to control mucosa. Upper panel Western blots of the indicated proteins (◀︎). As loading controls, separate aliquots of the indicated samples were run on a second blot and probed for β-actin (●) (n = 4 separate mice with matching adjacent and tumour tissue middle panel quantitative densitometry. lower panel IHC. A. ADAM17 *p < 0.005 WT-T compared to WT-ctl; †p < 0.05 WT-adj compared to WT-T; ‡p < 0.005 WT-T compared to Tg-adj; §p < 0.05 WT-T compared to Tg-T. B. K-ras *p < 0.05 WT-T compared to WT-ctl. C. SET *p < 0.05 WT-T compared to WT-ctl, †p < 0.05 WT-T compared to Tg-T D. XPO5 *p < 0.05 WT-T compared to WT-ctl
In addition to K-Ras and ADAM17, we next sought to identify other potential targets of miR-143 and miR-145 that might contribute to tumour suppression in the Tg mouse model. We first obtained a list of 1,338 unique miR-143 and miR-145 targets using Targetscan [Citation12]. Since the seed sequences of miR-143 and miR-145 are identical in mouse and human, the species are expected to share many predicted targets for these miRNAs. For a given miRNA, we searched for targets of either 3p or 5p miRNA species. We made the assumption that if miR-143 and miR-145 were downregulated in a cancer phenotype, then the translation of their miRNA targets might be increased. Using Ingenuity Pathway Analysis (IPA), we queried the list of predicted miRNA targets to determine diseases and biological functions predicted to have increased or decreased activation based on down-regulation of miR-143 and miR-145. The Diseases and Functions Analysis revealed a broad list of 128 phenotypes or functions with predicted increased activations, including several hallmarks of cancer, such as cell survival, cell viability, cell movement, invasion or migration, and angiogenesis (see Supplemental Table S3). Additionally, the analysis uncovered 32 pathways predicted to have decreased activations, including those associated with more indolent growth phenotypes such as apoptosis, growth failure, and cell death (see Supplemental Table S3).
We were struck by the prevalence of ADAM17 and K-RAS listed among the miRNA targets in many of the cancer-related pathways in the IPA results (see Supplemental Table S4 and supplemental Fig S8). Additionally, we selected XPO5 [Citation41–43] and SET (I2PP2A) [Citation41,Citation42] as two targets also represented in the predicted pathways with prior evidence of being up-regulated in experimental and human colon tumours [Citation44–46]. Both XPO5 and SET are predicted miR-143 targets, and SET is also a predicted miR-145 target. We first confirmed that the 3’-UTRs for both mouse and human SET and XPO5 contain complementary sequences to the seed sequences in miR-143 and/or miR-145/miR-145a. Proteins encoded by these genes have been implicated as driving oncogenic signals in colon cancer or other tumours [Citation44–46]. As shown in , compared to normal mucosa from wt mice, SET and XPO5 were increased in tumours from wt mice but not in tumours from Tg mice. Representative blots and immunostains are shown. The occurrences of SET and XPO5 in pathways regulated by miR-143 and miR-145 are shown in Supplemental Table S4. Shown in supplemental Fig S8 are the percentage of pathways predicted to be activated or suppressed by upregulations of SET, XPO5, ADAM17, and K-Ras in response to downregulations of miR-143/miR-145. We postulate that the net effect of changes in activation of these pathways would be to promote tumour growth.
Discussion
In this study, we showed that transfection with mimics of miR-145, miR-148a or miR-152 suppressed ADAM17 protein expression in human colon cancer cells and provided evidence that these miRNAs interact directly with ADAM17 3’UTR. Since many RNA-binding proteins modulate miRNA–mRNA interactions and furthermore expression levels of RNA-binding proteins are cell-context specific, our results extend to colon cancer cells the findings of prior reports in other cancer cell types [Citation19,Citation31,Citation32,Citation47–52]. It is important to define particular miRNA–mRNA interactions in specific cell types [Citation53]. In this regard, we found, for example, that miR-338 did not suppress ADAM17 in colon cancer cells, although this miRNA was reported to regulate ADAM17 in gastric cancer cells [Citation54]. We also showed that up-regulation of pre-miR-143 and pre-miR-145 in colonocytes regulated by a murine villin promoter increased resistance to DSS-colitis and inhibited AOM/DSS-induced tumour development. To our knowledge, this is the first direct demonstration of tumour suppressor roles for these miRNAs in an in vivo model of colonic tumorigenesis. We also showed their down-regulation by a Western diet. The suppression of DSS colitis in our Tg mouse model, moreover, is consistent with prior studies showing that deletion of miR-143 and miR-145 exacerbated DSS colitis [Citation37].
