
ABSTRACT
This study evaluated the hypothesis that the maternal metabolic stressed status could be inherited to their F1 daughters via epigenetic mechanism. The maternal cow blood β-hydroxybutyric acid (BHB) level (≥0.9 mM/L) was used as an indicator of maternal metabolic stress. Eight newborn daughters’ blood cells were used for methylation comparison and analysis. By Whole Genome Bisulphite Sequencing (WGBS), a total of 1,861 Differentially Methylated Regions (DMRs), including 944 differentially methylated cytosines (DMCs), were identified. Most DMRs were distributed in intronic and intergenic regions, and most of the DMR in promoter regions were hypermethylated. Differentially methylated genes (DMGs) with DMR methylation differences higher than 20% were mainly enriched in metabolism-related pathways. These results suggest that newborn calves’ metabolic pathways were altered, with 64 DMGs being clustered with metabolic signalling by KEGG analysis. Our study revealed the whole epigenetic landscape of calf blood cells and suggested that the maternal metabolic status can affect the embryo’s epigenetic status and metabolic-related pathways in offspring, providing further evidence for epigenetic intergenerational inheritance of metabolic stress in domestic animals. Besides, this study also contributed more evidence to support the Developmental Origins of Health and Disease (DOHAD) theory in large animals.
Introduction
Epigenetic markers such as histone modifications, chromatin remodelling, DNA methylation, and some small non-coding RNA (tsRNA [tRNA derived small RNA], piRNA [Piwi-interacting RNA], and microRNA) had been reported to be passed on to future generations via the germ cells [Citation1]. Among them, DNA methylation might plays an essential role in transgenerational inheritance of metabolic status [Citation2].
In milk production, the cows are expected to begin another cycle by conceiving again (artificial insemination) as soon as their reproductive function is reactivated. Usually, the cow’s reproductive tract has recovered and is ready for the first service 50–60 days postpartum, but this period often corresponds to the peak of lactation [Citation3]. High-yielding dairy cows during this period will be inseminated artificially to achieve a 365-day calving interval [Citation4]. The metabolic state of cows is affected at this stage as the body needs to mobilize more energy to produce milk. The blood glucose deficiency at this stage is often accompanied by many biochemical processes, including a high level of free unsaturated fatty acids, which will be converted into ketone compounds (beta-hydroxybutyric acid (BHB), acetate, acetone) [Citation5]. At this stage, the blood concentration of BHB is one of the factors that can be used as an indicator of metabolic status [Citation6].
Previous studies showed that high ketone body concentrations might cause metabolic status changes [Citation7]. Beta-hydroxybutyric acid is a vital metabolism regulator in early lactation [Citation8], and concentrations higher than 1.2 mmol/L in the blood are generally considered subclinical ketosis [Citation8]. Our previous study suggested that when the blood BHB concentration was used as an indicator of maternal metabolic stress, the cows with higher BHB levels produced embryos with significant transcriptome changes for genes related to energy metabolism [Citation3]. The potential mechanism might be that, to adapt to the maternal metabolic environment fluctuation, the embryos performed a corresponding metabolic change. The main impacted signalling pathways in this process, including mammalian target of rapamycin (mTOR) signalling and sirtuins, were both associated with metabolism. Additionally, dysregulation of mitochondrial function was also observed [Citation3]. This adaptive metabolic programming may determine the future adult phenotype, which is consistent with the Developmental Origins of Health and Disease (DOHAD) hypothesis [Citation9]. The DOHAD hypothesis states that environmental factors, including nutrient deficiencies or overnutrition during the parental period, might lead to embryo/foetus adaptive programming and finally influence the phenotype of future generations, including the development of diseases such as diabetes, cardiovascular disease, and metabolic stress syndrome [Citation10,Citation11].
In this study, we aimed to explore whether the cow’s metabolic status could be passed on to her daughter via an epigenetic mechanism. A portion of the embryos collected during the previous study [Citation3] was transferred to young recipients raised in the same conditions for reducing the influence of confounding factors. All cows (F0) were housed at the same farm as well.
We generated a single-base resolution blood DNA methylation landscape of calves from mothers of different metabolic status by WGBS. We also studied the differentially methylated regions (DMRs) and differentially methylated cytosines (DMCs) of these calves. Moreover, we investigated the DMR and DMC distribution in genomic elements such as intergenic, promoter regions, and others. This study will be a reference for a better understanding of complex metabolic disease epigenetic inheritance. Besides, in line with our previously published article [Citation3], this study provided good evidence to support the DOHAD hypothesis in domestic animals. It enabled a better understanding of the inter-generational inheritance of metabolic information. More importantly, our research results could provide better guidance on dairy cow management, which might help improve cow fertility.
