
ABSTRACT
Neural tube defects (NTDs) are a group of common and severe congenital malformations. The PI3K-AKT signalling pathway plays a crucial role in the neural tube development. There is limited evidence concerning any possible association between aberrant methylation in PI3K-AKT signalling pathway genes and NTDs. Therefore, we aimed to investigate potential associations between aberrant methylation of PI3K-AKT pathway genes and NTDs. Methylation studies of PI3K-AKT pathway genes utilizing microarray genome-methylation data derived from neural tissues of ten NTD cases and eight non-malformed controls were performed. Targeted DNA methylation analysis was subsequently performed in an independent cohort of 73 NTD cases and 32 controls to validate the methylation levels of identified genes. siRNAs were used to pull-down the target genes in human embryonic stem cells (hESCs) to examine the effects of the aberrant expression of target genes on neural cells. As a result, 321 differentially hypermethylated CpG sites in the promoter regions of 30 PI3K-AKT pathway genes were identified in the microarray data. In target methylation analysis, CHRM1, FGF19, and ITGA7 were confirmed to be significantly hypermethylated in NTD cases and were associated with increased risk for NTDs. The down-regulation of FGF19, CHRM1, and ITGA7 impaired the formation of rosette-like cell aggregates. The down-regulation of those three genes affected the expression of PAX6, SOX2 and MAP2, implying their influence on the differentiation of neural cells. This study for the first time reported that hypermethylation of PI3K-AKT pathway genes such as CHRM1, FGF19, and ITGA7 is associated with human NTDs.
Introduction
Neural tube defects (NTDs) are congenital malformations of the central nervous system, annually affecting approximately 18.6 infants per 10,000 live births worldwide [Citation1]. They are caused by disruptions during neurulation that result in incomplete closure or opening of the spinal cord or cranial regions of the developing neural tube [Citation2]. NTDs are often divided into three common clinical subtypes according to the stage, location, and severity of the defect: craniorachischisis (CRAN), anencephaly (AN), and spina bifida (SB) [Citation3].
Both genetic and non-genetic factors are reported to be risk modifiers and causative of NTDs. Maternal health and nutritional status are essential to a healthy pregnancy, and numerous studies have indicated that folic acid supplementation during the periconceptional period can reduce the risk of NTDs [Citation4,Citation5]. Unfortunately, while folate is the single largest modifier of NTD risk, there are still NTD-affected infants born to women who took folic acid supplements during the periconceptional period. These cases are reported to be folic acid-resistant or non-folate responsive NTDs [Citation6]. Both genetic and environmental factors, that are not folate responsive, are proposed to modify essential molecular, biochemical, or cellular pathways involved in the aetiology of this subclass of NTDs [Citation7–13].
One carbon metabolism (OCM) is of particular interest regarding NTD risk and prevention given that select OCM genes inactivated in mouse models result in defects that are folate resistant, suggesting variants of these genes may contribute to non-folate responsive human NTD cases [Citation14]. While genetic variants in genes involved in OCM and the planar cell polarity (PCP) pathway are among the most commonly reported genetic factors contributing to NTDs [Citation15,Citation16], there are hundreds of other gene mutations in mice that produce NTDs, but these are not associated with cases or risk of NTDs in humans. Therefore, these data or lack of supporting data indicate that other mechanisms and pathways are also involved in the aetiology of folate-resistant NTDs.
Epigenetic modification has been recently proposed as a contributing factor in the occurrence of NTDs. DNA methylation is one of the most important epigenetic mechanisms involved in chromatin organization and gene expression [Citation17]. Reports have indicated that aberrant methylation of LINE-1, an indicator of genome methylation, is associated with NTDs [Citation18]. Furthermore, aberrant methylation of several specific genes is reported to modify the risk of NTDs, including imprinted genes [Citation19], transposon genes [Citation20], folate receptor genes [Citation21], HOX genes [Citation22], and PCP pathway genes [Citation23]. Our data from a genome-wide DNA methylation microarray study also supports that hypermethylation of the CTNNA1 and MYH2 genes, in the tight junction pathway may contribute to the aetiology of NTDs [Citation24]. We have also reported that hypomethylation of GRHL3 increased risk for NTDs by interfering with the convergent extension elongation processes during the development of the embryonic neural tube [Citation25].
The phosphatidylinositol 3’-kinase-protein kinase B (PI3K-AKT) signalling pathway is one of the most important biological pathways. It can be activated by multiple types of cellular stimuli or toxicants and regulates fundamental cellular functions such as transcription, translation, proliferation, growth, and survival. As depicted in Table S1, several animal models found that PI3K-AKT pathway genes are reported to be associated with risk for NTDs. A previous study reported that PI3K regulates intraepithelial cell positioning during the development of neural tube [Citation26]. Knocking-out of Grhl2 in mice abnormally regulated Lamc2 and caused NTD phenotypes by impairing the movement of the neural folds [Citation27]. Abnormal gene expression of Fgf19 adversely affected the early activity of neural progenitor cells and caused NTDs in mice [Citation28]. Moreover, loss of function of several genes from other pathways may also regulate the expression of Pi3k-Akt signalling pathway genes and lead to NTDs in animal models [Citation29–32]. All these studies strongly suggest that PI3K-AKT pathway genes play key roles in neural tube development. To date, the role of PI3K-AKT pathway genes on NTD risk is supported by animal models, but human clinical evidence supporting the role of PI3K-AKT signalling pathway gene variants in NTD formation is completely lacking.