With respect to cell specificity, many studies of colon cancer-associated changes in miRNAs involved analysis of whole tissue that contains a mixture of stromal and epithelial cells. However, to identify potential targets of miRNAs, it is important to determine the cell of origin for a given miRNA [Citation53]. We observed that the cell types most enriched in predicted ADAM17-regulating miRNAs differed, with mature miR-145 (and polycistronic miR-143) abundantly expressed in stromal cells in the colon. In contrast, mature miR-148a was enriched in colonocytes and mature miR-152, while greater in stromal cells was expressed in colonocytes at nearly 40% of levels in stromal cells. With the exception of human pre-miR-148a, the other pre-miRNAs were readily detected in mouse or human stromal cells and colonocytes (Supplemental Fig S4). This might suggest that hsa-pre-miR-148a is relatively less stable than mature hsa-miR-148a.
We also demonstrated that the mature forms of these miRNAs were down-regulated in both human and experimental colon tumours. Overexpression of these miRNAs in tumour xenograft studies, using colon cancer cells or other cancer cell lines, has supported their tumour suppressor roles [Citation8,Citation55,Citation56]. Consistent with these observations, global knock out of miR-148a increased AOM/DSS-induced colon cancer [Citation57]. Cell-type-specific differences in expression levels of these miRNAs suggest that as autocrine regulators, miR-145 might play a more important role in regulating ADAM17 in stromal cells, whereas miR-148a likely contributes more to ADAM17 regulation in colonocytes. While we emphasize the importance of focusing on the same cell type for miRNAs and their mRNA targets, there is increasing evidence that miRNAs can regulate events by shuttling between cells via exosomes, for example from fibroblasts to cancer cells [Citation58–60]. Taken together with results from our transgenic mouse studies, loss of miR-143 and miR-145 in stromal cells would be predicted to reduce their shuttling to epithelial cells and could contribute to the role of the microenvironment in promoting tumour growth [Citation61].
Numerous putative targets have been identified for miRNAs. We used the miRNA database Target Scan to suggest potential targets of miR-143 and miR-145 [Citation11,Citation12]. Relevant to our investigations in the Tg mouse, Pagliuca et al., in an in vitro study showed that upregulated co-expression of polycistronic miR-143 and miR-145 in colon cancer cells orchestrated a multi-gene program that inhibited proliferation and enhanced apoptosis [Citation39]. Pathways involved in miR-143-miR-145 effects included a growth factor receptor-mitogen-activated protein kinase network and a p53 signalling pathway. In another in vitro study, Bauer and Hummon showed by microarray analysis and mass spectrometry that both individual targets as well as shared targets of miR-143 and miR-145 mediated the antiproliferative effects of this cluster [Citation38]. Furthermore, TargetScan, used for target prediction for the current study, was in good agreement with Bauer and Hummon’s experimental findings [Citation38].
