
ABSTRACT
Organisms exposed to endocrine disruptors in early life can show altered phenotype later in adulthood. Although the mechanisms underlying these long-term effects remain poorly understood, an increasing body of evidence points towards the potential role of epigenetic processes. In the present study, we exposed hatchlings of an isogenic lineage of the self-fertilizing fish mangrove rivulus for 28 days to 4 and 120 ng/L of 17-α-ethinylestradiol. After a recovery period of 140 days, reduced representation bisulphite sequencing (RRBS) was performed on the liver in order to assess the hepatic genome-wide methylation landscape. Across all treatment comparisons, a total of 146 differentially methylated fragments (DMFs) were reported, mostly for the group exposed to 4 ng/L, suggesting a non-monotonic effect of EE2 exposure. Gene ontology analysis revealed networks involved in lipid metabolism, cellular processes, connective tissue function, molecular transport and inflammation. The highest effect was reported for nipped-B-like protein B (NIPBL) promoter region after exposure to 4 ng/L EE2 (+ 21.9%), suggesting that NIPBL could be an important regulator for long-term effects of EE2. Our results also suggest a significant role of DNA methylation in intergenic regions and potentially in transposable elements. These results support the ability of early exposure to endocrine disruptors of inducing epigenetic alterations during adulthood, providing plausible mechanistic explanations for long-term phenotypic alteration. Additionally, this work demonstrates the usefulness of isogenic lineages of the self-fertilizing mangrove rivulus to better understand the biological significance of long-term alterations of DNA methylation by diminishing the confounding factor of genetic variability.
Introduction
Early-life exposure to environmental stressors, encountered during the sensitive period of embryogenesis or in juveniles, can be critical in shaping the long-term control of tissue physiology and homoeostasis. Although this paradigm, referred to as the Developmental Origins of Health and Disease (DOHaD) [Citation1], is now widely recognized, the molecular mechanisms by which early exposures influence the propensity of disease and phenotype later in life remain elusive. Identifying these mechanisms has become extremely important to understand the long-term effects of toxicants in human and wildlife and ensure the proper risk assessment of xenobiotic exposure [Citation2].
Nowadays, endocrine disrupting chemicals (EDCs) – xenobiotics able to interfere with the proper functioning of the endocrine system – are ubiquitous in the environment and our everyday life. The timely release and tightly regulated concentrations of hormones in early life are crucial for the proper development of the organism, including the reproductive, nervous, and immune systems. Therefore, early-life exposure to EDCs can have dramatic consequences on homoeostasis and physiology. In fact, mounting epidemiological evidence link exposures to EDCs to the increased incidence of metabolic diseases, immune diseases, neurological disorders, cancer and alteration of fertility in humans [Citation3–6]. Of the many EDCs present in the aquatic environment, 17-α-ethinylestradiol (EE2) – a synthetic derivative of oestradiol used in oral contraceptives – is of particular concern due to its high potency and resistance to degradation [Citation7]. In treated seawage, EE2 is often the major compound with oestrogenic activity and its impact on aquatic wildlife has been deeply investigated (reviewed in [Citation8]). In aquatic species, field studies and laboratory experiments show that exposure to EE2 affects fecundity, fertility, reproductive behaviour and induces intersex, endangering natural populations [Citation9].
Potential long-term and persistent effects of early-life EDC exposure involve the stable alteration of gene expression. This is possible through epigenetic regulation, involving histone modifications, non-coding RNAs, and DNA methylation. The latter refers to the transfer of a methyl group from a methyl-donor, S-adenosyl-methionine (SAM), to a cytosine in the context of CpG dinucleotides, forming a methylcytosine [Citation10,Citation11]. This process is catalysed by two families of DNA methyltransferases (DNMTs): whereas DNMT3 enzymes are responsible for de novo methylation during development and differentiation, the DNMT1 family is maintaining methylation through each cell division by copying hemimethylated DNA, making DNA methylation a stable and heritable modification. On the other hand, the addition of a hydroxyl group to methylcytosine by ten-eleven translocation (Tet) enzymes is one of the most common mechanisms of active demethylation [Citation12,Citation13]. In mammals, most DNA methylation occurs in the symmetric CpG context, while only a small amount of non-CpG methylation is observed [Citation10,Citation11]. DNA methylation regulates gene expression by altering chromatin state and accessibility to CpG sites. For instance, methylation occurring at promoter regions can prevent the binding of transcription factors, resulting in gene silencing. Yet, the relationship between methylation and gene expression is more complex, as methylation also occurs at intragenic sites, enhancers or suppressor elements [Citation13,Citation14]. DNA methylation is dynamically regulated throughout life: during gametogenesis and embryogenesis, two waves of demethylation/remethylation deeply reprogram the methylome to produce a totipotent zygote [Citation14–16]. Then, DNA methylation plays an important role throughout development as it directs cellular differentiation after reprogramming. During early development and juvenile stages, DNA methylation is thus thought to be particularly sensitive to environmental factors [Citation17].
An increasing number of studies highlight the potential roles of DNA methylation in mediating long-term effects of sub-toxic developmental exposure to xenobiotics and roles in the aetiology of diseases including cancer, obesity, cardiovascular disease, diabetes, hypertension, and neurodegenerative disorders [Citation18–22]. Indeed, DNA methylation could act as a long-term memory of past exposures mediating stable changes in gene expression [Citation23,Citation24]. In ecotoxicology, the role of epigenomics is receiving more and more attention to explain the long-term delayed and potential transgenerational effects of xenobiotics [Citation25–27]. In particular, accumulating evidence indicates that hormones and endocrine disrupting compounds can alter the epigenome [Citation21,Citation28,Citation29]. For instance, the regulation of DNMT transcription by ESR1 (an oestrogen receptor that acts as a transcription factor) may represent one of the possible mechanisms by which hormones influence methylation [Citation30]. Other potential mechanisms may involve the reduction of SAM availability [Citation31], the alteration of histone activity as a result of membrane receptor oestrogenic signalling [Citation32,Citation33], the expression of miRNAs, or direct interactions between oestrogen receptors and enzymes involved in the methylation machinery, such as thymine-DNA glycosylase (TDG) [Citation34].
So far, relatively few studies have examined the effects of endocrine disruptors on DNA methylation in aquatic species. Using bisulphite conversion and pyrosequencing, Strömqvist et al. [Citation35] have shown decreased methylation levels of three CpG sites located in the 5' flanking region of the vitellogenin I gene, a known biomarker of oestrogenic exposure, in the liver of male and female zebrafish, following a 14-day exposure to 100 ng/L EE2. Using Methylated DNA Immunoprecipitation (MeDIP) and high-throughput sequencing, followed by validation using bisulphite sequencing PCR (BSP) and RT-PCR, Mirbahai et al. [Citation36] investigated the whole-genome methylation and gene expression in tumours in the liver of the Common dab (Limanda limanda), sampled in various sites in English rivers. Genes involved in pathways related to cancer, including apoptosis, wnt/β-catenin signalling and genomic and non-genomic oestrogen responses, were altered both in methylation and transcription. In that case the exact mixture of environmental contaminants causing the liver tumours was not identified but the molecular responses point towards oestrogenic disruption. On zebrafish early life stages, Falisse et al. [Citation37] showed that exposure to the antibacterial agent and EDC triclosan modified the methylome after 7 days exposure, as well as the expression of related genes, using Reduced Representation Bisulphite Sequencing (RRBS).
The identification of environmentally-induced alterations in the epigenome ideally requires the use of individuals with no existing or very low genetic variability to rule out the confounding factors of the genotype on the observed phenotype [Citation38]. Such a model species is provided by the mangrove rivulus, Kryptolebias marmoratus, one of the only known self-fertilizing vertebrates (the other one being its sister species, K. hermaphroditus). Although males and hermaphrodites are present in natural populations, most reproduction occurs by self-fertilization of hermaphrodites. Exclusive selfing of hermaphrodites during several generations results in highly homozygous and virtually ‘clonal’ lineages that can occur naturally or be bred in a laboratory setting [Citation39,Citation40]. A naturally clonal and sexually reproducing vertebrate species allows the study of epigenetic mechanisms ruling out the confounding factor of genetic variability, which is especially useful in ecotoxicology experiments in the sublethal range and/or dealing with delayed effects where non-dramatic molecular changes are expected. Little is known about the epigenetic mechanisms in K. marmoratus but it has been reported that the sex-ratio can be modulated by temperature-sensitive DNA methylation [Citation41]. Recent studies described the DNA methylation reprogramming event during embryogenesis [Citation42], as well as the time course expression of enzymes involved in epigenetics during its development [Citation43,Citation44]. Another study has shown that parasite load can modify DNA methylation [Citation45], while these authors also found a higher differentiation of the methylation landscape between different genotypes than between environments [Citation46].
