
ABSTRACT
Previous genome-wide association studies (GWAS) have identified potential genetic variants involved in the risk of Alzheimer’s dementia, but their underlying biological interpretation remains largely unclear. In addition, the effects of DNA methylation and gene expression on Alzheimer’s dementia are not well understood. A network summary data-based Mendelian randomization (SMR) analysis was performed integrating cis- DNA methylation quantitative trait loci (mQTL) /cis- gene expression QTL (eQTL) data in the brain and blood, as well as GWAS summarized data for Alzheimer’s dementia to evaluate the pleiotropic associations of DNA methylation and gene expression with Alzheimer’s dementia and to explore the complex mechanisms underpinning Alzheimer’s dementia. After correction for multiple testing (false discovery rate [FDR] P < 0.05) and filtering using the heterogeneity in dependent instruments (HEIDI) test (PHEIDI>0.01), we identified dozens of DNA methylation sites and genes showing pleiotropic associations with Alzheimer’s dementia. We found 22 and 16 potentially causal pathways of Alzheimer’s dementia (i.e., SNP→DNA methylation→Gene expression→Alzheimer’s dementia) in the brain and blood, respectively. Approximately two-thirds of the identified DNA methylation sites had an influence on gene expression and the expression of almost all the identified genes was regulated by DNA methylation. Our network SMR analysis provided evidence supporting the pleiotropic association of some novel DNA methylation sites and genes with Alzheimer’s dementia and revealed possible causal pathways underlying the pathogenesis of Alzheimer’s dementia. Our findings shed light on the role of DNA methylation in gene expression and in the development of Alzheimer’s dementia.
Introduction
Alzheimer’s dementia, the most common form of dementia in elderly people and a major cause of disability, is a severe public health burden worldwide[Citation1], [Citation2]. Alzheimer’s dementia is a complex neurodegenerative disease, with contributions from a variety of neuropathological, genetic, and environmental risk factors [Citation3–5]. However, there are considerable gaps in our understanding of the nosology and aetiological complexity of Alzheimer’s dementia.
Alzheimer’s dementia is a highly heritable disease, and genome-wide association studies (GWAS) have revealed multiple genetic variants that are associated with susceptibility of Alzheimer’s dementia [Citation4,Citation6,Citation7]; however, the exact functions of the identified genetic variants remain uncharacterized. Because most genetic variants associated with complex traits are in non-coding regions of the genome, it is likely that such variants influence disease susceptibility through mechanisms other than proteins coding [Citation8–12]. Previous research found that DNA methylation regulating gene expression participates in the molecular mechanism of Alzheimer’s dementia [Citation13]. However, the causal relationship between genetic variants, DNA methylation and gene expression in influencing Alzheimer’s dementia has not been systematically explored.
Previous studies using summary data-based Mendelian randomization (SMR) analysis that integrated cis- DNA methylation quantitative trait loci (mQTL) or cis- gene expression QTL (eQTL) data with GWAS summarized data found that both DNA methylation and gene expression resided along the causal pathway from genetic variation to complex diseases, such as cardiovascular disease and Alzheimer’s dementia [Citation10,Citation12,Citation14–16]. The network Mendelian randomization (MR) aims to disentangle complex networks underlying the pathogenesis of a disease by utilizing multi-omics data in a multi-step SMR analysis [Citation17]. This approach has been successful in revealing the potentially causal pathways of several diseases, such as schizophrenia, coronary artery disease, and multiple sclerosis [Citation17,Citation18]. In the present paper, we performed a network SMR study integrating mQTL, eQTL, and GWAS summarized data for Alzheimer’s dementia to explore the pleiotropic association of DNA methylation and gene expression with Alzheimer’s dementia. In a previous study, we conducted a MR analysis integrating GWAS and mQTL data to explore the role of DNA methylation in the neuropathogenesis of Alzheimer’s disease (AD) [Citation16]. The current study is different from our previous one in that: 1) The network SMR approach adopted in the present study includes eQTL as well as mQTL data, enabling us to explore multiple potentially causal pathways; 2) The network SMR analyses use data from multiple studies and have much larger sample sizes, compared with the previous study which only used data from the Religious Orders Study and the Rush Memory and Ageing Project (ROSMAP) [Citation19]; and 3) We performed SMR analysis in both blood and brain, compared with the previous research in which we only analysed data from brain. We performed separate SMR analyses using data from the brain and blood because DNA methylation and gene expression are tissue specific. If findings from the two analyses are different, they may imply different pathways in the brain and blood underlying the pathogenesis of Alzheimer’s dementia. By contrast, overlapping results could provide more convincing evidence of the identified pathways underpinning Alzheimer’s dementia. The findings of the present study thus provide important insights into the complex mechanisms of DNA methylation and gene expression for Alzheimer’s dementia.