Several putative miR-143 or miR-145 targets were up-regulated in AOM tumours and/or in Apc mutant mouse adenomas (see accompanying GEO datasets in the references), including SET (I2PP2A) [Citation41] and XPO5 [Citation43,Citation62]. These targets are represented in the predicted pathways (Supplemental Table S4). SET is a putative target of both miR-143-3p and miR-145-5p, while XPO5 is a putative target of miR-143-3p. XPO5 encodes the transporter exporting pre-miRNA from the nucleus to the cytoplasm and functions as an oncogene in colon cancer [Citation44]. SET (I2PP2A) encodes an inhibitor of protein phosphatase 2A, a tumour suppressor [Citation63]. Our transgenic mouse model, with upregulation of miR-143 and miR-145 in colonocytes, gave us a unique opportunity to assess the effects of these polycistronic miRNAs in an intact organism on expression levels of predicted targets in colonic tumorigenesis. Both XPO5 and SET were up-regulated in tumours in WT mice, consistent with their putative oncogenic roles [Citation44,Citation45]. Also, consistent with a predicted identification of SET and XPO5 as targets of miR-143 and miR-145, tumours in Tg mice did not show up-regulation of these genes in contrast to tumours from WT mice. Supplemental Tables 3 and 4 show predicted targets of miR-143 and/or miR-145 for each pathway and indicate whether pathway activation is increased or decreased following miR-143/miR-145 down-regulation. These Tables provide a road map for future studies into mechanisms of tumour promotion modulated by loss of miR-143 and miR-145 in colon cancer. Additional studies, such as gene arrays and proteomic experiments will be needed to confirm these bioinformatic predictions.
Since pre-miR-143 and pre-miR-145 appear to be preserved in colon cancer, whereas the mature forms are downregulated [Citation3], in the current study, we asked if cancer cells might lack a factor required for pre-miRNA processing or express a factor that inhibits pre-miR-143 processing. As miR-143 and miR-145 are polycistronic and initially transcribed as a single pri-miRNA [Citation64], differences in expression between mature miR-143 and miR-145 in normal colonocytes could reflect sequence-specific differences in RT-PCR assays; or miRNA processing or stability differences as suggested for other polycistronic miRNAs [Citation65–67]. In this regard, mass spectrometry studies of RNA-binding proteins interacting with miRNAs identified distinctive partners for miR-143 and miR-145 that differentially regulate their stability or biogenesis [Citation68–70]. In agreement with the study by Michael et al. [Citation3], we observed that colon cancer cells (as well as bladder cancer cells) were unable to process pre-miR-143. In contrast, normal mouse colonocytes efficiently processed precursor to mature miR-143. In addition, our mixing experiments suggested that impaired processing reflects the loss of an activating factor, rather than the presence of an inhibitor. Potential mechanisms include tumour-associated changes in RNA-binding proteins, RNA editing, and modulation of miRNA decay [Citation71]. Deficiencies in Dicer1 in HCT116 cells to account for impaired processing were excluded by our real-time PCR and Western blotting assays (Supplemental Fig S6). While precursor miRNA levels are low in HCT116 cells (Supplemental Fig S5), this should not impact the assay shown in as exogenous labelled pre-miR-143 was added as a substrate for the assay.
We showed that mature forms of miR-143/145a, 148a, and -152 were reduced in the colon by Western diet and significantly down-regulated in tumours induced by AOM or by Apc deletion. These miRNAs were also down-regulated in human colon cancers, whereas ADAM17 was increased in colon tumours [Citation3,Citation35,Citation72,Citation73]. We speculate that increases in ADAM17 in tumours at least in part reflect down-regulation of these miRNAs [Citation73]. Other investigators have reported that diet influences miRNAs in human colorectal tissue and other cancerous tissues [Citation74,Citation75].
While miR-143 and miR-145 are expressed at relatively low levels in colonocytes, they are further down-regulated in cancer. For this reason, we asked whether the putative tumour suppressor roles of these miRNAs, as suggested by reports of their widespread downregulations in many tumour-types and supported by their anti-growth effects in tumour xenograft studies, could be tested in an in vivo model of tumorigenesis that proceeds from normal colonocytes to fully transformed colonocytes. This study has shown for the first time that expressing miR-143 and miR-145 in colonocytes inhibits tumour development. In this study, we also observed that ADAM17, K-Ras, XPO5, and SET that are demonstrated or predicted targets of miR-143 and miR-145 with oncogenic potential were upregulated in tumours from WT mice, but not in tumours from Tg mice. Upregulations of these miRNA targets (accompanying miR-143/miR-145 down-regulation) are predicted to drive pathways promoting colonic tumorigenesis. Thus, strategies limiting miR-143 and miR-145 down-regulation in colonocytes (and in colonic stromal cells) might prove useful for colon cancer prevention [Citation76].