Herein, we hypothesized that aberrant DNA methylation of PI3K-AKT pathway genes is associated with increased risk for NTDs in humans. To test our hypothesis, we first examined the methylation levels of PI3K-AKT signalling pathway genes in our genome microarray dataset obtained using neural tissues obtained from 10 terminated NTD cases and 8 controls. Candidate genes that harbour differentially methylated CpG sites between cases and controls were identified. We subsequently validated the candidate genes in a larger cohort of NTD cases and controls. Finally, we used human embryonic stem cells (hESCs) and induced hESC cells into neural cell aggregate and the expression of different markers to examine the effect of decreased expression of candidate target genes on neural tube development.
Methods
Participants and samples
The subjects were recruited from five rural counties in Shanxi province of northern China during the years 2011 and 2014. This is a region where the NTD prevalence is one of the highest regions in the world [Citation33]. Cases were electively terminated pregnancies following prenatal diagnosis of an NTD. Controls were terminated foetuses with no gross congenital malformations. Tissue samples from the lesion site of the residual brain or spinal cord were collected from NTD cases, while normal, non-diseased brain or spinal cord tissue was collected from control foetuses. Samples were stored at −80°C until utilized in various analyses. For the genome microarray, 10 NTD cases (6 anencephalics, 4 spina bifidas) and 8 non-malformed controls were included. In a subsequent validation study, 73 NTDs cases and 23 controls were involved. This study was approved by the institutional review board of Peking University and all of the participating mothers provided written informed consent.
Genome-wide DNA methylation assay
In the discovery stage, Infinium HumanMethylation450 (HM450K) BeadChip assay (Illumina, San Diego, CA, USA) was used to investigate genome-wide DNA methylation. The detailed process and quality control methods that were part of the analysis have been previously described elsewhere [Citation24,Citation25]. Briefly, DNA from neural tissues was extracted with the aid of the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). DNA concentration was determined with the NanoDrop2000 Ultra-micro spectrophotometer (Thermo Fisher Scientific, USA). An aliquot of 500 ng DNA was undergone bisulphite conversion with the EZ DNA methylation kit (Zymo Research, CA, USA). Bisulphite conversion efficiency was determined (Supplemental Method.c). Pooled bisulphite-treated DNA was used for subsequent array analysis. Arrays were processed according to the manufacturer’s protocol. Cluster analysis and component analysis with the methylation array data showed that the NTD case group and the non-malformed group could be distinguished from each other, except two controls that were mis-clustered into the case group (Supplementary Figures S1&S2). The methylation data of the PI3K-AKT pathway genes were extracted from the genome-wide array.
Target methylation sequencing assay
In the validation stage, the target-gene methylation sequencing assay was carried out by using Sequenom EpiTYPER (Sequenom, San Diego, USA) platform. In this stage, we chose the target genes based on the following criteria: (i) There were methylated sites that differed significantly and were all hypermethylated or all hypomethylated. (ii) The percentage of the significantly different methylated sites in the total detected CpG sites was greater than 20%.
An aliquot containing 1 μg DNA was used to perform bisulphite conversion with the EZ DNA Methylation Kit (Zymo Research, CA, USA). Then, the bisulphite-treated DNA was amplified by PCR. The PCR primers were designed by the online tool Epidesigner (http://www.epidesigner.com). The amplicons for each gene were targeted to aberrant CpG sites in the HM450K array and CpG islands. The primer sequences are presented in Table S2. The PCR products were incubated with shrimp alkaline phosphatase for 20 minutes and digested by the T cleavage enzyme (T Cleavage Mix) at 37°C for 3 hours. Subsequently, DNA methylation was analysed by the MassARRAY system. The results were analysed using EpiTyper software. Positive controls (completely methylated) and negative controls (non-methylated) were used to ensure the quality of the methylation assay.
H9 hESC cell culture and transfection
The cell line used in this study was H9 human embryonic stem cells (hESCs). For culturing this cell line, we used 100uL Matrigel (BD Biosciences, San Jose, CA), which was added to 48 mL of the DMEM/F12 medium. This media mixture was used to coat the plate with 2 ml/well one night before thawing the cells. hESCs were obtained as frozen vials in mTeSR1 medium (STEMCELL Technologies, Vancouver, BC, Canada). The cells were then cultured in E8 medium, which included supplementary E8 (Life Technologies, Carlsbad, CA) [Citation34–36] with ROCK inhibitor (1 mM) (Y-27,632; R&D Systems, Minneapolis, MN). ROCK inhibitor was removed 24 hours after the cells were attached to the plates. The H9 hESCs were then maintained in E8 medium at 37°C with 5% CO2.