Methods
Study design
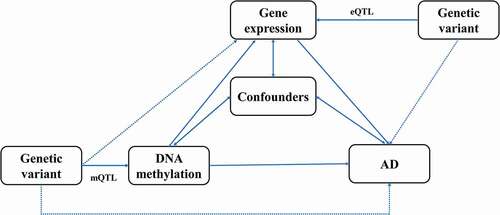
In the present study, we conducted SMR analysis integrating summarized GWAS data for Alzheimer’s dementia, cis-mQTL and cis-eQTL data in the blood and brain to prioritize DNA methylation sites and genes that were pleiotropically or potentially causally associated with the risk of Alzheimer’s dementia. Our analysis consisted of three different MR tests, as described below. First, the pleiotropic associations of genetically determined DNA methylation with Alzheimer’s dementia were analysed considering cis-mQTL as the instrumental variables (IVs), DNA methylation as the exposure, and Alzheimer’s dementia as the outcome. Second, the pleiotropic associations of genetically determined gene expressions with Alzheimer’s dementia were analysed considering cis-eQTL as the IVs, gene expression as the exposure, and Alzheimer’s dementia as the outcome. Third, the pleiotropic associations of genetically determined DNA methylation with gene expression were analysed considering cis-mQTL as the IVs, DNA methylation as the exposure, and gene expression as the outcome. The design of the network SMR analysis is presented in . Analyses were performed separately for brain and blood.
Figure 1. Study design of the network Mendelian randomization.
Data sources
mQTL data
The brain mQTL data (named brain-mQTL hereafter) were obtained from a meta-analysis of ROSMAP [Citation19], the study by Hannon et al. [Citation20], and the study by Jaffe et al. [Citation21]. The brain-mQTL data [Citation22] included 1,160 participants (Supplementary Table 1). In the ROSMAP study, only SNPs within 5 Kb from each CpG were available. In the study by Hannon et al., only SNPs within 500 Kb of each CpG with PmQTL < 1 × 10−10 were available. In the study by Jaffe et al., only SNPs within 20 Kb from each CpG with a false discovery rate (FDR [Citation23]) < 0.1 were available. The blood mQTL data (named blood-mQTL hereafter) were obtained from a meta-analysis of the study by McRae et al. and Wu et al. [Citation17,Citation24], which included a total of 1,980 participants and were generated using peripheral blood in two cohorts, the Brisbane Systems Genetics Study [Citation25] (BSGS) and the Lothian Birth Cohorts [Citation26] (LBC). DNA methylation data of all the samples were generated using Illumina HumanMethylation450 chips. Only CpG probes with at least a cis-mQTL at P < 5 × 10−8 and SNPs within 2 Mb from each CpG were retained. The mQTL data can be downloaded at https://cnsgenomics.com/data/SMR/#mQTLsummarydata.
eQTL data
The brain eQTL data (named brain-eQTL hereafter) were obtained from the meta-analysis of three studies including a total of 1,194 participants [Citation22]: GTEx [Citation27], the Common Mind Consortium [Citation28], and ROSMAP [Citation19]. The blood eQTL data (named blood-eQTL hereafter) were obtained from the CAGE eQTL summarized data [Citation29], which included 2,765 participants. Only SNPs within 1 Mb from each mRNA were available. The eQTL data can be downloaded at https://cnsgenomics.com/data/SMR/#eQTLsummarydata.
GWAS data for Alzheimer’s dementia
The GWAS summarized data for Alzheimer’s dementia were based on the meta-analysis of four large GWASs, including the Psychiatric Genomics Consortium (PGC-ALZ), the International Genomics of Alzheimer’s Project (IGAP), the Alzheimer’s Disease Sequencing Project (ADSP), and UK Biobank (UKB) [Citation6]. The summarized data included 455,258 individuals (71,880 cases and 383,378 controls) and 13,367,301 variants. The GWAS summarized data can be downloaded at https://ctg.cncr.nl/software/summary_statistics/.
Detailed information on Alzheimer’s dementia status of subjects included in the mQTL and eQTL data is not available. Many of the included studies, such as ROSMAP and LBC, are longitudinal studies in which subjects who were initially healthy are followed for many years. Therefore, some of the included subjects very likely had developed Alzheimer’s dementia or mild cognitive impairment during the follow-up.