Abbreviations
| ADAM17 | = | A Disintegrin and Metalloproteinase17 |
| AOM | = | Azoxymethane |
| CCC | = | Colon cancer cells |
| DSS | = | Dextran sulphate sodium |
| EGFR | = | Epidermal growth factor receptor |
| EV | = | Empty vector |
| IHC | = | Immunohistochemistry |
| Scr | = | Scrambled control |
| Tg | = | Transgene |
| XPO5 | = | Exportin5 |
| SET | = | Nuclear proto-oncogene |
| 3’-UTR | = | Three prime untranslated region |
| WT | = | Wild type |
Supplemental Material
Download Zip (5 MB)Acknowledgments
Grant Support: This work was supported by the National Institutes of Health Grants [R01 CA164124-01 and R01 CA180087-01 (YCL), R01 CA226303 (TCH)] and the Samuel Freedman GI Cancer Laboratory Fund at the University of Chicago and the University of Chicago Cancer Research Foundation (UCCRF) Women’s Board. The transgenic mouse was created by The Transgenic Mouse Core at the University of Chicago that receives financial support from NIH NCI Cancer Center Support Grant P30CA014599.
The miR-143 and miR-145 null mice were generously provided by Dr Eric Olson, Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA.
Disclosure statement
The authors disclose that they have no potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337.
- Duchaine TF, Fabian MR. Mechanistic insights into MicroRNA-mediated gene silencing. Cold Spring Harb Perspect Biol. 2019;11.
- Michael MZ, SM OC, Van Holst Pellekaan NG, et al. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891.
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866.
- Slaby O, Svoboda M, Michalek J, et al. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer. 2009;8:102.
- Slattery ML, Herrick JS, Pellatt DF, et al. MicroRNA profiles in colorectal carcinomas, adenomas and normal colonic mucosa: variations in miRNA expression and disease progression. Carcinogenesis. 2016;37:245–261.
- Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269.
- Zhu H, Dougherty U, Robinson V, et al. EGFR signals downregulate tumor suppressors miR-143 and miR-145 in Western diet-promoted murine colon cancer: role of G1 regulators. Mol Cancer Res. 2011;9:960–975.
- Nedaeinia R, Sharifi M, Avan A, et al. Inhibition of microRNA-21 via locked nucleic acid-anti-miR suppressed metastatic features of colorectal cancer cells through modulation of programmed cell death 4. Tumour Biol. 2017;39:1010428317692261.
- Mustafi R, Dougherty U, Mustafi D, et al. ADAM17 is a tumor promoter and therapeutic target in Western diet-associated colon cancer. Clin Cancer Res. 2017;23:549–561.
- Friedman RC, Farh KK, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105.
- Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4.
- Johannessen C, Moi L, Kiselev Y, et al. Expression and function of the miR-143/145 cluster in vitro and in vivo in human breast cancer. PLoS One. 2017;12:e0186658.
- Zaman MS, Chen Y, Deng G, et al. The functional significance of microRNA-145 in prostate cancer. Br J Cancer. 2010;103:256–264.
- Borralho PM, Simoes AE, Gomes SE, et al. miR-143 overexpression impairs growth of human colon carcinoma xenografts in mice with induction of apoptosis and inhibition of proliferation. PLoS One. 2011;6:e23787.
- Ibrahim AF, Weirauch U, Thomas M, et al. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011;71:5214–5224.
- Dougherty U, Cerasi D, Taylor I, et al. Epidermal growth factor receptor is required for colonic tumor promotion by dietary fat in the azoxymethane/dextran sulfate sodium model: roles of transforming growth factor-{alpha} and PTGS2. Clin Cancer Res. 2009;15:6780–6789.
- Wu J, Yin L, Jiang N, et al. MiR-145, a microRNA targeting ADAM17, inhibits the invasion and migration of nasopharyngeal carcinoma cells. Exp Cell Res. 2015;338:232–238.