Neural differentiation and siRNA transfection
The hESC cell culture and differentiation process was followed by the previous protocol [Citation34–36]. hESC colonies were rinsed with 1% phosphate-buffered saline (PBS; Life Technologies) and were subsequently treated with Accutase (Life Technologies) for 3 minutes. The cells were harvested after centrifugation for 2 minutes, at which point the cells were re-plated on a Matrigel-coated 24-well with 1.5 high-performance cover glass-bottom plate with a density of 2x103/cm2 in E8 medium with 10 mM ROCK inhibitor and incubated overnight. The E8 medium was replaced the next day by the E6 medium, which contained 10 mM SB431542 (Cellagentech, San Diego, CA (. collagen tech.com) which served to initiate cell differentiation. The medium was changed daily until the cells were used for analysis. On the first day of differentiation, 25pmol siRNAs of si-CHRM1, si-FGF19, si-ITGA7, and si-Scramble were transfected into cells by using Lipofectamine2000 (InvitrogenTM, CA, USA) according to the manufacturer’s protocol. The siRNAs were obtained from IDT and Sangon Biotech (Coralville, IA, USA), for each target gene, three si-RNAs were designed. The sequences are shown in Table S3.
Reverse transcriptase and quantitative PCR (RT-qPCR)
The RT-qPCR assay was performed to examine the knocking-down efficiencies and mRNA levels of target genes. For RT-qPCR analysis, total mRNA was extracted with an RNA extraction kit (Zymo Research). qPCR was carried out with designed primers as seen in Table S4. mRNA levels were determined by using Applied Biosystem SYBR 2X master Mix on an ABI QuantStudio Flex 7. The RT-qPCR assay was performed three times.
Immunofluorescence
Cells were washed twice with PBS and fixed with 4% paraformaldehyde (PFA) for 15 minutes at room temperature. After additional washes with PBS, the cells were blocked and permeabilized in tris-buffered saline (TBS)-DT for one hour, which contained 5% donkey serum and 0.3% Triton X-100. Cells were then incubated in the primary antibodies overnight at 4°C. The primary antibodies are: Anti-SOX2 (Sangon Biotech, D264316-0025, 1:100), Anti-MAP2 (2a+2b) (Sigma-Aldrich, M1406*, 1:100), Anti-PAX6 (ABclonal, A19099, 1:100), Anti-N cadherin (Proteintech, 66,219–1, 1:100). On the following day, the plates were washed with TBS containing 0.3% Triton X-100 (TBST) five times, each time for 5 minutes. Secondary antibodies (1:1,000; Alexa Fluor® 488-conjugated AffiniPure Goat Anti-Rabbit IgG, Alexa Fluor® 594-conjugated AffiniPure Goat Anti-Rabbit IgG, Jackson) were used to stain the cells for 1 hour at room temperature. The cells were finally stained by 300 nM DAPI for 2 minutes. Rosette-like cell aggregates were observed and images were captured by deconvolution microscope (Nikon T2). Fluorescent stained images were taken using a confocal (Leica TCS SP8). Quantitative analysis was performed by ImageJ. We randomly chose five fields of vision and measured the numbers of progenitors and differentiated neurons population. For each si-group, the percentages of each type of cells in the total cells were calculated. The percentages of different types of cells were compared between groups by Pearson’s χ2 test.
Statistical analysis
Pearson’s χ2 test was performed to test the difference in the distribution of population characteristics between NTD cases and controls. In the discovery stage, the original data were preprocessed using the Minfi package of R software. The process of analysing DMPs and DMRs used the IMA package of R software according to the protocol provided by the company (Supplemental Method.a). The quality control and calculation of P and β, which represents the difference of the methylation level between the case and the control groups, were as described in our previous publication [Citation25]. The data were normalized by Subset-quantile Within Array Normalization (SWAN) method (Supplemental Method.b). Independent t-tests were used to identify differentially methylated CpG sites. The P-value was corrected by Benjamini-Hochberg FDR methods to control for false discovery rate. In the validation stage, the differences in proportions of population characteristics between groups were examined using Pearson’s χ2 test. The distributions of methylation values of cases and controls were tested by the Shapiro-Wilk test. Independent samples t-test was used to compare the methylation level of each CpG site between cases and controls. Odds ratios (ORs) and 95% confidence intervals (CI) were estimated by logistic regression, adjusting for maternal education, occupation, and periconceptional folic acid supplementation. In the RT-qPCR assays, the relative expression level of mRNA to GAPDH was calculated. A Student t-test was performed to compare the expression levels between the control group (si-Scramble) and the target gene knock-down group. A two-tail P-value of < 0.05 was considered statistically significant. Statistical analyses were conducted with SPSS 20.0 software (IBM Company, USA).
Results
DNA methylation of PI3K-AKT signalling pathway genes in microarray analysis
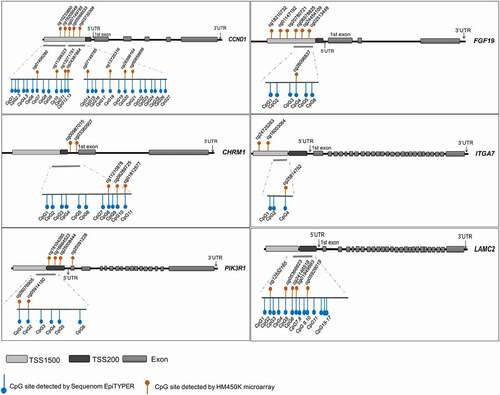
Methylation data of PI3K-AKT signalling pathway genes were extracted from the dataset obtained by Infinium HumanMethylation450 BeadChip assay (HM450K; Illumina, San Diego, CA, USA), using DNA extracted from neural tissues from ten NTD cases and eight controls [Citation24]. As seen in , a total of 31 PI3K-AKT signalling pathway genes with 1008 CpG sites were identified. These CpG sites were primarily located within the TSS1500, TSS200, and 5’-UTR regions. Among these CpG sites, 321 (31.8%) CpG sites were differentially methylated between cases and controls (Adjusted Ps<0.05), of which 203 (63.3% in total differentially methylated sites) were hypermethylated, and 118 (36.7% in total differentially methylated sites) were hypomethylated in the NTD samples.