In previous studies, we successfully used the mangrove rivulus to assess the delayed effects of BMAA (beta-N-Methylamino-L-alanine) [Citation47] and 17-α-ethinylestradiol (EE2) [Citation48,Citation49] in adults after an early-life stage exposure. Hatchlings were exposed to 4 and 120 ng/L EE2 for 28 days, and allowed to recover for 140 additional days in clean water. The 28 days exposure has been chosen to include a 4 weeks period of exposure during early life stages that would be long enough to potentially induce endocrine effects and to be environmentally relevant. At 168 dph, all the fish are sexually mature. This design represents a ratio 1:5 between the exposure and the recovery periods. We reported delayed and persistent effects of EE2 on growth, reproduction and steroid levels as well as liver, ovotestis and brain molecular phenotypes assessed by label-free quantitative proteomics. Effects of EE2 were tissue and dose dependent, with most effects occurring at the environmentally relevant concentration (4 ng/L) in brain and liver, and at the higher concentration in the gonads. Among the three studied organs, the liver showed the most sensitive response to EE2. In this organ, EE2 affected known oestrogen-responsive pathways such as lipid, fatty acid and steroid metabolism, apolipoproteins, innate immune system and inflammation. Interestingly, several proteins involved in SAM metabolism were affected in the liver, providing further indications of the potential effect of EE2 on DNA methylation. The liver serves an essential and conserved role for metabolic homoeostasis in all vertebrates, and reproduction in the case of oviparous species. Several studies have shown that the liver is an important target of endocrine disruption in mammals [Citation50] and fish [Citation36,Citation51–53]. Using an RRBS approach, the present study aimed at characterizing the hepatic methylation landscape in adults and investigating the genome-wide changes in DNA methylation in the liver of 168 dph adults that were exposed to EE2 as hatchlings to test the hypothesis of the potential involvement of DNA methylation in the delayed effects of EDCs.
Materials and methods
Experimental fish, EE2 exposure and sample collection
Individuals used in this study were from the same experiment reported in Voisin et al. [Citation48], which also described the generation of breeding stock population and collection of eggs. Upon hatching, mangrove rivulus were transferred to 300 mL glass jars filled with 100 mL of 25 ppt reconstituted salt water (Instant Ocean™ sea salt), and the corresponding treatment: vehicle control (0.000012% ethanol), 4 ng/L and 120 ng/L 17-α-ethinylestradiol (EE2) (Sigma-Aldrich E4876-1 G). Fish were raised at a temperature of 26°C and a 12:12 h photoperiod. Detailed preparation of EE2 solutions and the measurement of actual exposure concentrations by ELISA are reported in Voisin et al. [Citation48]. Average measured concentration of the nominal 120 ng/L exposure solution was 132.3 ± 14.7 ng/L. Since 4 ng/L is situated below the detection limit (20 ng/L), we refer to the measure of the stock solution, which was 10.15 ± 0.15 µg/L for the nominal 10 µg/L stock solution. Throughout the paper, we will refer to the nominal concentrations of 4 and 120 ng/L. Fish were exposed for a period of 28 days with a 100% water renewal 3 times a week. At 28 dph, fish were transferred to individual 1.2 L plastic containers filled with 400 mL of 25 ppt clean salt water and reared until 168 dph. Rivulus were fed 1 mL of concentrated newly hatched brine shrimp (Artemia nauplii) until 56 dph, 2 mL starting at 56 dph and 4 mL starting at 91 dph. At 168 dph, fish were sacrificed by a rapid transfer to 4°C 25 ppt salt water and sectioning the spinal cord. For each experimental condition, livers from five individuals were dissected out at 168 dph, snap-frozen in liquid nitrogen and stored at −80°C until DNA extraction. All rivulus husbandry and experimental procedures were performed in accordance with the Belgian animal protection standards and were approved by the University of Namur Local Research Ethics Committee (UN KE14/230). The agreement number of the laboratory for fish experiments is the LA1900048.
DNA extraction, RRBS library preparation and post-processing
Genomic DNA from individual livers was extracted using the NucleoSpin Tissue XS kit (Macherey-Nagel, Germany) following the manufacturer’s protocol. DNA concentration and quality were assessed using the NanoDrop 8000 spectrophotometer (Thermo Scientific, Waltham, MA) and 1% agarose gel electrophoresis. A total of 15 RRBS libraries (5 for each experimental condition) were prepared following established protocols [Citation54,Citation55]. In brief, genomic DNA was digested with MSPI (New England Biolabs, Ipswich, MA) followed by end repair, addition of 3′ A overhangs and addition of methylated adapters (Illumina, San Diego, CA) to the digested fragments. Following adapter ligation, size selection was performed by cutting 40–220 bp DNA fragments (pre-ligation size) from a 3% (w/v) NuSieve GTG agarose gel (Lonza, Basel, Switzerland). Subsequently, libraries were bisulphite converted using the EZ DNA methylation kit (Zymo Research, Irvine, CA) with an extended incubation time of 18–20 h. Bisulphite converted libraries were amplified by PCR and sequenced (100-bp single ended reads) by New Zealand Genomics Limited (University of Otago, New Zealand) on an Illumina HiSeq2000 sequencer. Sequences were obtained in FASTQ format.
The quality of sequenced reads was performed using the FastQC application (Babraham Institute, Cambridge, UK). Adaptor sequences were removed from the reads using the cleanadaptors program of the DMAP package [Citation56]. Single-ended bisulphite reads were aligned against the Kryptolebias marmoratus assembly (GCF_001649575.1_ASM164957v1) using the Bismark software v0.17.0 (default mode: alignments to complementary strands were ignored – i.e., not performed) [Citation57]. Proximal genes and genomic positions of the fragments were identified using the identgeneloc programme from the DMAP package [Citation58]. Fragments were linked to the nearest protein-coding gene and the genomic position was attributed as follows: internal (within the gene body), promoter (0–5 kb upstream of the transcription start site), upstream (5–10 kb upstream of the TSS) and intergenic (>10 kb). The annotation of intergenic fragments or fragments located upstream of genes (5–10 kb) and in intergenic regions (>10 kb) is provided as an indication and should be considered with caution as methylation of these regions may not have any influence on the corresponding gene expression. Fragments with a methylation level ≥ 80% are qualified as highly methylated, while those with a methylation level ≤ 20% are qualified as lowly methylated.
Analysis of differential DNA methylation
Base resolution differences in DNA methylation following developmental exposure to EE2 were assessed based on MSPI fragments (40–220 bp range) as a unit of analysis. To filter the fragments suitable for differential methylation analysis, we selected the fragments with ≥ 2 CpG sites, ≥10 reads per fragment (CpG10) and present in at least three of five individuals for each experimental condition. These fragments are referred to as ‘analysed fragments’. The 102,018 analysed fragments form our reduced representative genome (RR genome) and corresponds to a total of 14,679 unique RefSeq identifiers, as more than one fragment often maps to a same gene. Differential methylation analysis was performed as previously described using the diffmeth program from DMAP [Citation56,Citation58] between each experimental group, resulting in three group comparisons: Ctl versus 4 ng/L EE2 exposed individuals, Ctl versus 120 ng/L and 4 versus 120 ng/L. Determination of significantly differentially methylated fragments (DMFs) was based on ≥ 10% differential methylation between the groups and P-value <0.01, and are qualified as hyper- or hypomethylated fragments.
Network analysis
RefSeq identifiers were retrieved using BLAST for all analysed DMFs. To establish a reference gene list for the network analysis, a list including all DMFs was filtered to include only gene body and promoter genomic locations and contained 68,762 fragments mapped to rivulus RefSeq and human RefSeq identifiers. As more than one fragment often mapped to a same gene, this list corresponded to 10,440 unique human RefSeq identifiers. The list, containing P-values and methylation difference for each of the three treatment comparisons was uploaded in the Ingenuity Pathway Analysis software (IPA, QIAGEN, www.qiagen.com/ingenuity). To gain an overall view of the pathways and functions associated with promoter and gene body DMFs, as well as the connections between differentially abundant proteins, we performed a network analysis for each group comparison.