Results
The whole genome methylation profile of blood cells from calves
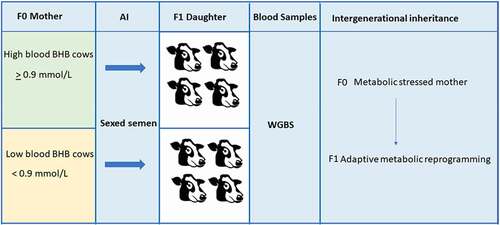
To identify whole-genome methylation differences between calves born from cows of different metabolic status, we analysed the whole genome methylation of blood cells at the single-site resolution level. Using WGBS, we analysed blood samples from eight female calves, and all the newborn calves are descended from sexed XX type sperm since different gender of the offspring could also be a confounding factor.
The bisulphite conversion rate is 96.9875%. Four samples originated from calves born from cows with low BHB levels (< 0.9 mmol/L), and another four samples were descended from cows with high BHB levels (≥0.9 mmol/L) (, Supplementary Table 1). After filtering out the adapter and low-quality reads, the clean reads were mapped to a synthetic converted bovine reference genome (ARS-UCD1.2). The bsseq package was used to analyse CpG methylation following removal of CpG with low coverage to eliminate false positive DMRs. The absolute methylation difference of all the DMRs was > 0.048 and the CpG number < 10. We thus obtained a total of 1,861 DMRs which were mainly enriched in chromosomes 1, 4, and 5 (Supplementary Figure 2). By using the bsseq package, all the DMCs and DMRs extracted will be considered significant, and the corresponding p-value could not be obtained by using the package.
Figure 1. Schematic diagram of the experimental design.The concentrations of blood BHB were measured at 45 days postpartum to determine the maternal metabolic status of eight cows. Regarding of result, cows were classed into low (< 0.9 mmol/L) and high (≥ 0.9 mmol/l) BHB levels. For each cow, two doses of sexed semen were used for artificial insemination. Then the embryos were transferred to nulliparous recipients of the same age and condition. The daughters of these cows (four for each treatment) were then used for DNA methylation analysis after collecting their blood.
Using the methykit package, the DMRs were delineated via defined window and step sizes. Also, every DMCs and DMRs we extracted via this package provided us the corresponding q value.
After removing duplicated and bisulphite non-conversion reads, we extracted 742,978 CpG contexts with an average coverage of 25.235 reads. As shown in the Manhattan plot (Supplementary Figure 1), the significantly methylated cytosines were distributed on 29 autosomes and the X chromosome. The cut-off was defined as -log10(q) = 20, and all the cytosine methylation differences were higher than 40% (Supplementary Figure 1). These results indicated that calves born from mothers of different metabolic status present significant differences in their whole genome methylation.
Changes in the methylome of white blood cells from calves
The volcano plots depicted for the significant CpG sites based on q values and methylation differences between calves from cows with high BHB (≥ 0.9 mmol/L) group and calves from cows with low BHB (< 0.9 mmol/L) group()). Analysis via bsseq package revealed that the extracted DMRs were mainly distributed in distal intergenic regions (53.14%) and introns (34.39%). Only a few DMRs were found in downstream (0.6%) and 3ʹUTR (0.43%) regions ()). Different methylation cut-offs were used to describe the DMR distribution on the whole genome, namely, 20% and 40% (Supplementary Figure 2(b)). A total of 26 DMRs with methylation difference levels superior to 40% were located in introns and distal intergenic regions. Among them, 6 were hypermethylated and 20 were hypomethylated. As shown in Supplementary Figure 2(c), 1,408 DMRs were located in protein-coding regions, while 7% of the DMRs were located in long non-coding RNA (lncRNA), and 7% were in small nuclear RNA (snRNA). Considering the bsseq could not help identify the corresponding p-value of each DMC and DMR, although all the DMC and DMR obtained via this package were considered significant. Here we also used another package for analysis.
Figure 2. Distribution of overall methylation. (a) Volcano plot of DNA methylation changes based on WGBS results. Calves born from mothers with high BHB (>0.9 mmol/L) levels were compared to calves born from mothers with low BHB (0.9 mmol/L) levels. Red dots represent significant hypermethylation (upper right) and significant hypomethylation (upper left) of cytosines.The vertical green lines indicate a 40% methylation difference, and a cut-off of 10 was adopted for -log10(q-value). (b) Distribution of differentially methylated regions of calves born from high BHB (>0.9 mmol/L) and low BHB (<0.9 mmol/L) mother cows. (c) Venn diagram of the differentially methylated regions (using 2000 base pairs as a window and 100 base pairs as a moving step). UTR: Untranslated Region.