Table 1. DNA methylation analysis of PI3K-Akt genes detected by microarray: Discovery
Among the detected CpG sites, the differentially methylated CpG sites in 10 PI3K-AKT signalling pathway genes were all hypermethylated in NTD cases. These 10 genes included CCND1, CDK4, CHRM1, KRAS, LAMC2, MCL1, FGF19, ITGA7, PIK3R1, and PRKCA. The percentages of differentially hypermethylated CpG sites to the total number of detected sites were calculated. In six genes, the percentages of significantly hypermethylated sites were greater than 20% ().
The locations of the differentially hypermethylated sites in the six genes are shown in (). The CpG sites in CHRM1 are mainly located in the TSS200 and 5’-UTR region. For the PI3KR1 gene, the differentially hypermethylated CpG sites are in the TSS200 region and first exon. Detailed information on each significantly hypermethylated CpG site is shown in Table S5.
Figure 1. Locations of CpG sites detected by HM450K and Sequenom EpiTYPER.
Targeted DNA methylation analysis in a larger sample
To validate the findings from the above discovery stage, we further examined differentially methylated regions using Sequenom EpiTYPER in 73 NTD cases and 36 controls. We focused on the six genes with more than 20% differentially hypermethylated sites to the total detected sites in the microarray dataset. The demographic characteristics of the subjects are described in (). Occupation, education, and folic acid supplementation showed significantly different distributions between the control and NTD case groups (Ps<0.05), thus these variables were considered as confounding factors in subsequent multivariate analyses.
Table 2. Characteristic of the validation population
As depicted in (), we primarily amplified the DNA regions that covered the CpG sites detected in the microarray. The methylation levels of each CpG site are shown in (). In the CCND1 and LAMC2 genes, none of the detected CpG sites showed a significant increase in NTD cases compared to controls. In the PI3KR1 gene, four significantly hypermethylated sites were replicated. A total of 11 CpG sites were confirmed in CHRM1, of which 10 sites showed significant differences between cases and controls. Three out of six CpG sites in the FGF19 gene exhibited significantly higher methylation levels in cases than in controls. One CpG site in the ITGA7 gene showed significant hypermethylation in NTD cases. All these findings support that hypermethylation of these PI3K-AKT signalling genes is associated with human NTDs. The detailed methylation levels and comparisons between NTD cases and controls are presented in Table S6.
Figure 2. Methylation pattern of target genes using Sequenom EpiTYPER.
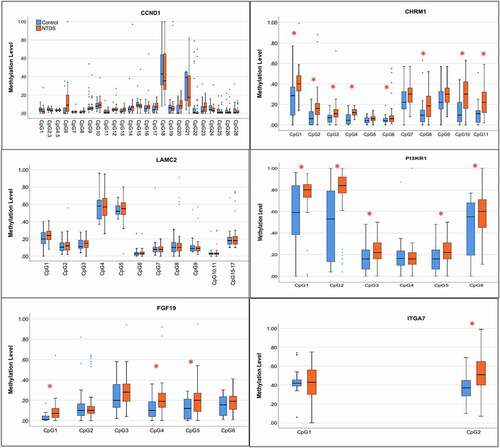
Methylation levels of those genes in the NTD group and the control group were compared (). The mean methylation of CHRM1, FGF19, and ITGA7 in NTDs and controls were significantly different (CHRM1: 21.08% vs 13.73%; FGF19: 19.76% vs 12.56%; ITGA7: 47.23% vs 40.34%), while the mean methylation of CCND1, PI3KR1, and LAMC2 did not show significant differences between NTD cases and controls. Logistic regression analysis demonstrated that hypermethylation of CHRM1, FGF19, and ITGA7 genes were risk factors for NTDs [CHRM1: aOR = 1.19 (95% CI, 1.08–1.30), FGF19: aOR = 1.10 (95% CI, 1.03–1.17), and ITGA7: aOR = 1.04 (1.01–1.08)] after adjusting for confounding factors. We further compared the differences of mean methylation levels between subtypes of NTDs, namely spina bifida and anencephaly, and controls. As shown in (), the mean methylation levels of CHRM1 and FGF19 were significantly higher in both spina bifida and anencephaly than controls. The mean methylation level of ITGA7 was significantly higher only in the spina bifida subgroup, while methylation levels of PI3KR1, CCND1, and LAMC2 showed no significant differences in the subgroup analysis.