Results
Characterization of the hepatic DNA methylation landscape in K. marmoratus
Using RRBS, we generated 15 K. marmoratus liver methylomes with an average of 29.5 million sequenced reads (between 19.7 and 40 million reads) (, Supplementary Table 1). Unique alignment efficiency of these libraries ranged from 40.3% to 46.8% (except the library 23–4 with 28.5%, which was discarded in further analyses), with an average of 44.4%. This corresponds to a total of 102,018 fragments suited for the analysis after MSPI digestion of genomic DNA and 40–220 bp size-selection (). The total size of the RR genome was 9.37 Mb, which accounted for 1.4% of the published full genome (GCF_001649575.1_ASM164957v1) [Citation59]. Within this RR genome, we counted 701,862 CpG dinucleotides, representing 6.1% of the 11.5 million CpG in the reference genome. Consequently, the enrichment factor in CpGs of the RR genome was 4.3 fold. Of all the analysed fragments, the majority was located within gene bodies (47%), followed by intergenic regions (30%), promoter (16%) and upstream regions (7%) (). The distribution of CpGs followed exactly the same pattern as the fragments (data not shown). We consider this dataset of about 6% of all genomic CpGs, albeit not uniformly distributed across the genome, as a representative sample that permits the investigation of changes in DNA methylation in liver due to exposure to toxicants. Global CpG methylation levels in liver tissue ranged from 63.5% to 66.4% in all samples. On the contrary, methylation level of non-CpG cytosines remained low between 1.7 and 2.4%. It should be noted that non-CpG methylation calculated here included the true genomic non-CpG methylation and the CpGs that failed to convert during bisulphite treatment.
Table 1. Characteristics of the reduced representative genome and comparisons with the genome
Figure 1. Hepatic DNA methylation landscape in K. marmoratus . (a) Frequency distribution of MSPI fragment size. Mean fragment size was 95 bp and median 88 bp; (b) Overall distribution of analysed fragments in genomic elements; (c:)) Frequency distribution of DNA methylation % for MSPI fragments. Distribution of MSPI fragments follows a bimodal distribution, which is expected given the binary character of DNA methylation in cells; (d) Boxplot of DNA methylation (%) of fragments according to their location in genomic elements (including medians and quantile 25–75%).
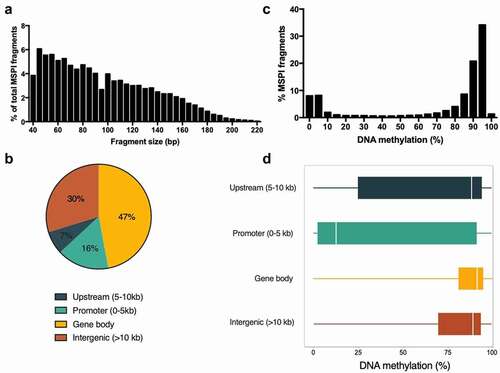
For the following description, we took only the control group into consideration, in order to avoid any possible effect of the EE2 treatment. The average DNA methylation level of most MSPI fragments in mangrove rivulus were above 80% (67% of all fragments – considered as highly methylated) or below 20% of methylation (20% of all fragments – considered as lowly methylated) (). The level of DNA methylation was the lowest in fragments located in promoter regions (mean = 42.3%; median = 12.8%) compared to upstream (mean = 66.2%; median = 88.4%), intergenic (mean = 72.6%; median = 89.1%) and gene body (mean = 78.0%; median = 91.5%). depicts the distribution and spread (25–75 quantiles) of DNA methylation level for fragments within each region. We can observe that the methylation range was the highest for fragments located in the promoter region, showing that even if the majority of these fragments were lowly methylated (52%), a considerable part was highly methylated (38%). Despite the fact that fragments in promoter regions represented only 16% of the total analysed fragments, they accounted for 42% of all lowly methylated fragments (), compared to fragments located in gene bodies (27%), intergenic (23%) and upstream (8%) regions. In contrast, only 9% of all highly methylated fragments were located in the promoter regions, compared to 53% in gene bodies, 31% in intergenic regions and 7% in upstream regions (). Supplementary Figure 1 depicts the coefficient of variability of the methylation level within each genomic region, for lowly methylated fragments on the one hand, and for highly methylated fragments on the other hand. Highly methylated fragments presented a low level of variability (< 3%), in opposition to lowly methylated fragments for which the coefficient of variability was higher than 33%. This pattern was the same for all the fragments, independently of the genomic region.
Figure 2. Distribution of lowly methylated (<20% methylation) (a) and highly methylated (> 80% methylation) (b) fragments among the genomic regions (gene body, promoter, upstream, intergenic) of the liver reduced representative genome of the mangrove rivulus.

Changes in DNA methylation following developmental exposure to EE2
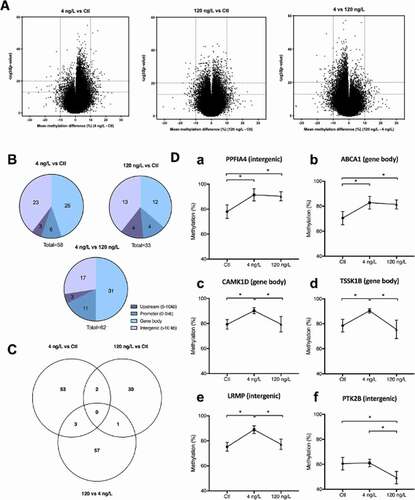
Global DNA methylation levels of rivulus liver averaged 65.1 ± 1.0% in Ctl samples, 66.0 ± 0.2% in 4 ng/L and 65.5 ± 0.6% in 120 ng/L exposed individuals and were not significantly different (Anova on arcsin squareroot transformed values). Compared to the Ctl group, 58 DMFs were reported for the low EE2 concentration of 4 ng/L, 33 for the higher concentration of 120 ng/L, while 62 DMFs were significant between 120 and 4 ng/L groups. The majority of DMFs had a higher methylation level at 4 ng/L compared to Ctl and 120 ng/L groups, and at 120 ng/L compared to Ctl (, ). depicts the DMF location within the RR genome and showed a similar pattern as the general distribution of the analysed fragments () with the DMFs located in the order gene body > intergenic > promoter > upstream regions. None of the significant DMF was common to all three group comparisons (). However, 2 DMFs were significant at both EE2 treatments compared to Ctl: one located in an intergenic region, in proximity of the liprin-alpha-4 (PPFIA4) gene, and one located in the gene body of the ATP-binding cassette sub-family A member 1 gene (ABCA1) (). In addition, there were three common DMFs to 4 ng/L vs Ctl and 4 ng/L vs 120 ng/L comparisons, of which two were found within gene bodies: calcium/calmodulin-dependent protein kinase type 1D (CAMK1D), testis-specific serine/threonine-protein kinase (TSSK1B) and one in the intergenic region in proximity of the lymphoid-restricted membrane protein-like gene (LRMP). Finally, one fragment was significantly differentially methylated in both 120 vs Ctl and 120 vs 4 ng/L, located in the intergenic region in proximity to the PTK2B (protein-tyrosine kinase 2) gene. report the DMFs for the three comparisons: 4 ng/L vs Ctl, 120 ng/L vs Ctl, and 4 ng/L vs 120 ng/L, respectively. Among all group comparisons, the maximum effect size (difference in mean methylation) observed for hypermethylation was 21.9% (promoter region of nipped-B-like protein B, NIPBL) and −19.0% for hypomethylation (intergenic region, eukaryotic translation initiation factor 4 gamma 3 EIF4G3). Both DMFs were significant between 4 ng/L and Ctl groups. It is also important to report the fact that very few DMFs were classified as lowly methylated fragments. When comparing exposed groups to Ctl, only NIPBL was lowly methylated in Ctl at 19.6% and increased to 41.5% after exposure to 4 ng/L EE2.
Table 2. Number of analysed fragments, analysed CpGs and number of DMFs in each group comparison
Table 3. Differentially methylated fragments (DMFs) between 4 ng/L EE2 and control individuals. Methylation difference represents the mean methylation at 4 ng/L minus the mean methylation in controls. Fragments discussed in the text are marked with *
Table 4. Differentially methylated fragments (DMFs) between 120 ng/L EE2 and control individuals. Methylation difference represents the mean methylation at 120 ng/L minus the mean methylation in controls. Fragments discussed in the text are marked with *
Table 5. Differentially methylated fragments (DMFs) between 120 ng/L EE2 and 4 ng/L EE2 exposed individuals. Methylation difference represents the mean methylation at 120 ng/L minus the mean methylation at 4 ng/L. Fragments discussed in the text are marked with *
Figure 3. (a) Volcano plot representations of significance and differential methylation in analysed fragments. Dotted lines represent thresholds of significance: −10LogP20, corresponding to P0.01 and mean methylation difference 10% (a) 4 ng/L vs control; B:120 ng/L vs control; C: 4 ng/L vs 120 ng/L. (b) Overall distribution of DMFs in genomic elements; (c) Venn Diagram showing specific and common DMFs between group comparisons; (d) Methylation levels (%) of common fragments between 4 vs Ctl and 120 vs Ctl comparisons (Da,Db), between 4 vs Ctl and 4 vs 120 comparisons (c–e) and between 120 vs Ctl and 4 vs 120 ng/L comparisons (f). For clearer representation of methylation differences, y axes start at 40%.