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20.
- Dougherty U, Mustafi R, Haider HI, et al. Losartan and vitamin D inhibit colonic tumor development in a conditional Apc-deleted mouse model of sporadic colon cancer. Cancer Prev Res (Phila). 2019;12:433–448.
- Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249.
- Untergasser A, Cutcutache I, Koressaar T, et al. Primer3 – new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2exp(-ΔΔ CT) Method. Methods. 2001;25:402–408.
- Yuan JS, Reed A, Chen F, et al. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85–97.
- Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682.
- Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357.
- Ghasemi A, Zahediasl S. Normality tests for statistical analysis: a guide for non-statisticians. Int J Endocrinol Metab. 2012;10:486–489.
- Rossi S, Di Narzo AF, Mestdagh P, et al. microRNAs in colon cancer: a roadmap for discovery. FEBS Lett. 2012;586:3000–3007.
- Doberstein K, Steinmeyer N, Hartmetz AK, et al. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia. 2013;15:218–230.
- Liu Y, Wu C, Wang Y, et al. MicroRNA-145 inhibits cell proliferation by directly targeting ADAM17 in hepatocellular carcinoma. Oncol Rep. 2014;32:1923–1930.
- Baek D, Villen J, Shin C, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71.
- Chan LY, Mugler CF, Heinrich S, et al. Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. Elife. 2018;7.
- Blanchot-Jossic F, Jarry A, Masson D, et al. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207:156–163.
- Faber C, Horst D, Hlubek F, et al. Overexpression of Dicer predicts poor survival in colorectal cancer. Eur J Cancer. 2011;47:1414–1419.
- Chivukula RR, Shi G, Acharya A, et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell. 2014;157:1104–1116.
- De Robertis M, Massi E, Poeta ML, et al. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9.
- Wang Z. The guideline of the design and validation of miRNA mimics. Methods Mol Biol. 2011;676:211–223.
- Bauer KM, Hummon AB. Effects of the miR-143/-145 microRNA cluster on the colon cancer proteome and transcriptome. J Proteome Res. 2012;11:4744–4754.
- Pagliuca A, Valvo C, Fabrizi E, et al. Analysis of the combined action of miR-143 and miR-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene. 2013;32:4806–4813.
- Chen X, Guo X, Zhang H, et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28:1385–1392.
- Satoh K, Yachida S, Sugimoto M, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci U S A. 2017;114:E7697–E706.
- Giardina C, Nakanishi M, Khan A, et al. Regulation of VDR expression in Apc-mutant mice, human colon cancers and adenomas. Cancer Prev Res (Phila). 2015;8:387–399.
- Kaiser S, Park YK, Franklin JL, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131.
- Shigeyasu K, Okugawa Y, Toden S, et al. Exportin-5 functions as an oncogene and a potential therapeutic target in colorectal cancer. Clin Cancer Res. 2017;23:1312–1322.
- Cristobal I, Torrejon B, Rubio J, et al. Deregulation of SET is associated with tumor progression and predicts adverse outcome in patients with early-stage colorectal cancer. J Clin Med. 2019;8.
- Cristobal I, Rincon R, Manso R, et al. Deregulation of the PP2A inhibitor SET shows promising therapeutic implications and determines poor clinical outcome in patients with metastatic colorectal cancer. Clin Cancer Res. 2015;21:347–356.
- Lu Y, Chopp M, Zheng X, et al. MiR-145 reduces ADAM17 expression and inhibits in vitro migration and invasion of glioma cells. Oncol Rep. 2013;29:67–72.
- Liu Y, Wu C, Wang Y, et al. Expression of miR-224, miR-145, and their putative target ADAM17 in hepatocellular carcinoma. Acta Biochim Biophys Sin (Shanghai). 2014;46:720–722.
- Su Y, Wang Y, Zhou H, et al. MicroRNA-152 targets ADAM17 to suppress NSCLC progression. FEBS Lett. 2014;588:1983–1988.