Table 3. Mean methylation level and the risk for neural tube defects (NTDs)
Table 4. Mean methylation level and the risk for anencephaly and spina bifida
Knock-down of PI3K-AKT signalling pathway genes impaired neuronal differentiation and inhibited rosette-like cell aggregates formation
In this study, we tested the impact of abnormal expression of target genes on neural differentiation and on the development of neural tube by culturing hESCs and induced the cells into rosette-like cell aggregates [Citation34,Citation35,Citation36]. Based on the results of the above microarray study, we selected three of the most significantly hypermethylated genes in NTD case samples, i.e., CHRM1, FGF19, and ITGA7. The process of hESCs culture and neural differentiation is shown in (). On the first day of differentiation, the siRNAs of CHRM1, FGF19, and ITGA7, as well as the Scrambled control were transfected into cells. The cells were harvested on day 6 post-transfection. RT-qPCR was performed to examine the knock-down effect of the siRNAs and we chose the siRNA of each gene with the best knocking-down efficiency to perform staining assay. Compared with the si-Scrambled group, all the targeted siRNAs significantly decreased the expression of target mRNA (). As () shows, in the negative control of the si-Scrambled group, the regular rosette-like neural cell aggregates were observed, while that was not observed in the siRNA treatment groups, indicating that decreased-expression of the FGF19, CHRM1, or ITGA7 genes impairs regular neural rosette-like cell aggregates formation.
Figure 3. Rosette-like cell aggregate formation in siRNA-treated culture. (a) Culture scheme for establishing neural rosettes. (b) RT-qPCR assay to examine the effect of siRNA knock-down. *: P < 0.05. (c) Immuno-staining of PAX6 and N-cadherin on cells. (d) RT-qPCR assay to examine the expression of PAX6 in each group. *: P< 0.05.
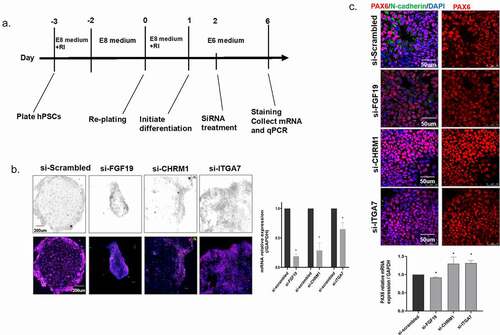
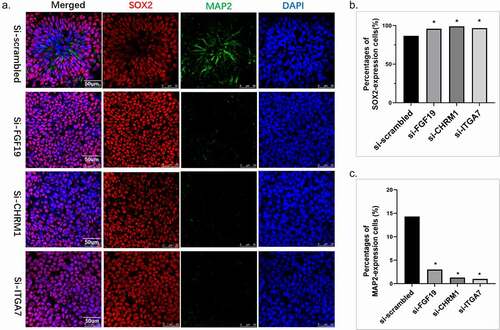
Several markers were subsequently stained to explore the underlying mechanisms. The structure was stained by neural progenitor marker, neuronal cadherin (N-cadherin), and Pax6 (Paired box transcription factor 6), a major driver of neuroectodermal cell fate determination. PAX6 is known to restrict the expression of several pluripotency factors. Both mRNA and the percentage of cells that expressing PAX6 were measured in si-RNAs cells. As a result, the mRNA of PAX6 was significantly elevated in the si-ITGA7 and si-CHRM1 groups (P< 0.05), while the mRNA of PAX6 in si-FGF19 slightly decreased (). We further investigate the neural differentiation by calculating the percentage of numbers that express the neural precursors’ cell marker, SOX2, and the cells express the differentiated neuron marker, MAP2 (). As depicts, the percentages of cells that express SOX2 were increased in the si-RNA groups. The percentages of cells that express MAP2 significantly decreased in si-RNA groups, indicating that the down-regulation of FGF19, CHRM1, and ITGA7 could impair the process of cell differentiation in neural tube development.
Figure 4. Neuronal cell differentiation. (a) Immuno-staining of SOX2 and MAP2 on cells. (b) Quantitative analysis was performed. We randomly chose five fields of vision and calculated the percentages of cells that express SOX2 in the total cells. The percentages of SOX2-expression cells of each si-RNA group were compared with the si-scrambled group. (c) Quantitative analysis was performed. We randomly chose five fields of vision and calculated the percentages of cells that express MAP2 in the total cells. The percentages of MAP2-expression cells of each si-RNA group were compared with the si-scrambled group.
Discussion
The PI3K-AKT signalling pathway is an important regulator of critical facets during early embryonic development. In this study, we evaluated the methylation status of several key genes in the PI3K-AKT signalling pathway, with a two-stage design, to examine whether aberrant methylation of PI3K-AKT pathway genes and associated CpG islands is associated with human NTDs. In the discovery stage, we initially identified a total of 321 differentially hypermethylated CpG sites and 10 genes that differed significantly between cases and controls concerning methylation status, and all were hypermethylated in the NTD cases. In the validation stage, CHRM1, FGF19, and ITGA7 were confirmed as being significantly hypermethylated in NTD patients. The neural rosette assay revealed that down-regulation of FGF19 and CHRM1 impaired neural rosette formation.