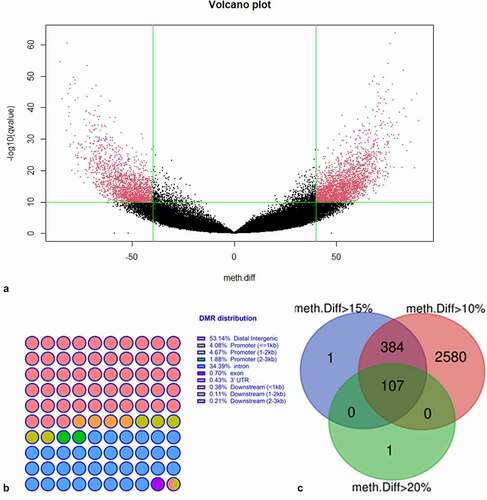
By using methykit package, a 2,000 base-pair window and a 100 base pair step were adopted to characterize DMRs. As indicated in the Venn diagram in ), by using the defined windows, the 108 DMRs methylation difference was higher than 20%, while the methylation difference for 492 DMRs was superior to 15%. In general, by defined windows, we obtained 3,071 significant DMRs for which the methylation difference was higher than 10%.
Functional classification analysis and KEGG analysis
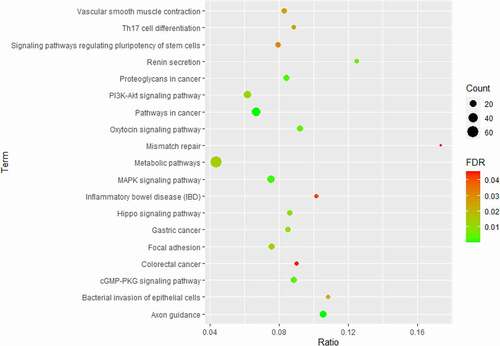
For further insight into the biological relevance of the differentially methylated genes, Functional classification and KEGG analysis was done. For the Functional classification analysis, the data were uploaded to PANTHER. The most enriched biological process GO terms were cellular process (GO:0009987) and metabolic process (GO:0008152) (), and the most enriched molecular function GO terms were binding (GO:0005488) and catalytic activity (GO:0003824). As for KEGG analysis, the most enriched KEGG pathway was the ‘metabolic pathway,’ which had the highest number of DMG hits (); however, this pathway was not the most significant one (p < 0.02). Other significantly enriched pathways included PI3K-AKT and MAPK, which were associated with metabolism [Citation12–14].
Figure 3. Biological and Molecular function summary from the functional classification analysis of DMG in PANTHER. The functional classification enrichment analysis of genes with altered methylation levels in the genome of calves. The horizontal axis indicates the number of genes with altered methylation levels. The black and red columns represent biological and molecular processes. GO, Gene Ontology. DMGs, Differentially methylated genes.
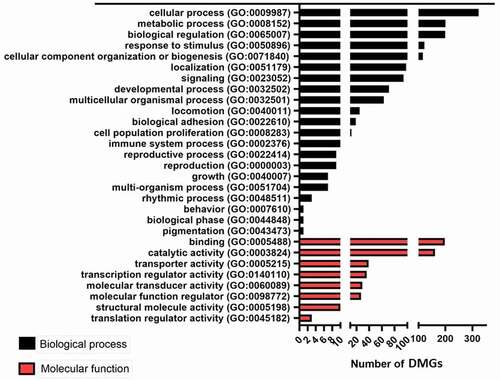
Figure 4. KEGG Pathway analysis of genes with altered DNA methylation. The count is the number of hits in the selected pathway; FDR, false discovery rate.