- Wu Y, Huang A, Li T, et al. MiR-152 reduces human umbilical vein endothelial cell proliferation and migration by targeting ADAM17. FEBS Lett. 2014;588:2063–2069.
- Su Y, Wang Y, Zhou H, et al. MicroRNA-152 targets ADAM17 to suppress NSCLC progression. FEBS Lett. 2014;588:1983–1988.
- Vila-Casadesus M, Vila-Navarro E, Raimondi G, et al. Deciphering microRNA targets in pancreatic cancer using miRComb R package. Oncotarget. 2018;9:6499–6517.
- Kent OA, McCall MN, Cornish TC, et al. Lessons from miR-143/145: the importance of cell-type localization of miRNAs. Nucleic Acids Res. 2014;42:7528–7538.
- Chen JT, Yao KH, Hua L, et al. MiR-338-3p inhibits the proliferation and migration of gastric cancer cells by targeting ADAM17. Int J Clin Exp Pathol. 2015;8:10922–10928.
- Li B, Xie Z, Li B. miR-152 functions as a tumor suppressor in colorectal cancer by targeting PIK3R3. Tumour Biol. 2016;37:10075–10084.
- Cao H, Liu Z, Wang R, et al. miR-148a suppresses human renal cell carcinoma malignancy by targeting AKT2. Oncol Rep. 2017;37:147–154.
- Zhu Y, Gu L, Li Y, et al. miR-148a inhibits colitis and colitis-associated tumorigenesis in mice. Cell Death Differ. 2017;24:2199–2209.
- Bhome R, Goh RW, Bullock MD, et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: role in driving cancer progression. Aging (Albany NY). 2017;9:2666–2694.
- Donnarumma E, Fiore D, Nappa M, et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget. 2017;8:19592–19608.
- Savardashtaki A, Shabaninejad Z, Movahedpour A, et al. miRNAs derived from cancer-associated fibroblasts in colorectal cancer. Epigenomics. 2019;11:1627–1645.
- Isella C, Terrasi A, Bellomo SE, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319.
- Pleiman JK, Irving AA, Wang Z, et al. The conserved protective cyclic AMP-phosphodiesterase function PDE4B is expressed in the adenoma and adjacent normal colonic epithelium of mammals and silenced in colorectal cancer. PLoS Genet. 2018;14:e1007611.
- Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–11062.
- Rangrez AY, Massy ZA, Metzinger-Le Meuth V, et al. miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet. 2011;4:197–205.
- Natera-Naranjo O, Aschrafi A, Gioio AE, et al. Identification and quantitative analyses of microRNAs located in the distal axons of sympathetic neurons. RNA. 2010;16:1516–1529.
- Chawla G, Sokol NS. ADAR mediates differential expression of polycistronic microRNAs. Nucleic Acids Res. 2014;42:5245–5255.
- Heo I, Joo C, Kim YK, et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708.
- Heo I, Joo C, Cho J, et al. Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol Cell. 2008;32:276–284.
- Vizcaino JA, Csordas A, del-Toro N, et al. Update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–56.
- Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A. 2012;109:3347–3352.
- Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol. 2014;24:R762–76.
- Chen Y, Song Y, Wang Z, et al. Altered expression of MiR-148a and MiR-152 in gastrointestinal cancers and its clinical significance. J Gastrointest Surg. 2010;14:1170–1179.
- Gaedcke J, Grade M, Camps J, et al. The rectal cancer microRNAome–microRNA expression in rectal cancer and matched normal mucosa. Clin Cancer Res. 2012;18:4919–4930.
- Slattery ML, Herrick JS, Mullany LE, et al. Diet and lifestyle factors associated with miRNA expression in colorectal tissue. Pharmgenomics Pers Med. 2017;10:1–16.
- Nara T, Narita S, Mingguo H, et al. Altered miRNA expression in high-fat diet-induced prostate cancer progression. Carcinogenesis. 2016;37:1129–1137.
- Melo S, Villanueva A, Moutinho C, et al. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc Natl Acad Sci U S A. 2011;108:4394–4399.