Statistical and bioinformatics analysis
The MR analyses were performed using the method implemented in the software SMR. Detailed information regarding the SMR method has been described previously [Citation30]. Briefly, SMR uses the principles of MR to test for pleotropic associations between the exposure and the outcome due to a shared and potentially causal variant at a locus. The heterogeneity in dependent instruments (HEIDI) test was done to explore the existence of linkage in the observed association. Rejection of the null hypothesis indicates that the observed associations might be due to two distinct genetic variants in high linkage disequilibrium (LD) with each other. We adopted a threshold of 0.01 as the criteria for rejection of the null hypothesis because recent research indicated that using PHEIDI <0.05 often leads to false positive findings [Citation17].
We adopted the default settings in SMR (e.g., PQTL <5 × 10−8, minor allele frequency [MAF] > 0.01, removing SNPs in very strong LD [r2 > 0.9] with the top associated QTL, and removing SNPs in low LD or not in LD [r2 <0.05] with the top associated QTL), and used FDR to adjust for multiple testing. Only SNPs with available SNP-exposure and SNP-outcome association data were retained. For the network SMR, pleotropic associations between genes and DNA methylation sites that were in less than 2 Mb of the corresponding genes were tested.
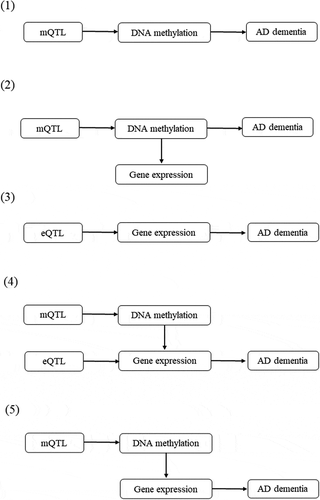
The pathways from genetic variants to Alzheimer’s dementia were delineated into five potential categories (). Annotations of the transcripts were based on the Affymetrix exon array S1.0 platform. To functionally annotate putative transcripts, we conducted a functional enrichment analysis using the functional annotation tool ‘Metascape’ [Citation31] for the genes identified in the pathways from genetic variants to Alzheimer’s dementia. Gene symbols corresponding to significant genes (FDR P <0.05) were used as the input of the gene ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment analysis. The information on DNA methylation can be obtained at (http://bigd.big.ac.cn/methbank) [Citation32].
Figure 2. Potential pathways linking genetic variants with Alzheimer’s dementia.
Data cleaning and statistical/bioinformatical analysis were performed using R version 4.0.0 (https://www.r-project.org/), PLINK 1.9 (https://www.cog-genomics.org/plink/1.9/) and SMR (https://cnsgenomics.com/software/smr/).
Results
Pleiotropic associations between DNA methylation and Alzheimer’s dementia
Using brain-mQTL data and GWAS summary data for Alzheimer’s dementia, we performed SMR analysis to evaluate the associations between 94,537 CpGs probes and Alzheimer’s dementia. We found 68 CpGs that showed pleiotropic associations with Alzheimer’s dementia after correction for multiple testing and application of the HEIDI test (PHEIDI >0.01; Supplementary Table 2). Using blood-mQTL data and GWAS summary data for Alzheimer’s dementia, we evaluated the associations between 93,087 CpG probes and Alzheimer’s dementia. We found 95 CpGs that showed pleiotropic associations with Alzheimer’s dementia after correction for multiple testing and application of the HEIDI test (Supplementary Table 3), of which 22 CpGs overlapped with the brain-mQTL results, including cg03526776, cg12672189, cg15837308, cg10846853, cg13778567, cg20172563, cg13210467, cg05868365, cg18648645, cg08729755, cg03887787, cg19792802, cg08416661, cg16757332, cg09251291, cg08898775, cg26312935, cg02220965, cg06985993, cg06015834, cg09495303, and cg10553748.
Pleiotropic associations between gene expression and Alzheimer’s dementia
Using brain-eQTL data and GWAS summary data for Alzheimer’s dementia, we performed SMR analysis to evaluate the associations between 7,466 mRNA probes and Alzheimer’s dementia. We found 15 mRNA probes tagging 15 genes that showed pleiotropic associations with Alzheimer’s dementia after correction for multiple testing and application of the HEIDI test (Supplementary Table 4). Using blood-eQTL data and GWAS summary data for Alzheimer’s dementia, we tested the associations between 8,519 mRNA probes and Alzheimer’s dementia. We found 15 mRNA probes tagging 13 genes that showed pleiotropic associations with Alzheimer’s dementia after correction for multiple testing and application of the HEIDI test (Supplementary Table 5), of which only one gene FIBP overlapped with the brain-eQTL result.