Metabolism related DMGs and pathway exploration
Differentially methylated genes (DMGs) were defined as genes with differentially methylated regions distributed in their promoters or gene bodies. The Cytoscape software was used for the analysis of metabolism-related pathways. The selection was limited to the DMGs associated with metabolic-related pathways after KEGG analysis; therefore, 64 genes were uploaded into Cytoscape for detailed analysis. The different colours in represent different metabolic pathways. The more colours the node was connected to, the more likely the DMG has a pivotal role. As shown in , overlapping target DMGs mainly included diacylglycerol kinase 1 (DGK1), diacylglycerol kinase beta (DGKB), phospholipid phosphatase 2 (PLPP2), phospholipase A2 group IVA (PLA2G4A), and phosphoglycerate mutase 2 (PGAM2). The PGAM2 is related to the metabolism of pyruvate and catalyzes the reversible reaction of 3-phosphoglycerate (3-PGA) to 2-phosphoglycerate (2-PGA) in the glycolytic pathway. The PGAM is a dimeric enzyme that presents in different tissues. Interestingly, two of its CpG methylation sites in buccal epithelial cells from adolescents were reported to be associated with maternal stress [Citation15].
Figure 5. Analysis of metabolism-related pathways. Each colour represents one specific metabolic pathway. The more colours the node was connected to, the more likely the DMG has a pivotal role.
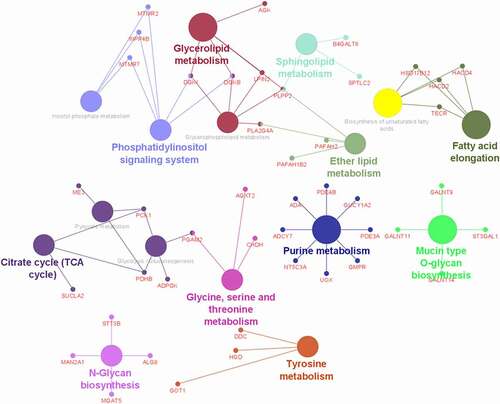
The Phosphatidic Acid Phosphatase Type 2 C (PLPP2) is involved in three metabolism-related pathways and contained two sites with methylation differences higher than 60% in our study. Interestingly, in human research, famine exposure during maternal gestation was also reported to be associated with PLPP2 CpG dinucleotides cg27370573 methylation in adult blood [Citation16].
Discussion
These results allow us to determine the DNA methylation signature of dairy calves born from mothers with different metabolic status. As mentioned in the introduction part, a previous study from our team suggested that metabolic stress in cows, as defined by high BHB levels at conception, impacted the metabolism of in vivo generated embryos. The transcriptomic and epigenetic analysis have shown that the most important signalling pathways affected by the maternal BHB status were mTOR and sirtuins signalling associated with metabolism and mitochondrial function dysregulation[Citation3]. Previous findings also found that the concentration of BHB in cows correlates with changes in the follicular transcriptome and affected specific signalling pathways, including the oestradiol-related signalling pathway, the PI3 Kinase controlling p38 expression, and the retinoic acid response pathway [Citation17].
Whole-genome bisulphite sequencing was performed on blood samples from newborn daughters. Blood cells were selected for study mainly because blood collection is not an invasive procedure, which is easier to obtain and less harmful to the animals. Although white blood cells are a mixture of different leucocyte populations and may blur the epigenetic signals [Citation18], this study aimed to find the conserved epigenetic and phenotypic characteristics of daughters born from mothers under metabolic stress.
In humans, evidence showed that undernutrition during pregnancy leads to changes in the metabolic status of the future generation, causing diabetes, obesity, and cardiovascular diseases [Citation19]. However, studies about this topic in cattle are limited, only a few studies have focused on the link between maternal nutrition and the fertility of offspring reported [Citation20,Citation21].
In this study, we found that the main signalling pathways affected by the maternal metabolic status included metabolic pathways, PI3K-AKT signalling, and the cGMP-PKG pathway. The PI3K-AKT pathway is associated with glucose metabolism, and AKT along with mTOR, is the downstream regulator of PI3K [Citation22]. Knock-out of PI3K-AKT-mTOR signalling in mice is associated with metabolic deficiencies and insulin sensitivity changes in embryos [Citation12,Citation23]. Interestingly, mTOR signalling was also enriched in our previous research [Citation3]. Furthermore, protein kinase G is believed to be the primary transduction regulator of cGMP [Citation24], and the cGMP-PKG pathway is reported to regulate a large spectrum of processes, including metabolism [Citation25]. These results further indicated that the maternal metabolic status promotes adaptive metabolic changes in embryos, and these changes could be maintained after birth.
Further analysis of metabolic-related pathways showed that glycerolipid metabolism, fatty acid elongation, purine metabolism, and mucin-type O-glycan biosynthesis pathways were most significantly enriched. Among them, DGK1, DGKB, PLA2G4A, PGAM2, and other DMGs are involved in two metabolic pathways, PGAM2 is involved in pyruvate metabolism, and PLPP2 is involved in three metabolic-related pathways.
