
ABSTRACT
Animal domestication is a process of environmental modulation and artificial selection leading to permanent phenotypic modifications. Recent studies showed that phenotypic changes occur very early in domestication, i.e., within the first generation in captivity, which raises the hypothesis that epigenetic mechanisms may play a critical role on the early onset of the domestic phenotype. In this context, we applied reduced representation bisulphite sequencing to compare methylation profiles between wild Nile tilapia females and their offspring reared under farmed conditions. Approximately 700 differentially methylated CpG sites were found, many of them associated not only with genes involved in muscle growth, immunity, autophagy and diet response but also related to epigenetic mechanisms, such as RNA methylation and histone modifications. This bottom-up approach showed that the phenotypic traits often related to domestic animals (e.g., higher growth rate and different immune status) may be regulated epigenetically and prior to artificial selection on gene sequences. Moreover, it revealed the importance of diet in this process, as reflected by differential methylation patterns in genes critical to fat metabolism. Finally, our study highlighted that the TGF-β1 signalling pathway may regulate and be regulated by several differentially methylated CpG-associated genes. This could be an important and multifunctional component in promoting adaptation of fish to a domestic environment while modulating growth and immunity-related traits.
Introduction
Animal domestication is a process of modifying a population phenotype through anthropic selection [Citation1] and concerns species chosen for their high value from a human perspective. It starts with wild animals that are kept under controlled conditions and selectively bred or cross-bred for traits of interest, such as improved growth. The environmental conditions during domestication are very distinct of those to which the organism is exposed in the wild. For example, farm-like conditions often include captivity and high density rearing, regular and ad libitum feeding, highly homogenous diet and stable abiotic conditions (temperature, photoperiod), assisted health care and the absence of predatory pressure [Citation2], but also continuous contact with humans. Such drastic changes in the environment can lead to strong phenotypic reaction at an early stage, which, if genetically encoded and repeated over many generations, can eventually give grounds for adaptation. Environmentally-induced changes in phenotype over generations are accompanied and often fostered by strong artificial selection, especially when phenotypic traits sensitive to the environment are also simultaneously targeted by selective breeding [Citation3].
Interestingly, since the farm environments are alike and target traits that are often the same among highly unrelated species, their domestication leads to a set of convergent phenotypic traits, often referred to as the domesticated phenotype [Citation4]. Among the most known traits of domesticated phenotype, we can cite altered behaviours such as increased tameness or reduced stress responsiveness, body proportion and size modifications with reduced locomotory capacities and physiological changes related to increased growth potential [Citation5]. Domestication can also lead to unintentional phenotypic traits, such as decreased immunological resistance [Citation6], a consequence of rearing in relatively sterile conditions or co-selection with targeted traits such a growth [Citation7].
The presence of unwelcome traits in artificially generated domestic phenotype suggests that the process of domestication is not yet fully controlled, and its mechanisms not completely understood. For example, various studies highlight an extremely fast pace of stable phenotypic modifications in a population under ongoing domestication, i.e., within an evolutionary time too short to be explained only by classical genetics. Standing genetic variation can be low in a limited-size founder population F0 [Citation8] and natural occurrence of mutations and subsequent artificial selection can be accounted only in trans-generational intervals, while adaptive changes can occur even within the first generation of organisms submitted to a domestic environment [Citation9,Citation10]. Rapid changes in gene expression can be due to epigenetic mechanisms such as DNA methylation, that not only modulate the response to the new environment but can also be stable over generations. In comparison with genetic variation, epigenetic variation is more likely to have higher rates of spontaneous mutation [Citation11] and to mediate a more sensitive reaction to environmental changes [Citation12,Citation13]. Therefore, in case of environmental changes too fast for an appropriate genetic response, epigenetic mechanisms can have a crucial role in modulating genetically driven phenotypic plasticity and by rapidly generating phenotypic variants that match new environmental conditions and that could be subsequently used in the process of adaptation or artificial selection.