Pleiotropic associations between DNA methylation and gene expression
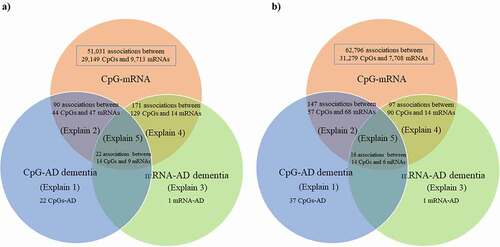
Using brain-mQTL data and brain-eQTL data in the SMR, we found 51,031 associations between 29,149 CpGs and 9,713 mRNAs after correction for multiple testing and application of the HEIDI test (). Using blood-mQTL data and blood-eQTL data, we found 62,796 associations between 31,279 CpGs and 7,708 mRNAs after correction for multiple testing and application of the HEIDI test (), of which 2,886 associations between 2,203 CpGs and 1,207 mRNAs overlapped with results obtained in the brain.
Figure 3. Venn diagram of the 5 different pathways linking genetic variants with Alzheimer’s dementia.
Pathways from genetic variants to Alzheimer’s dementia
In the brain, we found 22 pathways in which genetic variants exerted their effect on Alzheimer’s dementia mediated completely by DNA methylation. We discovered 90 pathways involving 44 CpGs and 47 mRNAs tagging 47 genes in which genetic variants exerted their effect on Alzheimer’s dementia mediated by DNA methylation regulating gene expression; only one pathway in which genetic variants exerted their effect on Alzheimer’s dementia mediated completely by gene expression; and 171 pathways involving 129 CpGs and 14 mRNAs tagging 14 genes in which gene expression influenced the susceptibility of Alzheimer’s dementia regulated by DNA methylation (Supplementary Table 2, Supplementary Table 4, and ). Finally, we found 22 pathways involving 14 CpGs regulating 9 mRNAs tagging 9 genes in linking genetic variants with the risk of Alzheimer’s dementia (i.e., SNP→DNA methylation→Gene expression→Alzheimer’s dementia; ).
Table 1. Prioritizing genes and DNA methylation involved in potentially causal pathways linking genetic variants with Alzheimer’s dementia in brain.
In the blood, we found 37 pathways in which genetic variants exert their effect on Alzheimer’s dementia mediated completely by DNA methylation; 147 pathways involving 57 CpGs and 68 mRNAs tagging 62 genes in which genetic variants exert their effect on Alzheimer’s dementia mediated by DNA methylation regulating gene expression; only one pathway in which genetic variants exert their effect on Alzheimer’s dementia mediated completely by gene expression; and 97 pathways involving 90 CpGs and 14 mRNAs tagging 12 genes in which gene expressions influence the susceptibility of Alzheimer’s dementia regulated by DNA methylation (Supplementary Table 3, Supplementary Table 5, and ). Finally, we found 16 pathways involving 14 CpGs regulating 6 mRNAs tagging 6 genes in linking genetic variants with the risk of Alzheimer’s dementia (i.e., SNP→DNA methylation→Gene expression→Alzheimer’s dementia; ).
Table 2. Prioritizing genes and DNA methylation involved in potentially causal pathways linking genetic variants with Alzheimer’s dementia in blood.
Functionally relevant DNA methylation and target genes
Of the 68 identified CpGs in the brain, 46 (67.7%) were associated with the expression of 56 mRNAs tagging 56 genes comprising 112 CpG-mRNA pairs (Supplementary Table 6). GO enrichment analysis of biological process, molecular function, and cellular component pathways showed that the genes that were cis associated with the CpGs identified in the pathways from genetic variants to Alzheimer’s dementia were involved in eight GO terms, such as antigen processing and presentation (GO: 0019882) and regulation of innate immune response (GO:0045088; Supplementary Figure 1 and Supplementary Table 7). GO enrichment analysis of the genes identified in the pathways from genetic variants to Alzheimer’s dementia revealed one significant GO term of neutrophil mediated immunity (GO:0002446; Supplementary Figure 2 and Supplementary Table 8).
Of the 95 identified CpGs in blood, 58 (61.05%) were associated with 74 mRNAs tagging 66 genes comprising 163 CpG-mRNA pairs (Supplementary Table 9), of which 16 associations between 14 CpGs and 6 genes, including BCKDK, CTSW, FIBP, FCER1G, APH1B and PILRA, overlapped between brain and blood. The genes that were cis associated with the CpGs identified in the pathways from genetic variants to Alzheimer’s dementia were involved in fourteen GO terms, such as antigen processing and positive regulation of leukocyte degranulation (GO:0043302; Supplementary Figure 3). GO enrichment analysis of the genes identified in the pathways from genetic variants to Alzheimer’s dementia revealed three significant GO terms: immune response-regulating signalling pathway (GO:0002764), transmembrane receptor protein tyrosine kinase signalling pathway (GO:0007169), and regulated exocytosis (GO:0045055; Supplementary Fig. 4). More information can be found in Supplementary Figures 3–4 and Supplementary Tables 10–11.