Fibroblast growth factor (FGF) protein family plays important roles in multiple functions, including angiogenesis, mitogenesis, pattern formation, cellular differentiation, metabolic regulation, tissue repair, and oncogenesis [Citation37]. Several studies demonstrated that FGF proteins were important for neural tube development by controlling neural crest cell migration [Citation38] and promoting neurulation [Citation39]. The FGFs interact with the Sonic Hedgehog [Citation40], WNT [Citation41], and BMP pathways [Citation42], which are key contributors to neural tube development. It has previously been reported that folate deficiency during early development leads to disturbance of the FGF pathway and results in encephalocele phenotypes in mice [Citation43]. FGF19 is primarily expressed in the foetal brain during embryogenesis [Citation44]. A study showed that FGF19 and WNT8C could mediate mesodermal and neural signals, respectively [Citation45]. A previous study generated a mir-302/367 knockout mouse model and found that the deletion of mir-302/367 resulted in abnormal expression of Fgf19 and disrupted the normal process of neural progenitor cell differentiation, thus leading to an early embryonic lethality and open neural tube defects [Citation28]. Other investigators determined that FGF19 promotes the spontaneous generation of dorsoventrally polarized neural-tube-like structures in cerebellar organoids [Citation46]. At present, no human evidence of the association between FGF19 and NTDs has been reported. Our study represents the first demonstration that hypermethylation of FGF19 was associated with an increased risk for human NTDs, suggesting that hypermethylation of FGF19 may impair embryonic neural tube closure.
Cholinergic receptor muscarinic 1 (CHRM1) modulates hippocampal and cortical functions, thus contributing to an organism’s cognitive makeup, including memory and attention functions. CHRM1 has been reported to be related to neural system disabilities, such as schizophrenia and severe problem in memory and sleep [Citation47–49]. However, the role of CHRM1 in the development of NTDs has not previously been reported. This study detected, for the first time, hypermethylation of CHRM1 in neural tissue of NTDs, and the association was validated in a larger cohort of subjects. We found that hypermethylation of CHRM1 was associated with both anencephaly and spina bifida. Our study provides novel evidence supporting the role of CHRM1 in human NTD cases.
Integrins are heterodimeric membrane glycoproteins that mediate a wide spectrum of cell-cell and cell-matrix interactions that have key roles in cell migration and morphological development, differentiation, and metastasis. The integrin alpha-7 (ITGA7) is specifically involved in differentiation and migration processes. ITGA7 has been well studied concerning the aetiology of muscular disorders and cancers. However, scant attention has been paid to congenital malformations. A study reported that ITGA7 promotes proliferation, invasion, and migration of papillary thyroid carcinoma cells through regulating epithelial-to-mesenchymal transition [Citation50]. Before our studies, there was no previous evidence linking hypermethylation of ITGA7 to an increased risk for human NTDs.
PI3KR1 is a subunit of phosphoinositide 3-kinase (PI3K), which is a lipid kinase that phosphorylates the inositol ring of phosphatidylinositol and related compounds at the 3-prime position [Citation51]. PI3KR1 plays a crucial role in the metabolism of glucose and inositol [Citation52]. In humans, variants in the PI3KR1 gene are associated with SHORT syndrome and immunodeficiency [Citation53,Citation54]. In this study, we found that methylation levels of PI3KR1 in NTD cases were significantly higher than in controls. But after adjusting for maternal occupation, education, and folic acid supplementation, the association turned out to be insignificant. Thus, more studies are needed to examine the role of this gene in the aetiology of NTDs.
Transcription start sites (TSS) and 5’-UTRs are important components promoter regions that control gene expression levels [Citation55]. Previous work has focused on DNA methylation in CpG islands at TSS and showed that aberrant methylation in TSS significantly impacted the regulation of gene expression [Citation56]. In this study, we focused on specific promoter regions with altered CpG methylation. The hypermethylation of gene promoters are inversely associated with gene expression, that is, the gene hypermethylation in the promoter region would down-regulate the expression of the gene in question [Citation57]. In this study we used siRNAs to knock down the expression of FGF19, CRHM1 and ITGA7 genes in hESCs to investigate whether low expression of the target genes impairs neural rosette development. We demonstrated that down-regulating the expression of those three genes could influence the differentiation of neural cells.
Many previous studies used blood as a source of DNA with which to investigate the impact of altered methylation and the aetiology of NTDs. However, DNA methylation is considered to be tissue-specific; therefore, it is crucial to detect the methylation level in the specific lesion site. If one wants to understand methylation issues that are relevant to NTD aetiology, then the nervous tissues at the site of failed neural tube closure should be examined. In the current study, we examined neural tissues from NTD foetuses and controls, which makes our findings uniquely relevant to the outcome in question. However, because of the timing of neural tube closure in human embryos, we were limited to obtaining the tissue weeks after the neural tube failed to close. We, therefore, cannot completely rule out the possibility that the observed aberrant methylation occurs secondary to the lesion site proper. Besides, considering that it is difficult to obtain the fresh tissues of the lesion site and neural system, mRNA expression levels of the human tissues are hard to be examined.
In conclusion, for the first time, we were able to show that hypermethylation in several genes in select key genes of the PI3K-AKT signalling pathway (e.g. FGF19 and CHRM1) may be involved in the occurrence of human NTDs. Our study provides new insight into epigenetic mechanisms underlying NTD formation. Further studies are warranted to explore whether the aberrant methylation of PI3K-AKT signalling pathway genes interacts with nutritional factors such as inositol, glucose, or folic acid, which are known nutritional modifiers of NTD risk.
Declarations
Ethics approval and consent to participate
This study was approved by the institutional review board of Peking University and all of the participating mothers provided written informed consent.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abions
FDR: False discovery rate; HM450K: HumanMethylation450 BeadChip; hESCs: Human embryonic stem cells; OR: Odds ratio; PCR: Polymerase chain reaction; NTDs: Neural tube defects; SB: Spina bifida; AN: Anencephaly; TSS: Transcription start sites; FGF: Fibroblast growth factor; CHRM1: Cholinergic receptor muscarinic 1; PI3K: Phosphoinositide 3-kinase.