A large spectrum of phenotypic traits associated with epigenetic patterns in domestic animals has been studied in several highly valuable species. For example, DNA methylation has been related to disease resistance [Citation6,Citation14], growth [Citation15], response to diet [Citation16] or rearing conditions [Citation17,Citation18]. These studies raise the hypothesis that this epigenetic mechanism has a rather ubiquitous role in modulating environmentally induced phenotypic traits in farm animals and, in case of commercially valuable traits, could provide a substantial help when establishing selective breeding schemes.
There has been an increasing interest in epigenetics research applied to aquaculture [Citation19], since it has become the fastest growing food production sector in the world [Citation20]. While some fish species such as carp have a known long history of domestication, many new aquatic species are currently at early stages of domestication [Citation21]. Thus, a deeper knowledge of epigenetic mechanisms related to the process of domestication can have a substantial impact on a further development of aquaculture field. In our study, we focused on one of the most important species in aquaculture, Nile tilapia (Oreochromis niloticus), to investigate changes that occur in the fast muscle methylome within a single generation domestication. We expected to detect epigenetic differences underlying fast adaptation to aquaculture conditions and to get an insight into methylation patterns contributing to the onset of ‘domestic phenotype,’ i.e., potential epigenetic marks of fish domestication.
Results
Experimental groups and library characterization
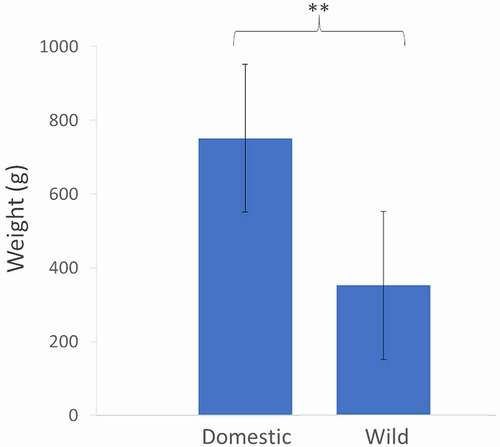
The number of eggs obtained from each female was 192.8 ± 66.1 (mean ± S.D., n = 6) and the overall mortality until the late larval stage was 9.0% ± 2.8% (mean ± S.D., n = 6) (Table S1). After a single generation of domestication, Nile tilapia females from the domestic group were 2-fold heavier than their wild counterparts, despite being of similar age and reproductive status (). For the sake of simplicity, the term ‘domestic’ is henceforth used to designate the group of fish undergoing domestication.
Figure 1. Average weight (g) of 12 females from the wild and domestic groups. Domestic females were statistically heavier than small females (paired t-test, p-value <0.01).
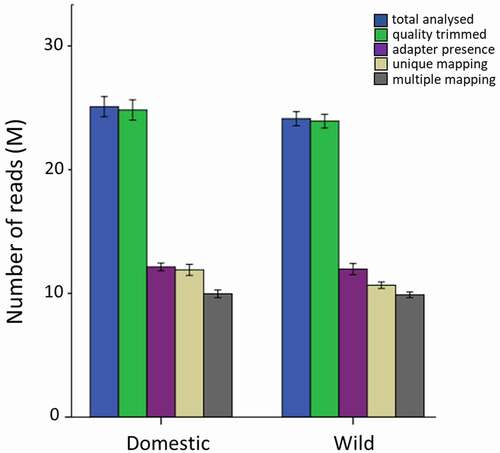
A mean of 25 M raw reads was obtained per library, of which 11 M were uniquely mapped (i.e., 44% mapping efficiency) and an additional 10 M reads could be multiple mapped (, Table S2). Neither individual samples nor compared groups differed with respect to their raw, trimmed or mapped reads. The mean CpG coverage of reads mapping uniquely to the fraction of the genome that was successfully aligned with reads was around 200-fold (Figure S1). The absence of a peak at the right side of the histogram indicated that the data did not contain PCR duplicates.
Figure 2. Number of total raw, quality-trimmed, adapter trimmed, uniquely mapped and multiply mapped reads in the domesticated- and wild female group.