Discussion
In this study, we performed network SMR analysis by integrating multi-omics data to explore the complex mechanisms underpinning Alzheimer’s dementia. We identified dozens of DNA methylation sites and genes showing pleiotropic associations with Alzheimer’s dementia. Approximately two-thirds of the identified CpGs had an influence on gene expressions and the expression of almost all the identified genes was regulated by DNA methylation. To the best of our knowledge, this is the first network SMR study to evaluate the pleiotropic associations of DNA methylation and gene expression with Alzheimer’s dementia and to identify the potential causal pathways of gene regulation linking genetic variants with Alzheimer’s dementia.
In our study, some of the identified causal DNA methylation sites were reported to be associated with the risk factors of Alzheimer’s dementia. For example, cg24465943 in FCER1G, cg02747950 in RAB8B, and cg05868365 and cg00411097 in TMEM184A were found to be associated with ageing, a major risk factor for Alzheimer’s dementia [Citation33,Citation34]. One methylation site, cg02747950 in RAB8B was associated with type 2 diabetes (T2D). Increasing evidence suggests epidemiological and pathological links between Alzheimer’s dementia and T2D [Citation35,Citation36]. In addition, cg05656486, cg08850169 and cg24049880 in NDUFS2, cg09251291 in RAB8B, and cg19739596 in MS4A3 were associated with systemic lupus erythematosus (SLE), which could increase the risk of Alzheimer’s dementia [Citation37]. Whether and how these identified methylation loci affect the risk of Alzheimer’s dementia via these risk factors warrants further investigation.
In our study, some of the causal genes identified in our network SMR, including KAT8, BCKDK, FCER1G, and PILRA, were reported to be associated with Alzheimer’s dementia [Citation6,Citation7,Citation38,Citation39]. KAT8, located on chromosome 16, encodes a member of the MYST histone acetylase protein family [Citation40]. MYST acetyltransferases are essential in transcription, and DNA replication, recombination and repair, and are involved in autophagy [Citation41,Citation42]. A previous study demonstrated that mutations in histone acetyltransferases, including MYST acetyltransferases, could lead to abnormal development of the nervous system and increased neurodegeneration [Citation43], indicating thatKAT8 might play a role in the aetiology of Alzheimer’s dementia by regulating MYST-family acetyltransferase activity. BCKDK encodes the branched-chain alpha-ketoacid dehydrogenase complex (BCKD), which is an important regulator of the valine, leucine, and isoleucine catabolic pathways [Citation44]. BCKDK mutations leads to a potentially treatable form of autism with intellectual disability and epilepsy [Citation45]. Another study found that variants in BCKDK were associated with Parkinson’s disease [Citation46]. These findings, together with ours, demonstrated the important role of BCKDK in neurological diseases and highlighted the potential of this gene as a promising target for the prevention and treatment of Alzheimer’s dementia. FCER1G encodes a high affinity IgE receptor, which plays an important role in allergic reactions. A previous study showed that FCER1G predicted the risk of Alzheimer’s dementia and was significantly upregulated when exposed to Aβ [Citation47]. PILRA, which encodes a cell surface inhibitory receptor that participates in the dynamic interaction between the activation and suppression of cell signal transduction [Citation48], may be involved in the pathogenesis of Alzheimer’s dementia due to its involvement in immune regulation. In addition, a whole exome sequencing study provided evidence that PILRA played an important role in the pathogenesis of Alzheimer’s dementia [Citation49]. Based on the above findings, future research needs to further explore the application of these genes in the development of Alzheimer’s dementia.
Some genes found by our network SMR didn’t overlap with those identified in previous GWASs of Alzheimer’s dementia, implying that these identified genes might be novel genes underlying the pathogenesis of Alzheimer’s dementia. These genes could exert their function through a variety of mechanisms underlying the aetiology of Alzheimer’s dementia. For example, FIBP encodes acidic fibroblast growth factor (FGF) intracellular binding protein (FIBP). FIBP is known to be implicated in the FGF signalling pathway [Citation50], which has been shown to affect cell proliferation, cell survival, chemotaxis, migration, and cell adhesion; it also plays an important role in the development of many diseases [Citation51–53]. A previous study suggested that abnormally increased FGF-2-associated dysregulation of dentate gyrus neurogenesis, especially neuronal polarity, was involved in the pathogenesis of Alzheimer’s dementia [Citation54]. Another study found that APOE interacted with FGF1 in leading to episodic memory deficits and hippocampus atrophy, thereby contributing to Alzheimer’s dementia [Citation55]. The identified genes in the brain and blood did not overlap except FIBP. These findings, together with ours, demonstrated the important role of FIBP in the pathogenesis of Alzheimer’s dementia and highlighted the potential of this gene as a promising target for the prevention and treatment of Alzheimer’s dementia.