Supplemental Material
Download MS Word (17.3 MB)“Availability of data and materials“
Aiguo Ren and Linlin Wang conceptualized the study and supervised the implementation of the study. Tian Tian conducted the methylation assay and functional study and drafted the manuscript. Xinyuan Lai performed the second hESC experiment. Kuanhui Xiang, Shengju Yin, Robert M. Cabrera and John W. Steele provided crucial guidance on the neural rosette study. Xiao Han conducted the RT-qPCR assay and helped with cell culture experiments. Richard H. Finnell, Robert M. Cabrera, John W. Steele, and Yunping Lei edited and critically revised the manuscript.
Disclosure statement
Dr. Finnell formerly held a leadership position with the now dissolved TeratOmic Consulting LLC. He also receives travel funds to attend editorial board meetings of the Journal of Reproductive and Developmental Medicine published out of the Red Hospital of Fudan University.
Supplemental material
Supplemental data for this article can be accessed here.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Wallingford JB, Niswander LA, Shaw GM, et al. The continuing challenge of understanding, preventing, and treating neural tube defects. Science. 2013;339(6123):1222002.
- Copp AJ, Greene ND. Genetics and development of neural tube defects. J Pathol. 2010;220(2):217–230.
- Salih MA, Murshid WR, Seidahmed MZ. Classification, clinical features, and genetics of neural tube defects. Saudi Med J. 2014;35(Suppl 1):S5–S14.
- Daly LE, Kirke PN, Molloy A, et al. Folate levels and neural tube defects. Implications for prevention. JAMA. 1995;274(21):1698–1702.
- Mills JL, Signore CC. Folic acid and the prevention of neural-tube defects. N Engl J Med. 2004;350(21):2209–2211. author reply 2209–2211.
- Copp AJ, Stanier P, Greene ND. Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol. 2013;12(8):799–810.
- Boyles AL, Hammock P, Speer MC. Candidate gene analysis in human neural tube defects. Am J Med Genet C Semin Med Genet. 2005;135C(1):9–23.
- Lei YP, Zhang T, Li H, et al. VANGL2 mutations in human cranial neural-tube defects. N Engl J Med. 2010;362(23):2232–2235.
- Chen Z, Lei Y, Cao X, et al. Genetic analysis of Wnt/PCP genes in neural tube defects. BMC Med Genomics. 2018;11(1):38.
- Lei Y, Kim SE, Chen Z, et al. Variants identified in PTK7 associated with neural tube defects. Mol Genet Genomic Med. 2019;7(4):e00584.
- Kibar Z, Bosoi CM, Kooistra M, et al. Novel mutations in VANGL1 in neural tube defects. Hum Mutat. 2009;30(7):E706–715.
- Kharfallah F, Guyot MC, El Hassan AR, et al. Scribble1 plays an important role in the pathogenesis of neural tube defects through its mediating effect of Par-3 and Vangl1/2 localization. Hum Mol Genet. 2017;26(12):2307–2320.
- Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol. 2010;88(8):633–652.
- Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr. 2010;30:57–81.
- Steele JW, Kim SE, Finnel RH. One-carbon metabolism and folate transporter genes: do they factor prominantly in the genetic etiology of neural tube defects? Biochimie. 2020;173:27–32.
- Juriloff DM, Harris M. A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2012;94(10):824–840.
- Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213(2):384–390.
- Wang L, Chang S, Guan J, et al. Tissue-Specific methylation of long interspersed nucleotide element-1 of homo sapiens (L1Hs) during human embryogenesis and roles in neural tube defects. Curr Mol Med. 2015;15(5):497–507.
- Wu L, Wang L, Shangguan S, et al. Altered methylation of IGF2 DMR0 is associated with neural tube defects. Mol Cell Biochem. 2013;380(1–2):33–42.
- Wang L, Wang F, Guan J, et al. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am J Clin Nutr. 2010;91(5):1359–1367.
- Farkas SA, Bottiger AK, Isaksson HS, et al. Epigenetic alterations in folate transport genes in placental tissue from fetuses with neural tube defects and in leukocytes from subjects with hyperhomocysteinemia. Epigenetics. 2013;8(3):303–316.
- Rochtus A, Izzi B, Vangeel E, et al. DNA methylation analysis of Homeobox genes implicates HOXB7 hypomethylation as risk factor for neural tube defects. Epigenetics. 2015;10(1):92–101.
- Shangguan S, Wang L, Chang S, et al. DNA methylation aberrations rather than polymorphisms of FZD3 gene increase the risk of spina bifida in a high-risk region for neural tube defects. Birth Defects Res A Clin Mol Teratol. 2015;103(1):37–44.
- Wang L, Lin S, Zhang J, et al. Fetal DNA hypermethylation in tight junction pathway is associated with neural tube defects: A genome-wide DNA methylation analysis. Epigenetics. 2017;12(2):157–165.
- Tian T, Wang L, Shen Y, et al. Hypomethylation of GRHL3 gene is associated with the occurrence of neural tube defects. Epigenomics. 2018;10(7):891–901.