Networks associated with gene body and promoter DMFs
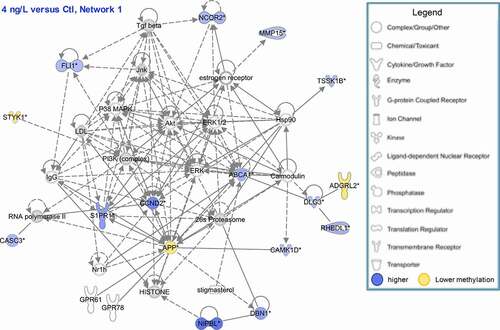
A network analysis was performed with the IPA software using genes corresponding to DMFs located in gene body and promoter regions. Only networks with the highest number of associated DMFs are reported (). The highest network scores were found in the 4 ng/L vs Ctl comparison: the first network involved 16 molecules, with functions related to organismal abnormalities, cell morphology and nervous system function and is represented in . The second network was composed of 15 molecules and was related to gene expression, drug metabolism and lipid metabolism. In 120 ng/L exposed individuals compared to Ctl, only one network with more than nine molecules was established in IPA, related to lipid metabolism, molecular transport and small molecule biochemistry. Finally, four networks were created when comparing 120 ng/L to 4 ng/L exposed individuals, endorsing the differences in the response at these two concentrations. The first network was related to antigen presenting, cell-to-cell signalling and the inflammatory response. The 2nd and 3rd networks were mostly associated with cellular processes (e.g., cellular organization and morphology, cell-to-cell signalling) and the 4th network was linked to cellular death and survival and connective tissue function.
Table 6. Top molecular networks and functions associated with our dataset. Only differentially methylated fragments situated in promoter or gene body and their corresponding annotated genes were taken into account
Figure 4. Network of genes corresponding to significant DMFs (located in promoter region or in gene body) at 4 ng/L compared to control. Blue: hypermethylation at 4 ng/L compared to control, Yellow: hypomethylation at 4 ng/L compared to control.
DNA methylation of genes involved in growth, steroidogenesis and reproduction
As mentioned above, a total of 14,679 unique RefSeq identifiers were attributed among the 102,018 analysed fragments. It is therefore possible to search for genes of interest regardless of their significance. Based on the effects observed on growth, steroid hormone levels and reproduction reported in Voisin et al. [Citation48] following EE2 exposure, we extracted DNA methylation levels of selected genes or biomarkers involved in the response to EE2 (oestrogen receptor), reproduction (vitellogenin), steroidogenesis (aromatase) and the somatotropic axis (insulin-like growth factor I and II, Igf1 receptor, Igf binding protein and Igf2 mRNA binding proteins) (). This list is non-exhaustive and contains 24 fragments located in gene bodies [Citation21] or promoter regions [Citation3]. The methylation difference for all the fragments is very low (below 5%). However, a fragment of 153 bp located in vitellogenin-1-like gene body showed a hypomethylation of 9.4% after exposure to EE2 at 120 ng/L compared to control.
Table 7. DNA methylation levels of fragments related to genes involved in growth, steroidogenesis or reproduction
Discussion
Characterization of the mangrove rivulus RR genome
Cytosine methylation is a predominant epigenetic regulation of gene expression. It occurs mainly on CpG dinucleotides and is classically associated with gene silencing when it occurs at CpG rich promoter regions [Citation60]. Whole genome bisulphite sequencing (WGBS) provides global genome coverage of DNA methylation at single-base resolution, and is consequently referred as the ‘gold standard’ method. Despite its benefits, including a high coverage of CpGs (>90% in human) and an unbiased representation, it remains expensive and difficult to use with large number of samples, mostly because the detection of changes in methylation between samples demands high sequencing depth [Citation61]. In the present study, we opted for reduced representation bisulphite sequencing (RRBS), an approach that also resolves single-base DNA methylation, combining the use of restriction enzymes and bisulphite sequencing. Unlike WGBS, RRBS can only sequence rich-CpG regions, and has limited coverage of the genome [Citation62]. However, the fact that it enriches the analysed fragments with a high density of CpGs makes this technique cost-effective and allows to work on a higher number of samples with a high sequencing depth [Citation63]. Here we reported the DNA methylation pattern of mangrove rivulus liver using RRBS. This species shows a very peculiar reproduction strategy as it is the only vertebrate which alternates between cross-fertilization and selfing, the latter being the most common. It results in several lineages of isogenic individuals, and epigenetic mechanisms have been proposed to explain the occurrence of phenotypic variability despite a low level of genetic diversity [Citation42]. We reported a RR genome representing 1.4% of the whole genome, which was comparable to what was obtained in mouse (1.4%) or in zebrafish (2.2%) brain [Citation54,Citation64]. The percentage of CpG covered in the RR genome reached 6.1% of the total number of genomic CpGs, reaching a 4.3-fold enrichment in CpG sites. This CpG coverage is similar to what was found in mouse and zebrafish (7.0% and 5.3%, respectively [Citation54],), but higher than what other studies reported for diverse fish species such as stickleback [1% [Citation65]], Atlantic salmon [2.75% [Citation66]], rainbow trout [<1% [Citation67]] and guppies [1.5–2% [Citation68]]. In mangrove rivulus brain, Berbel-Filho et al. [Citation46] also reported a lower CpG coverage at 1.2%. This might be astonishing considering that different tissues of the same species usually show the same methylation profile [Citation69] and methodological aspects can not be rejected to explain this discrepancy between those studies. Most noticeable is the fact that we obtained significantly more reads in the present study (explaining that fragments aligned to a higher proportion of the genome) but also a lower alignment efficiency (44.4% in average vs 62.9%) as many fragments could not be mapped. This relatively low level of alignment efficiency was also observed in zebrafish brain following the exact same technical workflow [Citation54]. RRBS reads are relatively short (40–220 bp with majority being < 100 bp) and its plausible that this contributes to large proportion of unmapped reads. Another possible explanation is the proportion of RRBS reads that are derived from repeat regions in the genome as the RRBS reads from repeat region aligns with less efficiency [Citation70,Citation71].
The distribution of the analysed fragments across the RR genome showed 16% located in promoter region. The analysed fragments in this region are also low in zebrafish liver at 12%, and even lower in Atlantic salmon (4%) [Citation66]. On the contrary, this percentage is higher in mammal species such as mouse liver (52%) [Citation69]. This proportion of analysed fragments located in promoter region can explain the observed difference of the mean global methylation level reported in fish species and in mammals, the latter having a lower value (28% in mouse vs 65% in the mangrove rivulus, 70% in zebrafish, 75% in Atlantic salmon). The promoter regions being less methylated on average than other regions in both fish and mammals, a higher proportion of fragments from this region decreases the average global methylation as observed in mouse RR genome. These differences between mammals and fish can also be reflected in the proportion of highly and lowly methylated fragments. If the majority of fragments are either highly or lowly methylated in all reported vertebrates, a larger proportion of fragments present a methylation level higher than 80% in fish than in mammals (67% of fragments in our study) [Citation69].