Characterization of the fast muscle methylome in Nile tilapia
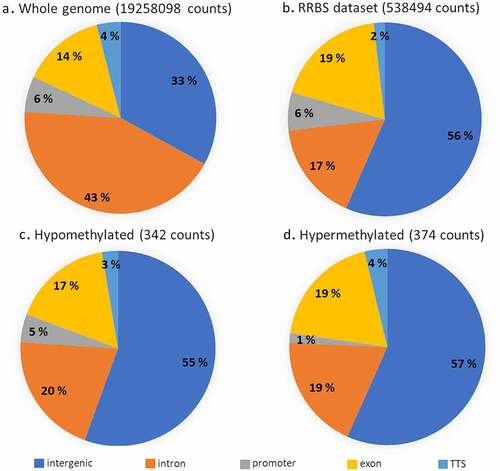
The global cytosine count was 177 million with a fold coverage of 2.2–2.4 (). Average methylation corresponded to 60% and did not differ within or between the two analysed groups (Table S3). The mean methylation levels per sample and per chromosome were homogenous, with the exception of chromosome LG3 showing 73% methylation (Table S4). However, the genomic context of the Reduced Representation Bisulphite Sequencing (RRBS) dataset differed from that in the whole Nile tilapia genome (). Indeed, intergenic-annotated cytosines were enriched almost two-fold in the reduced representation genome compared to whole genome (Pearson’s Chi-squared, p-value < 2.2e-16). A two-fold decrease in CpG sites within intronic regions was found in the RRBS dataset when compared to the whole genome () (Pearson’s Chi-squared, p-value < 2.2e-16). Different enrichments between reduced- and whole genome cytosines were also observed in exons and TTS (Pearson’s Chi-squared, p-value < 2.2e-16).
Table 1. Global cytosine methylation level in CpG, CHG and CHH contexts, number of cytosines analysed and cytosine coverage with respect to the whole Nile tilapia genome.
Figure 3. Genomic context of CpG sites found in the whole genome, our RRBS dataset and associated with hypo- and hypermethylated sites.
Unequal distribution functional relevance of differentially methylated (DM) CpG sites between wild fish and their progeny undergoing domestication
A total of 538,494 CpG positions that were covered at least 10 times and present in every sample at least once were used for analysis of differential methylation between the groups of wild (reference) and domestic group. Interestingly, the genomic context was similar between the sets of hyper- and hypomethylated CpG sites (), except for the promoter region, associated in the domestic group with 5 times more hypomethylated sites than the hypermethylated ones (Pearson’s Chi-squared, p-value = 0.002) (). Overall, the genomic context of DM CpG sites was similar to the one of all CpGs analysed in this study. In addition, a total of 6 differentially methylated regions (DMR) of 1000 bp (4 hypermethylated and 2 hypomethylated) have been found (Table S5), containing a minimum of 10 CpG each and each covered at least 3 times.
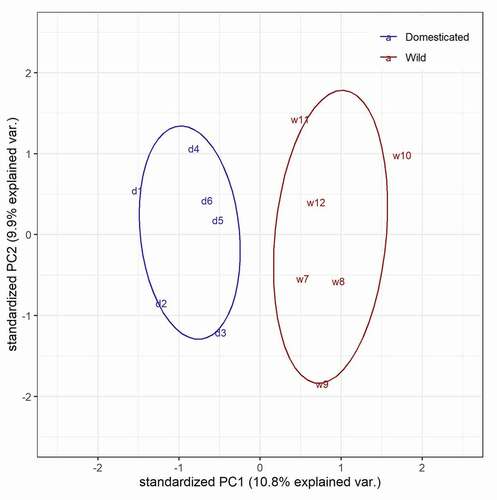
Out of a total number of 715 differentially methylated CpG sites (FDR < 0.01) (, Table S6), the number of hypo- and hypermethylated CpG sites was similar and represented on average 0.7‰ of all CpG sites analysed. The chromosomal distribution differed between hypo- and hypermethylated CpG sites (Table S7). For example, chromosome LG3 was represented by a similar number of hypo- and hypermethylated CpG sites, but chromosome LG18 contained 3 times more hypomethylated sites than hypermethylated ones. Within-chromosomes, the distribution of DM CpG sites did not show any specific pattern, except for the longest chromosome LG23, which contained all the DM CpG sites within the first 50% of its sequence (). The lack of DMCpG sites in the second half of chromosome LG23 can be explained by a low RRBS coverage of this region (only 1664 CpGs analysed were located further than 44 M nucleotides) compared to the first half of LG23 (28,310 CpGs located within the first 44 M nucleotides of LG23). The Principal Component Analysis on all cytosine levels available in the RRBS dataset showed a clear separation between wild and domestic groups (, Table S8).