The results suggest that the foetus may activate its metabolic mechanism to adapt to the mother’s metabolic status changes, which is consistent with the DOHAD theory [Citation26]. We have illustrated the association between maternal periconceptional metabolic stress to their embryo phenotypic, transcriptomic, and epigenetic alteration in our previous study, as mentioned in the introduction part. In this paper, we further studied their F1 daughter DNA methylation signature.
However, our study did not reveal any significant phenotypic differences between the F1 daughters, which may be explained by the limited number of animals. In a further study, we will continue to monitor the milk production after the parturition of these F1 daughters to assess the potential phenotype alterations, although a much larger number of animals would be required for statistical analysis. Further study would provide us more insights into the altered methylation signature of specific parameters such as milk production and fertility. In addition, although we screened many DMCs and DMRs at a genome-wide level, we still cannot completely rule out the possibility that genetic differences may contribute to these results. A more significant number of animals would need to be tested to rule out this possibility.
Overall, our study provided evidence for DNA methylation inter-generational inheritance associated with cow metabolism. Our study showed, for the first time, that different methylation patterns of metabolic-related genes such as DGK1, DGKB, PLA2G4A, and PGAM2 in daughters were possibly induced by metabolic status differences of their mothers at the peak of lactation, accompanied by fluctuations in metabolic-related signalling pathways, as well as significant epigenetic differences. Furthermore, our results supported the metabolic aspect in the DOHAD theory in large animals, especially in cows, for which there is only limited data now. Finally, this research also provided further guidance for the rational breeding of cows and provided reference to seek a balance between maximizing profits, breeding management, and dairy cow health.
Methods
Experimental design and sample collection
Selected cows were classified based on their BHB levels in the blood at 45 days postpartum. Cows with blood BHB levels under 0.9 mmol/L were considered low in BHB, and cows presenting at least 0.9 mmol/L of BHB in their blood were considered metabolic stressed. All cows were inseminated between 70 and74 days postpartum with sexed semen, for more details, refer to our published article [Citation3]. Embryos were collected seven days after the beginning of oestrus and transferred to synchronized recipient heifers. All newborn calves were females. Blood samples were collected from these eight calves within two days after birth.
Blood gDNA extraction
Whole blood samples from the tail vein were obtained from the newborn calves. Genomic DNA was then extracted with the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA). The purified gDNA was resuspended in 100 µL nuclease-free water. The concentration and integrity of the gDNA were assessed on a Tapestation 4200 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, USA).
Library preparation
Genomic DNA from calf blood underwent bisulphite conversion and the libraries were constructed with the Pico Methyl-SeqTMLibrary Prep Kit (Catalogue Nos.D5455). After purification of the PCR products, the Agilent 2200 Tapestation was used for validation and quantification of the libraries. The Pico Methyl-Seq libraries were then ready for sequencing on the Illumina HiSeq X sequencing platform to yield the 150 bp paired-end reads.
Whole genome bisulphite sequencing, data quantification and analysis
Adapter-removal and quality trimming of the libraries were performed with TRIMMOMATIC [Citation27]. BISMARK [Citation28] was then used to align the bisulphite-converted libraries to a synthetic converted bovine genome (ARS-UCD1.2, bosTau9) (https://www.ncbi.nlm.nih.gov/assembly/GCF_002263795.1/) and to infer methylation calls. The Bioconductor package BSseq [Citation29] was then used to perform methylation smoothing and identify differentially methylated cytosines and differentially methylated regions. The MethylKit package [Citation30] was also used for cytosine methylation analysis and searching of hypermethylated and hypomethylated regions with an adjusted p-value.
Functional enrichment analysis
Functional classification was performed by PANTHER(http://pantherdb.org/).KEGG analysis was performed by KOBAS.3 (http://kobas.cbi.pku.edu.cn/kobas3/?t=1), a web server for the enrichment analysis of gene set databases [Citation31]. The KEGG terms were considered significantly enriched when the q value was < 0.05.
Supplemental Material
Download MS Word (1.7 MB)Acknowledgments
The authors thank Isabelle Dufort for technical support and Janie Lévesque from CRSAD (Centre de Recherche en Sciences Animales de Deschambault, Deschambault, Quebec, Canada) for sample collection and Louis Picard for embryo transfer. The authors also thank Isabelle Laflamme for technical assistance.
Availability of the supporting data
WGBS data are available in the GEO under the GEO accession number GSE175855.