Regarding the mangrove rivulus, even if the majority of promoter fragments were lowly methylated, a significant proportion were highly methylated (38%), which indicates an elevated range of the methylation level in promoter regions, and consequently a regulatory activity of gene expression. In contrast, other genomic regions presented a narrower range in methylation levels, with the majority of fragments being highly methylated. When compared to zebrafish, an interesting difference appeared regarding the proportion of analysed fragments located in gene bodies. While it reached 47% in mangrove rivulus, zebrafish has only 25% of CpGs in introns and exons. Conversely, the proportion of analysed intergenic fragments is higher in zebrafish (63%) than in mangrove rivulus (37% including intergenic and upstream regions). A possible explanation could reside in the higher proportion of transposable elements (TEs) in zebrafish genome. There is increasing evidence that TEs are important targets of DNA methylation to silence their expression and consequently show a high level of CpG methylation. The zebrafish genome is rich in TEs as 52% of its genomic sequence is composed of the different classes of TEs [Citation72,Citation73], compared to 27.3% in the mangrove rivulus [Citation59]. This difference could explain the relatively lower proportion of analysed fragments located in intergenic regions in the mangrove rivulus compared to zebrafish. When considering other fish species, the part of TEs in genomes is very variable and values as low as 5% have been reported in Tetraodon nigroviridis [Citation73]. This variability of TE amount is explained by the large range of genome size among fish, and Chalopin et al. [Citation72] reported a positive correlation between TE content and genome size in teleosts species, which also applies to the mangrove rivulus. Contrary to what was advanced by Zhang et al. [Citation69], it is therefore unlikely that the pattern of high global methylation observed in fish RR genomes is the result of an enrichment in TEs. Nevertheless, the role of DNA methylation in TEs should be further investigated, including a precise look at the different types of TEs. For example, the rolling-circle transposons, known as Helitrons, are particularly abundant in the mangrove rivulus genome (0.65%) compared to other fish species including its phylogenetic relatives [Japanese medaka (0.03%) and Turquoise killifish (0.06%)]. From all studied fish species, only zebrafish showed a higher level of Helitrons at 1.5% [Citation73]. It is known that TEs in general, and the Helitron subfamily in particular, are mediator of host genes regulation and contribute to genome evolution and adaptation [Citation74]. It has been suggested that the mobilization of TEs and the changes in epigenetic pattern could be important in the process of organism’s rapid adaptation to environmental changes [Citation75]. The exact role of these elements and their methylation level in the mangrove rivulus is a promising perspective to investigate how this species adapt and evolve using a mixed-mating reproduction system and consequently a low genetic diversity within lineages.
Effects of EE2 on the liver methylome
Growing evidence suggests that EDCs can modify DNA methylation and consequently gene expression pattern. Recently, experiments on zebrafish embryos clearly established a link between exposure to bisphenol A, DNA methylation and behavioural impairments [Citation76]. Data also showed that EE2 can modify the methylation status of target genes [Citation35]. In a previous study, we showed that the liver proteome of adult mangrove rivulus was impaired by exposure to EE2 during early life stages (ELS) [Citation49]. It has been hypothesized that exposure to EE2 during ELS can impair DNA methylation and induce long-term consequences later in life as observed on the proteome and on the phenotype [Citation48]. To test this hypothesis, we aimed at detecting potential long-term changes in the hepatic DNA methylation of 168 dph adults following a 28-day early-life exposure of mangrove rivulus hatchlings. We obtained a total of 146 DMFs among all group comparisons, from which a majority were observed between the two EE2 treatments and between 4 ng/L exposed individuals and Ctl. The larger observed impacts of the lowest EE2 concentration on DNA methylation is in accordance with a non-monotonic dose–response relationship often reported for exposure to endocrine disruptors. Hormones are effective at very low concentration, having a high affinity for their receptors, and so are the endocrine disrupting chemicals [Citation77]. The present results are reinforced by a similar response detected in brain and liver protein expression profiles of the mangrove rivulus exposed in the same conditions to EE2 [Citation49]. To our knowledge, only one other study investigated the long-term effects of early-life exposure to chemicals on fish methylome [Citation78]. This study investigated the effects of two compounds, mono(2-ethylhexyl) phthalate (MEHP, 30 µM) and 5-azacytidine (5AC, 10 µM, an inhibitor of DNMT1) in zebrafish. They found that most methylation changes occurred at conserved non-genic regions with cis-regulatory functions (i.e., enhancers, silencers, transcription factor binding sites).
Among the differentially methylated fragments (DMFs) found in adult mangrove rivulus liver following early-life exposure to EE2, none was significantly affected in all three group comparisons (Ctl vs 4 ng/L; Ctl vs 120 ng/L; 4 vs 120 ng/L). Six DMFs were significant in two out of three comparisons, of which three were located in gene bodies. A DMF corresponding to ATP-binding cassette sub-family A member 1 (ABCA1) was hypermethylated at both 4 ng/L and 120 ng/L compared to Ctl. ABCA1 is a known oestrogen-responsive gene [Citation79]. It was included in networks in both comparisons and appeared to play a central role in the response to EE2, showing interactions with the oestrogen receptor and several other molecules (). It is a major regulator of cholesterol and phospholipid homoeostasis by regulating efflux of cholesterol and phospholipids outside of the cell, that are then taken up by apolipoproteins A1 and E to form high density lipoprotein (HDL) molecules. Namely, oestradiol exposure of smooth muscle cells has been shown to enhance cholesterol efflux to APOA1 and HDL, which was associated to ABCA1 overexpression via ESR2 and liver X receptor (LXR) activation [Citation80]. We suggest that the alteration of methylation level of ABCA1 gene might be related to the altered abundances of liver apolipoproteins and other downstream proteins involved in lipid metabolism previously reported [Citation49]. The fact that both concentrations of EE2 impacted this gene methylation level indicates that this mechanism might be a general mode of action of EE2, and could induce effects at environmental concentrations as low as 4 ng/L.
Two DMFs were significantly hypermethylated at 4 ng/L in comparison to both Ctl and 120 ng/L treatments: the calcium/calmodulin-dependent protein kinase type 1D (CAMK1D), and the testis-specific serine/threonine-protein kinase (TSSK1B). Both DMFs were included in the first network of 4 ng/L vs Ctl and 120 vs 4 ng/L comparisons. CAMK1D is a protein kinase that participates in the calcium-regulated calmodulin-dependent kinase cascade, regulating calcium-mediated granulocyte function and respiratory burst and activating the transcription factor CREB1. According to , CAMK1D interacts not only with calmodulin, but also with the amyloid precursor APP, which can be induced by oestrogens through extracellular-regulated kinase 1 and 2 (ERK1/2) phosphorylation [Citation81]. Finally, TSSK1B is essential for spermatid development and spermatogenesis. While TSSK1B is likely not expressed in the liver, it can be possible that an epigenetic alteration of this gene could be found in the ovotestis following EE2 exposure, with potential consequences on reproduction. Although no study to date has linked EE2 and TSSK1B, a study in rats has shown that bisphenol A exposure decreases the expression of TSSK1B mRNA [Citation82].
We must nevertheless be careful when interpreting the changes of methylation in these 3 DMFs. First, the observed differences in methylation, if significant, ranged between 10.3 and 14.6%. Further studies should determine whether these differences have a biological impact on gene expression. Moreover, these three DMFs occurred in gene bodies, as did the majority of DMFs in this study (and MSPI fragments). While methylation of DNA at promoters is well known to suppress transcription, the role of DNA methylation in gene bodies is more difficult to interpret [Citation83]. Gene body methylation is conserved among plants and animals, with a general preference for exons [Citation84]. Potential roles include the stimulation or inhibition of transcription elongation [Citation85] and the regulation of splicing [Citation86]. In mammals, most genes possess alternative transcription start sites that can be located within the gene bodies, complicating the link between methylation and expression [Citation86]. Nevertheless, it is now recognized that gene body methylation is positively correlated with transcriptional activity in most species [Citation12] and we should not disregard the possible implication of these observed changes in methylation after exposure to EE2.
Beside significant changes of DNA methylation within gene bodies, we also reported several DMFs belonging to intergenic regions. Interpreting alterations in DNA methylation in these regions is challenging as the gene annotation may not directly apply. Intergenic regions are typically highly methylated in all vertebrates and bear a conserved role of maintaining genome stability, by preventing the transposition of repetitive TEs and silencing cryptic promoters and cryptic splice sites [Citation87]. Moreover, TEs possess strong promoters that may be able to transcribe neighbouring genes (transcriptional interference). Finally, a compact chromatin state in these regions can prevent recombination. Most (>90%) methylcytosines occur in transposons, and low methylation of these regions could reduce genomic stability [Citation88]. As above mentioned, the diversity and composition of TEs in the mangrove rivulus could contribute to the genetic recombination and evolutionary adaptation in this self-fertilizing species [Citation59]. To gain further insights into the role of intergenic methylation, future research should investigate the overlapping of DMFs with potential TEs.