DNA methylation can influence gene expression, and its relationship with transcriptional activity plays an important role in influencing diseases [Citation11]. In our study, the expression of almost all the identified genes was regulated by DNA methylation, highlighting the important role of DNA methylation in the regulation of gene expression in leading to Alzheimer’s dementia. In addition, the presence of ‘vertical causal pathways’ underpinned the pathogenesis of Alzheimer’s dementia, linking genetic variants, DNA methylation, and gene expression to Alzheimer’s dementia. DNA methylation and gene expression of NDUFS2, STAG3, CTSW, and PRSS36 play an important role in the regulation of how genetic variants affect the development of Alzheimer’s dementia. NDUFS2, located on chromosome 1, was associated with mitochondrial complex I deficiency and was essential for oxygen-sensing [Citation56]. A previous study showed that mitochondrial dysfunction was present in degenerative disorders, including Alzheimer’s dementia and Parkinson’s disease [Citation57]. It is possible that NDUFS2 plays a role in the aetiology of Alzheimer’s dementia by regulating mitochondrial dysfunction. Moreover, CTSW encoded a cysteine proteinase as a member of the peptidase C1 family, which was found to be associated with the expression of natural killer and cytotoxic T-cells [Citation58]. A study found that the increased ratio of cytotoxic T cells was regarded as a preclinical biomarker of Alzheimer’s dementia and involved in the Aβ neuropathological mechanism [Citation59]. Both animal and in vivo human studies are needed to explore the physiological functions that these genes exert and the pathways that they are involved in influencing Alzheimer’s dementia.
Approximately two-thirds of the identified CpGs influenced gene expression, indicating that DNA methylation does not always influence gene expression. Future research should focus on exploring the role of DNA methylation in the pathogenesis of Alzheimer’s dementia and exploring the application of DNA methylation in the targeted intervention and prediction of Alzheimer’s dementia. The genetic risk score (GRS) is a powerful tool in assessing the risk of many diseases such as Alzheimer’s dementia [Citation4,Citation60]. However, GRS cannot capture the environmental contributions to the risk of a disease and often has limited clinical utility. Environmental exposure that affects disease progression can modify methylation patterns [Citation61,Citation62], which in turn can affect many biological processes and influence disease susceptibility [Citation63,Citation64]. Combining methylation risk score (MRS) and GRS can improve predictive accuracy in identifying high-risk populations of diseases [Citation65,Citation66]. Future studies with individual-level multi-omics data are needed to explore the utility of combining MRS and GRS using the identified DNA methylation sites and genes in assessing the risk of Alzheimer’s dementia.
Our study has some limitations. We performed the SMR and HEIDI analyses to detect DNA methylation – gene expression, DNA methylation – Alzheimer’s dementia, and gene expression – Alzheimer’s dementia associations separately and focused on association signals that were consistent across the three types of analyses at a locus. This strategy potentially loses power due to thresholding the results by P-values in the multiple steps. In addition, we adopted correction for multiple testing to reduce the false positive rate; however, we might have missed important SNPs, CpGs, and genes. The MR analyses were based on several assumptions, some of which cannot be tested directly. Our results may be biased if these assumptions are violated, and cautions should be executed in interpreting the potentially causal pathways from genetic variants to Alzheimer’s dementia. Finally, due to the scarcity of publicly relevant data, we did not examine how DNA methylation or gene expression of the identified genes changed in subjects who had Alzheimer’s dementia compared to the healthy controls. Examination of such changes would help researchers better understand the roles of these genes in the pathogenesis of Alzheimer’s dementia. We plan to pursue this in a future study when we have access to data that provide gene expression data and status of Alzheimer’s disease at the individual level.
In conclusion, our network SMR analysis provided evidence supporting the pleiotropic association of some novel DNA methylation sites and genes with Alzheimer’s dementia and revealed possible causal pathways underlying the pathogenesis of Alzheimer’s dementia. Our findings highlighted the important role of DNA methylation in the regulation of gene expression leading to Alzheimer’s dementia. Future studies are needed to explore the biological functions of the identified CpGs and genes in the development of Alzheimer’s dementia.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author contributions
Di Liu designed the study, performed the statistical analyses, wrote the first draft of the manuscript, and drew the figure.