Disclosure statement
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Szyf M. Nongenetic inheritance and transgenerational epigenetics. Trends Mol Med. 2015;21:134–144.
- Bianco-Miotto T, Craig JM, Gasser YP, et al. Epigenetics and DOHaD: from basics to birth and beyond. J Dev Orig Health Dis. 2017;8:513–519.
- Chaput, C, Sirard, M. A. (2020). Embryonic response to high beta-hydroxybutyrate (BHB) levels in postpartum dairy cows. Domestic animal endocrinology. 72:106431.
- Crowe MA, Hostens M, Opsomer G. Reproductive management in dairy cows - the future. Ir Vet J. 2018;71:1.
- Lucy M, Butler S, Garverick H. Endocrine and metabolic mechanisms linking postpartum glucose with early embryonic and foetal development in dairy cows. Animal. 2014;8:82–90.
- Xu W, van Knegsel AT, Vervoort JJ, et al. Prediction of metabolic status of dairy cows in early lactation with on-farm cow data and machine learning algorithms. J Dairy Sci. 2019;102:10186–10201.
- Zarrin M, Wellnitz O, van Dorland HA, et al. Induced hyperketonemia affects the mammary immune response during lipopolysaccharide challenge in dairy cows. J Dairy Sci. 2014;97:330–339.
- Zarrin M, Grossen-Rösti L, Bruckmaier R, et al. Elevation of blood β-hydroxybutyrate concentration affects glucose metabolism in dairy cows before and after parturition. J Dairy Sci. 2017;100:2323–2333.
- Dickinson H, Moss T, Gatford KL, et al. A review of fundamental principles for animal models of DOHaD research: an Australian perspective. J Dev Orig Health Dis. 2016;7:449–472.
- Haugen A, Schug T, Collman G, et al. Evolution of DOHaD: the impact of environmental health sciences. J Dev Orig Health Dis. 2015;6:55–64.
- Fleming T, Velazquez M, Eckert J. Embryos, DOHaD and david barker. J Dev Orig Health Dis. 2015;6:377–383.
- Ciraolo E, Iezzi M, Marone R, et al. Phosphoinositide 3-kinase p110β activity: key role in metabolism and mammary gland cancer but not development. Sci Signal. 2008;1:ra3–ra.
- Gehart H, Kumpf S, Ittner A, et al. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep. 2010;11:834–840.
- Lien EC, Lyssiotis CA, Cantley LC. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer R Fortschritte der Krebsforschung Progres dans les recherches sur le cancer. 2016;207:39–72.
- Essex MJ, Boyce WT, Hertzman C, et al. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. 2013;84:58–75.
- Tobi EW, Slieker RC, Stein AD, et al. Early gestation as the critical time-window for changes in the prenatal environment to affect the adult human blood methylome. Int J Epidemiol. 2015;44:1211–1223.
- Girard A, Dufort I, Sirard M-A. The effect of energy balance on the transcriptome of bovine granulosa cells at 60 days postpartum. Theriogenology. 2015;84:1350–61. e6.
- Dechow CD, Liu W-S. DNA methylation patterns in peripheral blood mononuclear cells from Holstein cattle with variable milk yield. BMC Genomics. 2018;19:744.
- Somer RA, Thummel CS. Epigenetic inheritance of metabolic state. Curr Opin Genet Dev. 2014;27:43–47.
- Rousseau-Ralliard D, Chavatte-Palmer P, Couturier-Tarrade A. DOHaD and fertility: maternal nutrition, placental functions and offspring fertility. Méd de la Reprod. 2020;22:78–89.
- Copping KJ, Ruiz-Diaz MD, Rutland CS, et al. Peri-conception and first trimester diet modifies reproductive development in bulls. Reprod Fertil Dev. 2018;30:703–720.
- Yu JSL, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–3060.
- Terauchi Y, Tsuji Y, Satoh S, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85α subunit of phosphoinositide 3–kinase. Nat Genet. 1999;21:230–235.
- Garcı́a-Villafranca J, Guillén A, Castro J. Involvement of nitric oxide/cyclic GMP signaling pathway in the regulation of fatty acid metabolism in rat hepatocytes. Biochem Pharmacol. 2003;65:807–812.
- Pfeifer A, Kilić A, Hoffmann L. Role of cGMP in fat and metabolism. BMC Pharmacol Toxicol. 2013;14:O25–O.
- Hanson M, Gluckman P. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev. 2014. DOI:https://doi.org/10.1152/physrev.00029.2013
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27:1571–1572.
- Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83.
- Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87.
- Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–22.