In addition, we pointed out the very low proportion of DMFs classified as lowly methylated fragment (< 20% methylation). The nipped-B-like protein B (NIPBL) is the exception as it showed 19.6% methylation in Ctl but increased to 41.5% in fish exposed to 4 ng/L EE2. It is remarkable that this fragment is also the one showing the highest effect size of our study (+ 21.9%) and is located in the promoter region. Few more lowly methylated fragments were significant when comparing the 2 treatments, but all others were either highly methylated or showed an intermediary methylation level. Two explanations can be advanced. First, the lowly methylated fragments were about 1/3 less numerous than the highly methylated ones in the RR genome as reported in . This is related to the proportionally low occurrence of fragments located in the promoter region, which have the lowest methylation level. Second, we also showed that lowly methylated fragments have a 10-fold higher coefficient of variability (33% vs 3% in highly methylated fragments). It results that the statistical power to detect changes of lowly methylated fragments is lower and that we can consequently report only DMFs with a high effect size, such as NIPBL. To overstep this limitation, one should include a higher number of replicates in future RRBS analyses. It is even more important for this kind of studies for which effects are searched long after the end of the exposure, which decrease the probability to observe high effect sizes. Nevertheless, our study is the first to report the effect of an endocrine disruptor exposure on the methylation level of NIPBL promoter region. This effect, which was significant while the exposure had stopped 140 days earlier, could have serious long-term consequences on adult fish. NIPBL is associated to cohesin in order to mediate chromatin cohesion, important for mitosis. This complex plays an important role in histone acetylation, chromosome architecture and therefore in regulation of gene expression [Citation89,Citation90]. In human, a mutation of NIPBL is associated to the ‘Cornelia de Lange’ syndrome, a developmental disorder [Citation91]. In zebrafish, the zygotic genome activation is facilitated by cohesin and was a key for the embryogenesis [Citation92]. Intriguingly, the cohesin complex has been proposed to modulate oestrogen-dependent gene transcription and a link has been established with breast cancer [Citation93]. Our results suggest that NIPBL hypermethylation could be a mode of action of EE2 exposure and could potentially modify gene regulation. Further studies should investigate the long-term links between the change in NIPBL methylation level and the overall changes in gene expression in adults, long after the exposure to EE2.
Additionally, we reported the methylation level of fragments corresponding to oestrogen receptors, the aromatase, vitellogenin and several genes involved in the somatotropic axis, which is the main regulator of growth in vertebrates. Most differences in mean methylation of these fragments between experimental conditions were comprised between 0 and 5% and were not significant. Consequently, we cannot link the previously observed effects of EE2 exposure on growth, steroid hormone levels and reproduction [Citation48] with the DNA methylation of these genes. However, one fragment located in gene body of vitellogenin 1 was 9.4% less methylated at 120 ng/L compared to control, while there was no significant effect at 4 ng/L. Vitellogenin is the precursor of egg yolk proteins, produced in the liver of sexually mature female fish in response to endogenous oestrogens. It is well known that it can also be induced by exogenous oestrogens such as EE2 [Citation94]. Stromqvist et al. [Citation35] reported a significant hypomethylation of three CpGs sites located between two potential predicted oestrogen responsive elements (EREs) upstream of the first vitellogenin exon, in the liver of adult zebrafish after exposure to 100 ng/L EE2 during 14 days. In mangrove rivulus, further studies should investigate whether the methylation changes of the vitellogenin 1 fragment could influence vitellogenin expression and further the reproduction of hermaphrodites. The different effect between the two tested concentrations can be associated with the reported impact on reproduction by Voisin et al. [Citation48] who showed fewer laid eggs by hermaphrodites after exposure to 4 ng/L, but not to 120 ng/L. The implication of some compensatory mechanisms at the highest tested concentration involving DNA methylation of vitellogenin 1 CpGs might be hypothesized.
As previously mentioned, DMFs reported as being significant in the present study mostly showed different methylation level between 10 and 20%. These effect sizes could be considered as low, mostly if we compare with experiments comparing normal and pathological tissue such as tumour [Citation95] which can be over 20%. Berbel-Filho et al. [Citation46] also reported higher effect sizes on the rivulus reared during 10 months in differentially enriched environments (up to 48.9% of hypermethylation). If it is necessary to question the biological relevance of changes in DNA methylation below 20%, it is not surprising to have low effect sizes using an experimental design where fish were reared in the same environment for most of their life after an initial exposure during 28 days. This is a similar approach as in studies searching for the role of pregnancy and early-life exposures on later-in-life health outcomes in human. These studies show small-magnitude epigenetic effect sizes below 5%, while it can still persist and be replicated across populations and time and be correlated with gene expression [Citation96]. The individuals used in the present study being issued from the same isogenic lineage, the confounding factor of genetic variability is reduced, which may help to reveal more subtle difference in DNA methylation level, and stress the usefulness of this species in epigenetic studies as model to investigate the developmental origin of health and diseases.
Little correspondence was found between the proteome previously published [Citation49] and the epigenome (Supplementary Figure 2). Although the number of identified genes was about 10 x greater in the methylome analysis, only about half of the total identified proteins in the liver was found in the methylome analysis. Similarly, we could retreive the methylation level of fragments belonging to approximately half (52%) of the differentially abundant proteins. None of these differentially abundant proteins could be related to a change in DNA methylation level of the promotor region. Conversely, only five of the significantly DMFs were also identified, but not significant, in the proteomic analysis in liver: G1/S-specific cyclin-D2-like (CCND2, promoter region, significant between 4 ng/L and Ctl), teneurin-1-like (TENM1, gene body, significant between 120 ng/L and Ctl), protein NDRG1-like (NDRG1, promoter, significant between 120 ng/L and Ctl), fibrinogen alpha chain (FGA, intergenic, significant between 120 ng/L and Ctl), and death-associated protein 1 (DAP, gene body, significant between 4 and 120 ng/L). Comparison between DNA methylation and protein expression pattern is rare but is fully justified. The proteome is the main functional product of gene expression and is closely related to the molecular phenotype [Citation97]. As such, the proteomic level can generate new hypothesis on the modes of action of xenobiotics and on the adaptive responses of organisms [Citation98]. Epigenetics being at the interface between the genotype and the environment, a more systematic link with the proteome would improve the understanding of the adverse outcome pathways (AOPs) and the outcomes in terms of adaptation to the environment. However, it is challenging to correlate these two levels, because of technical variability, and because differentially abundant proteins are not necessarily regulated by differential methylation, but rather the end product of upstream regulators, themselves differentially methylated. Also, protein abundance results not only from transcription, which can be influenced by DNA methylation, but also from mRNA stability, alternative splicing, translation, protein turnover and post-translational modifications. Nevertheless, comparison of proteome and methylation data can be eased by the analysis of gene ontology. Some biological processes were identified in both datasets, such as lipid metabolism, inflammation, connective tissue development and molecular transport, which makes their long-term impairment plausible mechanisms of EE2 toxicity.
Conclusions
In conclusion, our study provides further evidence on the capacity of EDCs to alter the methylome and shows that these changes can be apparent several months after the exposure, supporting the hypothesis of possible long-term modulation of gene expression through epigenetics. It also confirms the non-monotonic response to EDCs as most significant effects were observed at the environmental concentration of 4 ng/L. The DMFs belonging to promoter and gene bodies revealed networks involved in several biological functions potentially regulated by oestrogens, including lipid metabolism, cellular processes (death and survival), connective tissue function, molecular transport and inflammation, many of which were also identified in a previous proteomic analysis. Importantly, we reported that methylation of nipped-B-like protein B (NIPBL) might be an important mode of action of EE2 and, as involved in chromosome organization and gene expression, could have long-term impact on gene regulation and on organism’s general functions. To gain a more comprehensive view of the significance of DMFs in promoter, gene body, upstream and intergenic regions, future studies should investigate the overlapping of the DMFs with potential enhancers, insulators, transcription start sites, TEs, oestrogen-responsive elements and other transcription factor binding sites that may interact with oestrogen receptors. Among them, TEs, and more particularly helitrons, are potentially important targets of environmental stressors that could be involved in the response of organisms to environmental changes. Despite its limitations, RRBS has proved to be an efficient method to investigate effects of EE2 on DNA methylation in the mangrove rivulus. This self-fertilizing fish should be further considered as a model species to investigate the interplay between environmental stressors, epigenetics and adaptation. The possibility to naturally discard the confounding factor of genetic variability makes this species a top model to better understand the roles of epigenetics in the long-term response to environmental xenobiotics and in the developmental origin of health and diseases.
Data accessibility statement
All the data generated by the RRBS workflow will be submitted to Dryad repository and will be publicly accessible. The BAM files after Bismark alignment and the excel sheet containing the values for all analysed fragments have already been submitted and are available at this link: https://datadryad.org/stash/share/k7Lq6mHbxBMF5BHdk8cOp1EFGSGVK3tfJoBx4tPYnzc.
The DOI is: https://doi.org/10.5061/dryad.v41ns1rsv
The Fastq files will be submitted when a decision will have been given for the acceptance of the manuscript.