Youxin Wang designed the study, reviewed and commented on the manuscript.
Huiquan Jing reviewed and commented on the manuscript.
Qun Meng is the corresponding author, designed the study, reviewed and commented on the manuscript.
Jingyun Yang is the corresponding author, designed the study, took responsibility for the accuracy of the analysis, and had authority over manuscript preparation and the decision to submit the manuscript for publication.
Supplemental Material
Download Zip (601.4 KB)Acknowledgments
We would like to thank Dr. Jian Yang and the Complex Traits Genomics group who developed the SMR software and have made their code publicly available. We also would like to thank PGC-ALZ, IGAP, ADSP and UKB investigators for making their data publicly available.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All the data used for the analyses are publicly available, as indicated in the corresponding subsections. https://ctg.cncr.nl/software/summary_statistics/
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509.
- GBD. 2017 disease and injury incidence and prevalence collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392:1789–1858.
- Nelson PT, Elizabeth H, Schmitt FA, et al. Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121(5):571–587.
- Sims R, Hill M, Williams J. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci. 2020;23(3):311–322.
- Lourida I, Hannon E, Littlejohns TJ, et al. Association of lifestyle and genetic risk with incidence of dementia. Jama. 2019;322(5):430–437.
- Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. 10 .1038 /s41588-018- 0311-9.
- Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414–430.
- Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195.
- Wagner JR, Busche S, Ge B, et al. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15(2):R37.
- Richardson TG, Haycock PC, Zheng J, et al. Systematic Mendelian randomization framework elucidates hundreds of CpG sites which may mediate the influence of genetic variants on disease. Hum Mol Genet. 2018;27(18):3293–3304.
- Hannon E, Gorrie-Stone TJ, Smart MC, et al. DNA-Methylation quantitative-Trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am J Hum Genet. 2018;103(5):654–665.
- Huan T, Joehanes R, Song C, et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat Commun. 2019;10(1):4267.
- Semick SA, Bharadwaj RA, Collado-Torres L, et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer’s disease. Acta Neuropathol. 2019;137(4):557–569
- Richardson TG, Zheng J, Davey SG, et al. Mendelian randomization analysis identifies CpG sites as putative mediators for genetic influences on cardiovascular disease risk. Am J Hum Genet. 2017;101(4):590–602.
- Trajanoska K, Morris JA, Oei L, et al. Assessment of the genetic and clinical determinants of fracture risk: genome wide association and mendelian randomisation study. BMJ. 2018;362:k3225.
- Liu D, Wang Y, Jing H, et al. Mendelian randomization integrating GWAS and mQTL data identified novel pleiotropic DNA methylation loci for neuropathology of Alzheimer’s disease. Neurobiol Aging. 2021;97:18–27.
- Wu Y, Zeng J, Zhang F, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918.
- Jacobs BM, Taylor T, Awad A, et al. Summary-data-based Mendelian randomization prioritizes potential druggable targets for multiple sclerosis. Brain Comm. 2020;2(2):fcaa119.
- Ng B, White CC, Klein H-U, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 2017;20(10):1418–1426.
- Hannon E, Spiers H, Viana J, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2016;19(1):48–54.
- Jaffe AE, Gao Y, Deep-Soboslay A, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19(1):40–47.
- Qi T, Wu Y, Zeng J, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9(1):2282.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B (Methodological). 1995;57(1):289–300.
- McRae AF, Marioni RE, Shah S, et al. Identification of 55,000 replicated DNA methylation QTL. Sci Rep. 2018;8(1):17605.
- Powell JE, Henders AK, McRae AF, et al. The Brisbane SYSTEMS GENETICS Study: genetical genomics meets complex trait genetics. PLoS One. 2012;7(4):e35430.
- Deary IJ, Gow AJ, Pattie A, et al. Cohort profile: the Lothian birth cohorts of 1921 and 1936. Int J Epidemiol. 2012;41(6):1576–1584.
- Aguet F, Brown AA, Castel SE, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213.
- Fromer M, Roussos P, Sieberts SK, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19(11):1442–1453.
- Lloyd-Jones LR, Holloway A, McRae A, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet. 2017;100(2):228–237.
- Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487.
- Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
- Li R, Liang F, Li M, et al. MethBank 3.0: a database of DNA methylomes across a variety of species. Nucleic Acids Res. 2018;46:D288–d95.
- Florath I, Butterbach K, Muller H, et al. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014;23(5):1186–1201.
- Weidner CI, Lin Q, Koch CM, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15(2):R24.
- Zhang J, Chen C, Hua S, et al. An updated meta-analysis of cohort studies: diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract. 2017;124:41–47.