- Torroba B, Herrera A, Menendez A, et al. PI3K regulates intraepithelial cell positioning through Rho GTP-ases in the developing neural tube. Dev Biol. 2018;436(1):42–54.
- Ray HJ, Niswander LA. Grainyhead-like 2 downstream targets act to suppress epithelial-to-mesenchymal transition during neural tube closure. Development. 2016;143(7):1192–1204.
- Yang SL, Yang M, Herrlinger S, et al. MiR-302/367 regulate neural progenitor proliferation, differentiation timing, and survival in neurulation. Dev Biol. 2015;408(1):140–150.
- Wang B, Zhang Y, Dong H, et al. Loss of Tctn3 causes neuronal apoptosis and neural tube defects in mice. Cell Death Dis. 2018;9(5):520.
- Wilson NR, Olm-Shipman AJ, Acevedo DS, et al. SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci Rep. 2016;6:17735.
- Garlena RA, Lennox AL, Baker LR, et al. The receptor tyrosine kinase Pvr promotes tissue closure by coordinating corpse removal and epidermal zippering. Development. 2015;142(19):3403–3415.
- Pickett EA, Olsen GS, Tallquist MD. Disruption of PDGFRalpha-initiated PI3K activation and migration of somite derivatives leads to spina bifida. Development. 2008;135(3):589–598.
- Liu J, Zhang L, Li Z, et al. Prevalence and trend of neural tube defects in five counties in Shanxi province of Northern China, 2000 to 2014. Birth Defects Res A Clin Mol Teratol. 2016;106(4):267–274.
- Philipp K, Thoralf O, Julius AS, et al. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration.PNAS.2009;106(9):3225-3230.
- Lippmann ES, Estevez-Silva MC, Ashton RS. Defined human pluripotent stem cell culture enables highly efficient neuroepithelium derivation without small molecule inhibitors. Stem Cells. 2014;32(4):1032–1042.
- Fedorova V, Vanova T, Elrefae L, et al. Differentiation of neural rosettes from human pluripotent stem cells in vitro is sequentially regulated on a molecular level and accomplished by the mechanism reminiscent of secondary neurulation. Stem Cell Res. 2019;40:101563.
- Nishimura T, Utsunomiya Y, Hoshikawa M, et al. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim Biophys Acta. 1999;1444(1):148–151.
- Zhang XT, Wang G, Li Y, et al. Role of FGF signalling in neural crest cell migration during early chick embryo development. Zygote. 2018;26(6):457–464.
- Kindt LM, Coughlin AR, Perosino TR, et al. Identification of transcripts potentially involved in neural tube closure using RNA sequencing. Genesis. 2018;56(3):e23096.
- Morales AV, Espeso-Gil S, Ocana I, et al. FGF signaling enhances a sonic hedgehog negative feedback loop at the initiation of spinal cord ventral patterning. Dev Neurobiol. 2016;76(9):956–971.
- Turner DA, Hayward PC, Baillie-Johnson P, et al. Wnt/beta-catenin and FGF signalling direct the specification and maintenance of a neuromesodermal axial progenitor in ensembles of mouse embryonic stem cells. Development. 2014;141(22):4243–4253.
- Hegarty SV, O’Keeffe GW, Sullivan AM. BMP-Smad 1/5/8 signalling in the development of the nervous system. Prog Neurobiol. 2013;109:28–41.
- Chang S, Lu X, Wang S, et al. The effect of folic acid deficiency on FGF pathway via Brachyury regulation in neural tube defects. FASEB J. 2019;33(4):4688–4702.
- Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331(6024):1621–1624.
- Ladher RK, Anakwe KU, Gurney AL, et al. Identification of synergistic signals initiating inner ear development. Science. 2000;290(5498):1965–1967.
- Muguruma K, Nishiyama A, Kawakami H, et al. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10(4):537–550.
- Dean B, McLeod M, Keriakous D, et al. Decreased muscarinic-1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Molec Psychiat. 2002;7:1083–1091.
- Anagnostaras SG, Murphy GG, Hamilton SE, et al. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nature.Neurosci. 2003;6:51–58.
- Niwa Y, Kanda GN, Yamada RG, et al. Muscarinic acetylcholine receptors Chrm1 and Chrm3 are essential for REM sleep. Cell Rep. 2018;24(2231–2247):2018.
- Guan YBA, Xia E, Kong L, et al. Downregulating integrin subunit alpha 7 (ITGA7) promotes proliferation, invasion, and migration of papillary thyroid carcinoma cells through regulating epithelial-to-mesenchymal transition. Acta Biochim Biophys Sin. 2020;52(2):116–124.
- Whitman M, Kaplan DR, Schaffhausen B, et al. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature Neurosci. 1985;315(6016):239–242.
- Terauchi Y, Tsuji Y, Satoh S, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85-alpha subunit of phosphoinositide 3-kinase. Nature Genet. 1999;21:230–235.
- Elkaim E, Neven B, Bruneau J, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: a cohort study. J Allergy Clin Immun. 2016;138:210–218.
- Hauvin-Robinet C, Auclair M, Duplomb L, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93:141–149.
- PA J. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492.
- Farthing CRFG, Ng RK, Chan CF, et al. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS.Genet. 2008;4(6):e1000116.
- X Yj Z, Sundaresan A, Cokus S, et al. Genomewide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126(6):1189–1201.