Author contributions
Anne-Sophie Voisin: investigation, manuscript writing and editing, experimental design, data acquisition and analysis
Victoria Suarez Ulloa: data analysis, manuscript editing
Peter Stockwell: bioinformatic data analyses
Aniruddha Chatterjee: supervision, methodology, RRBS analyses, manuscript editing
Frédéric Silvestre: investigation, supervision, funding acquisition, manuscript writing and editing, experimental design, submission and revision
The authors report no conflict of interest
Supplemental Material
Download Rich Text Format File (11.5 MB)Acknowledgements
We thank the Department of Pathology at the University of Otago (New Zealand) for providing facilities for RRBS library preparation. Special thanks to Prof Mike Eccles, Jackie Ludgate and Anna Leichter for their advice on the workflow and sequencing. This study was supported by the FRS-FNRS (Belgian National Fund for Scientific Research) grant N°T.0174.14 (Epigenetics in the mangrove rivulus), including a PhD fellowship to A.-S. Voisin and a post-doc fellowship to Victoria Ulloa-Suarez. Travel support was provided by a credit for short stay abroad by FRS-FNRS (2017/V 3/5/079 – IB/MF). We are also thankful to Ivan Blanco for his help in data post-processing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Barker DJ. The developmental origins of adult disease. J Am Coll Nutr. 2004;23:588S–595S.
- Brockmeier EK, Hodges G, Hutchinson TH, et al. The role of omics in the application of adverse outcome pathways for chemical risk assessment. Toxicol Sci. 2017;158:252–262.
- Braun JM. Early-life exposure to EDCs: role in childhood obesity and neurodevelopment. Nat Rev Endocrinol. 2017;13:161.
- Heindel JJ, Newbold R, Schug TT. Endocrine disruptors and obesity. Nat Rev Endocrinol. 2015;11:653–661.
- Kajta M, Wójtowicz AK. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol Rep. 2013;65:1632–1639.
- Kuo CH, Yang SN, Kuo PL, et al. Immunomodulatory effects of environmental endocrine disrupting chemicals. Kaohsiung J Med Sci. 2012;28:S37–42.
- Clouzot L, Marrot B, Doumenq P, et al. 17α-Ethinylestradiol: an endocrine disrupter of great concern. Analytical methods and removal processes applied to water purification. A review. Environ Prog. 2008;27:383–396.
- Matthiessen P, Wheeler JR, Weltje L. A review of the evidence for endocrine disrupting effects of current-use chemicals on wildlife populations. Crit Rev Toxicol. 2018;48:195–216.
- Aris AZ, Shamsuddin AS, Praveena SM. Occurrence of 17α-ethynylestradiol (EE2) in the environment and effect on exposed biota: a review. Environ Int. 2014;69:104–119.
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220.
- Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 2009;19:959–966.
- De Mendoza A, Lister R, Bogdanovic O. Evolution of DNA methylome diversity in eukaryotes. J Mol Biol. 2020;432:1687–1705.
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38.
- Edwards JR, Yarychkivska O, Boulard M, et al. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. 2017;10. DOI:https://doi.org/10.1186/s13072-017-0130-8
- Guo H, Zhu P, Yan L, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–610.
- Smith ZD, Chan MM, Humm KC, et al. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615.
- Dorts J, Falisse E, Schoofs E, et al. DNA methyltransferases and stress-related genes expression in zebrafish larvae after exposure to heat and copper during reprogramming of DNA methylation. Sci Rep. 2016;6:34254.
- Eid A, Zawia N. Consequences of lead exposure, and it’s emerging role as an epigenetic modifier in the aging brain. NeuroToxicology. 2016;56:254–261.
- Gore AC, Walker DM, Zama AM, et al. Early life exposure to endocrine-disrupting chemicals causes lifelong molecular reprogramming of the hypothalamus and premature reproductive aging. Mol Endocrinol. 2011;25:2157–2168.
- Goyal D, Limesand SW, Goyal R. Epigenetic responses and the developmental origins of health and disease. J Endocrinol. 2019;242:T105–T119.
- Stel J, Legler J. The role of epigenetics in the latent effects of early life exposure to obesogenic endocrine disrupting chemicals. Endocrinology. 2015;156:3466–3472.
- Wadhwa PD, Buss C, Entringer S, et al. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–368.
- Barouki R, Melén E, Herceg Z, et al. Epigenetics as a mechanism linking developmental exposures to long-term toxicity. Environ Int. 2018;114:77–86.
- Mirbahai L, Chipman JK. Epigenetic memory of environmental organisms: a reflection of lifetime stressor exposures. Mutat Res Genet Toxicol Environ Mutagen. 2014;764-765:10–17.
- Brander SM, Biales AD, Connon RE. The role of epigenomics in aquatic toxicology. Environ Toxicol Chem. 2017;36:2565–2573.
- Vandegehuchte MB, Janssen CR. Epigenetics and its implications for ecotoxicology. Ecotoxicology. 2011;20:607–624.
- Vandegehuchte MB, Janssen CR. Epigenetics in an ecotoxicological context. Mutat Res Genet Toxicol Environ Mutagen. 2014;764-765:36–45.
- Walker CL. Minireview: epigenomic plasticity and vulnerability to EDC exposures. Mol Endocrinol. 2016;30:848–855.
- Zhang X, Ho SM. Epigenetics meets endocrinology. J Mol Endocrinol. 2011;46:R11–32.
- Shi JF, Li XJ, Si XX, et al. ERα positively regulated DNMT1 expression by binding to the gene promoter region in human breast cancer MCF-7 cells. Biochem Biophys Res Commun. 2012;427:47–53.
- Lee DH, Jacobs DR, Porta M. Hypothesis: a unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ Health Perspect. 2009;117:1799–1802.
- Anderson AM, Carter KW, Anderson D, et al. Coexpression of nuclear receptors and histone methylation modifying genes in the testis: implications for endocrine disruptor modes of action. PLoS One. 2012;7:e34158.
- Casati L, Sendra R, Sibilia V, et al. Endocrine disrupters: the new players able to affect the epigenome. Front Cell Dev Biol. 2015;3:37.
- Liu Y, Duong W, Krawczyk C, et al. Oestrogen receptor β regulates epigenetic patterns at specific genomic loci through interaction with thymine DNA glycosylase. Epigenetics Chromatin. 2016;9:7.
- Strömqvist M, Tooke N, Brunström B. DNA methylation levels in the 5' flanking region of the vitellogenin I gene in liver and brain of adult zebrafish (Danio rerio)–sex and tissue differences and effects of 17alpha-ethinylestradiol exposure. Aquat Toxicol. 2010;98:275–281.
- Mirbahai L, Yin G, Bignell JP, et al. DNA methylation in liver tumorigenesis in fish from the environment. Epigenetics. 2011;6:1319–1333.
- Falisse E, Ducos B, Stockwell PA, et al. DNA methylation and gene expression alterations in zebrafish early-life stages exposed to the antibacterial agent triclosan. Environ Pollut. 2018;243:1867–1877.
- Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109.
- Avise JC, Tatarenkov A. Population genetics and evolution of the mangrove rivulus Kryptolebias marmoratus, the world’s only self-fertilizing hermaphroditic vertebrate. J Fish Biol. 2015;87:519–538.
- Mackiewicz M, Tatarenkov A, Perry A, et al. Microsatellite documentation of male-mediated outcrossing between inbred laboratory strains of the self-fertilizing mangrove killifish (Kryptolebias Marmoratus). J Hered. 2006;97:508–513.
- Ellison A, Rodríguez López CM, Moran P, et al. Epigenetic regulation of sex ratios may explain natural variation in self-fertilization rates. Proc Biol Sci. 2015;282. DOI:https://doi.org/10.1098/rspb.2015.1900
- Fellous A, Voisin A-S, Labed-veydert T, et al. DNA methylation in adults and during development of the self- fertilizing mangrove rivulus, Kryptolebias marmoratus. Ecol Evol. 2018;8:6016–6033.
- Fellous A, Earley RL, Silvestre F. Identification and expression of mangrove rivulus (Kryptolebias marmoratus) histone deacetylase (HDAC) and lysine acetyltransferase (KAT) genes. Gene. 2019;691:56–69.
- Fellous A, Earley RL, Silvestre F. The Kdm/Kmt gene families in the self-fertilizing mangrove rivulus fish, Kryptolebias marmoratus, suggest involvement of histone methylation machinery in development and reproduction. Gene. 2019;687:173–187.
- Berbel-Filho WM, De Leaniz CG, Moran P, et al. Local parasite pressures and host genotype modulate epigenetic diversity in a mixed ‐ mating fish. Ecol Evol. 2019;9:8736–8748.
- Berbel-Filho WM, Rodríguez-Barreto D, Berry N, et al. Contrasting DNA methylation responses of inbred fish lines to different rearing environments. Epigenetics. 2019;14:939–948.