- Boccardi V, Murasecco I, Mecocci P. Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res Rev. 2019;54:100936.
- Wotton CJ, Goldacre MJ. Associations between specific autoimmune diseases and subsequent dementia: retrospective record-linkage cohort study, UK. J Epidemiol Community Health. 2017;71(6):576–583.
- Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry. 2018;8(1):99.
- Schwartzentruber J, Cooper S, Liu JZ, et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat Genet. 2021;53(3):392-402.
- Thomas T, Dixon MP, Kueh AJ, et al. Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol Cell Biol. 2008;28(16):5093–5105.
- Füllgrabe J, Klionsky DJ, Joseph B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy. 2013;9(10):1621–1623.
- Carrozza MJ, Utley RT, Workman JL, et al. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19(6):321–329.
- Wang Z, Zang C, Cui K, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138(5):1019–1031.
- Han AC, Goodwin GW, Paxton R, et al. Activation of branched-chain alpha-ketoacid dehydrogenase in isolated hepatocytes by branched-chain alpha-ketoacids. Arch Biochem Biophys. 1987;258(1):85–94.
- Novarino G, El-Fishawy P, Kayserili H, et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science. 2012;338(6105):394–397.
- Heckman MG, Kasanuki K, Diehl NN, et al. Parkinson’s disease susceptibility variants and severity of Lewy body pathology. Parkinsonism Relat Disord. 2017;44:79–84.
- Sierksma A, Lu A, Mancuso R, et al. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med. 2020;12(3):e10606.
- Wang J, Shiratori I, Uehori J, et al. Neutrophil infiltration during inflammation is regulated by PILRα via modulation of integrin activation. Nat Immunol. 2013;14(1):34–40.
- Patel T, Brookes KJ, Turton J, et al. Whole-exome sequencing of the BDR cohort: evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2018;44(5):506–521.
- Thauvin-Robinet C, Duplomb-Jego L, Limoge F, et al. Homozygous FIBP nonsense variant responsible of syndromic overgrowth, with overgrowth, macrocephaly, retinal coloboma and learning disabilities. Clin Genet. 2016;89(5):e1–4.
- Auguste P, Javerzat S, Bikfalvi A. Regulation of vascular development by fibroblast growth factors. Cell Tissue Res. 2003;314(1):157–166.
- Chen S, Li H, Gaudenz K, et al. Defective FGF signaling causes coloboma formation and disrupts retinal neurogenesis. Cell Res. 2013;23(2):254–273.
- Ma F, Zhu T, Xu F, et al. Neural stem/progenitor cells on collagen with anchored basic fibroblast growth factor as potential natural nerve conduits for facial nerve regeneration. Acta Biomater. 2017;50:188–197.
- Tatebayashi Y, Lee MH, Li L, et al. The dentate gyrus neurogenesis: a therapeutic target for Alzheimer’s disease. Acta Neuropathol. 2003;105(3):225–232.
- Chang YT, Kazui H, Ikeda M, et al. Genetic Interaction of APOE and FGF1 is associated with memory impairment and hippocampal atrophy in Alzheimer’s disease. Aging Dis. 2019;10(3):510–519.
- Dunham-Snary KJ, Wu D, Potus F, et al. Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and Hypoxic pulmonary vasoconstriction. Circ Res. 2019;124(12):1727–1746.
- Humphrey DM, Parsons RB, Ludlow ZN, et al. Alternative oxidase rescues mitochondria-mediated dopaminergic cell loss in Drosophila. Hum Mol Genet. 2012;21(12):2698–2712.
- Wex T, Wex H, Hartig R, et al. Functional involvement of cathepsin W in the cytotoxic activity of NK-92 cells. FEBS Lett. 2003;552(2–3):115–119.
- Yasuno F, Kazui H, Kajimoto K, et al. Mutual effect of cerebral amyloid β and peripheral lymphocytes in cognitively normal older individuals. Int J Geriatr Psychiatry. 2017;32(12):e93–e9.
- Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 2017;14(3):e1002258.
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330(6004):612–616.
- Murgatroyd C, Patchev AV, Wu Y, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12(12):1559–1566.
- Grundberg E, Meduri E, Sandling JK, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013;93(5):876–890.
- Gaunt TR, Shihab HA, Hemani G, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17(1):61.
- Clark SL, Hattab MW, Chan RF, et al. A methylation study of long-term depression risk. Mol Psychiatry. 2020;25(6):1334–1343.
- Guan Z, Raut JR, Weigl K, et al. Individual and joint performance of DNA methylation profiles, genetic risk score and environmental risk scores for predicting breast cancer risk. Mol Oncol. 2020;14(1):42–53.