- Carion A, Markey A, Hétru J, et al. Behavior and gene expression in the brain of adult self-fertilizing mangrove rivulus fish (Kryptolebias marmoratus) after early life exposure to the neurotoxin β-N-methylamino-L-alanine (BMAA). Neurotoxicology. 2020;79:110–121.
- Voisin A-S, Fellous A, Earley RL, et al. Delayed impacts of developmental exposure to 17-α-ethinylestradiol in the self-fertilizing fish Kryptolebias marmoratus. Aquatic Toxicol. 2016;180:247–257.
- Voisin A-S, Kültz D, Silvestre F. Early-life exposure to the endocrine disruptor 17-α-ethinylestradiol induces delayed effects in adult brain, liver and ovotestis proteomes of a self-fertilizing fish. J Proteomics. 2018;1–13.
- Foulds CE, Treviño LS, York B, et al. Endocrine-disrupting chemicals and fatty liver disease. Nat Rev Endocrinol. 2017;13:445–457.
- De Wit M, Keil D, Remmerie N, et al. Molecular targets of TBBPA in zebrafish analysed through integration of genomic and proteomic approaches. Chemosphere. 2008;74:96–105.
- Feswick A, Munkittrick KR, Martyniuk CJ. Estrogen-responsive gene networks in the teleost liver: what are the key molecular indicators. Environ Toxicol Pharmacol. 2017;56:366–374.
- Humble JL, Hands E, Saaristo M, et al. Characterisation of genes transcriptionally upregulated in the liver of sand goby (Pomatoschistus minutus) by 17α-ethinyloestradiol: identification of distinct vitellogenin and zona radiata protein transcripts. Chemosphere. 2013;90:2722–2729.
- Chatterjee A, Ozaki Y, Stockwell PA, et al. Mapping the zebrafish brain methylome using reduced representation bisulfite sequencing. Epigenetics. 2013;8:979–989.
- Chatterjee A, Stockwell PA, Horsfield JA, et al. Base-resolution DNA methylation landscape of zebrafish brain and liver. Genom Data. 2014;2:342–344.
- Chatterjee A, Stockwell PA, Rodger EJ, et al. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res. 2012;40:e79.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572.
- Stockwell PA, Chatterjee A, Rodger EJ, et al. DMAP: differential methylation analysis package for RRBS and WGBS data. Bioinformatics. 2014;30:1814–1822.
- Rhee JS, Choi BS, Kim J, et al. Diversity, distribution, and significance of transposable elements in the genome of the only selfing hermaphroditic vertebrate Kryptolebias marmoratus. Sci Rep. 2017;7:40121.
- Bock C, Beerman I, Lien WH, et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell. 2012;47:633–647.
- Wreczycka K, Gosdschan A, Yusuf D, et al. Strategies for analyzing bisulfite sequencing data. J Biotechnol. 2017;261:105–115.
- Meer A, Gnirke A, Bell GW, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877.
- Chatterjee A, Rodger EJ, Stockwell PA, et al. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. J Biomed Biotechnol. 2012;2012:741542.
- Smith ZD, Gu H, Bock C, et al. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48:226–232.
- Metzger DCH, Schulte PM. Persistent and plastic effects of temperature on DNA methylation across the genome of threespine stickleback (Gasterosteus aculeatus). Proc Biol Sci. 2017;284.
- Uren Webster TM, Rodriguez-Barreto D, Martin SAM, et al. Contrasting effects of acute and chronic stress on the transcriptome, epigenome, and immune response of Atlantic salmon. Epigenetics. 2018;13:1191–1207.
- Baerwald MR, Meek MH, Stephens MR, et al. Migration-related phenotypic divergence is associated with epigenetic modifications in rainbow trout. Mol Ecol. 2016;25:1785–1800.
- Hu J, Pérez-Jvostov F, Blondel L, et al. Genome-wide DNA methylation signatures of infection status in Trinidadian guppies (Poecilia reticulata). Mol Ecol. 2018;27:3087–3102.
- Zhang C, Hoshida Y, Sadler KC. Comparative epigenomic profiling of the DNA methylome in mouse and zebrafish uncovers high interspecies divergence. Front Genet. 2016;7:110.
- Chatterjee A, Rodger EJ, Stockwell PA, et al. Generating multiple base-resolution DNA methylomes using reduced representation bisulfite sequencing. In: Seymour G, Cullinan M, Heng N, editors. Oral biology. New York, N.Y: Humana Press; 2017. p. 279–298.
- Chatterjee A, Rodger EJ, Morison IM, et al. Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. In: Seymour G, Cullinan M, Heng N, editors. Oral biology. New York, N.Y: Humana Press; 2017. p. 249–277.
- Chalopin D, Naville M, Plard F, et al. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol Evol. 2015;7:567–580.
- Shao F, Han M, Peng Z. Evolution and diversity of transposable elements in fish genomes. Sci Rep. 2019;9:15399.
- Volff JN. Genome evolution and biodiversity in teleost fish. Heredity (Edinb). 2005;94:280–294.
- Lerat E, Casacuberta J, Chaparro C, et al. On the Importance to acknowledge transposable elements in epigenomic analyses. Genes (Basel). 2019;10:258.
- Olsvik PA, Whatmore P, Penglase SJ, et al. Associations between behavioral effects of bisphenol A and DNA methylation in zebrafish embryos. Front Genet. 2019;10:184.
- Vandenberg LN, Colborn T, Hayes TB, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33:378–455.
- Kamstra JH, Sales LB, Aleström P, et al. Differential DNA methylation at conserved non-genic elements and evidence for transgenerational inheritance following developmental exposure to mono(2-ethylhexyl) phthalate and 5-azacytidine in zebrafish. Epigenetics Chromatin. 2017;10:20.
- Srivastava RA. Estrogen-induced regulation of the ATP-binding cassette transporter A1 (ABCA1) in mice: a possible mechanism of atheroprotection by estrogen. Mol Cell Biochem. 2002;240:67–73.
- Wang H, Liu Y, Zhu L, et al. 17β-estradiol promotes cholesterol efflux from vascular smooth muscle cells through a liver X receptor α-dependent pathway. Int J Mol Med. 2014;33:550–558.
- Zhang S, Huang Y, Zhu YC, et al. Estrogen stimulates release of secreted amyloid precursor protein from primary rat cortical neurons via protein kinase C pathway. Acta Pharmacol Sin. 2005;26:171–176.
- Marmugi A, Ducheix S, Lasserre F, et al. Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology. 2012;55:395–407.
- Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257.
- Feng S, Cokus SJ, Zhang X, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 2010;107:8689–8694.
- Yang X, Han H, De Carvalho DD, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590.
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492.
- Zemach A, McDaniel IE, Silva P, et al. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919.
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–251.
- Gao D, Zhu B, Cao X, et al. Roles of NIPBL in maintenance of genome stability. Semin Cell Dev Biol. 2019;90:181–186.
- Zhu Z, Wang X. Roles of cohesin in chromosome architecture and gene expression. Semin Cell Dev Biol. 2019;90:187–193.
- Newkirk DA, Chen YY, Chien R, et al. The effect of Nipped-B-like (Nipbl) haploinsufficiency on genome-wide cohesin binding and target gene expression: modeling Cornelia de Lange syndrome. Clin Epigenetics. 2017;9:89.
- Meier M, Grant J, Dowdle A, et al. Cohesin facilitates zygotic genome activation in zebrafish. Development. 2018;145.
- Antony J, Dasgupta T, Rhodes JM, et al. Cohesin modulates transcription of estrogen-responsive genes. Biochim Biophys Acta. 2015;1849:257–269.
- Marin MG, Matozzo V. Vitellogenin induction as a biomarker of exposure to estrogenic compounds in aquatic environments. Mar Pollut Bull. 2004;48:835–839.
- Zhang Y-J, Wu H-C, Yazici H, et al. Global hypomethylation in hepatocellular carcinoma and its relationship to aflatoxin B1 exposure. World J Hepatol. 2012;4:169.
- Breton CV, Marsit CJ, Faustman E, et al. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: the children’s environmental health and disease prevention research center’s epigenetics working group. Environ Health Perspect. 2017;125:511–526.
- Silvestre F, Gillardin V, Dorts J. Proteomics to assess the role of phenotypic plasticity in aquatic organisms exposed to pollution and global warming. Integr Comp Biol. 2012;52:681–694.
- Diz AP, Martínez-Fernández M, Rolán-Alvarez E. Proteomics in evolutionary ecology: linking the genotype with the phenotype. Mol Ecol. 2012;21:1060–1080.