Figure 4. Volcano plot representing q-value and methylation difference in all CpG sites compared between wild and domesticated fish. Red dots represent differentially methylated CpG sites (methylation difference >25%, FDR <0.01). Several genes associated with most extreme CpG methylation differences are indicated. In addition, blue dots highlight all the 15 DM CpG sites associated specifically with stub-1 gene.
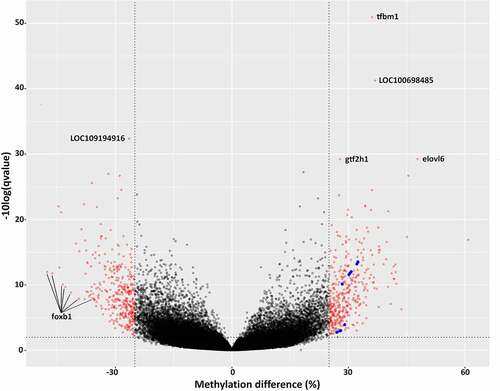
Figure 5. Chromosomal distribution of differentially methylated CpG sites between wild females and their progeny undergoing domestication (716 positions, FDR < 0.01, methylation difference >25%). Wild females are set as reference. Histograms pointing inwards (dark green) and outwards (dark red) represent hypomethylated and hypermethylated sites in domestic group, respectively.
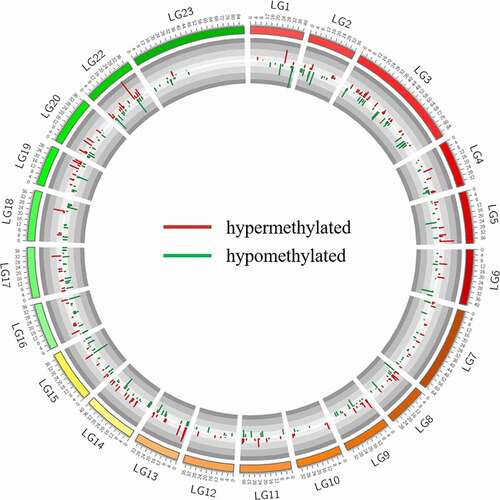
Figure 6. Principal component analysis of 12 fish separated in the domestic (d, blue) and wild group (w, red).
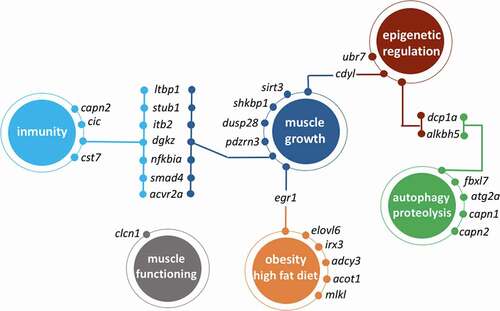
The functional annotation only showed a significant enrichment of functions related mostly to rRNA metabolism and modifications (Table S9). These limited results are due, at least in part, to the relatively small number of genes associated with DM CpGs. Among the 489 manually annotated genes associated with a total of 715 differentially methylated CpG sites (Table S6), we focused on genes linked to metabolism, muscle growth, immunity and epigenetics, since they are directly relevant to the domestication phenotype ().
Table 2. List of functionally relevant selected genes associated with DM CpG sites in Nile tilapia females. Six domesticated individuals were compared to six wild individuals. Only the results with q-value < 0.01 and methylation difference >25% are retained. Positive percentage values indicate hypermethylation in domesticated individuals when compared to wild individuals, whereas negative values indicate hypomethylation in the same comparison. Frequency Ratio corresponds to a number of unique gene-associated DM CpG sites divided by a total number of CpG sites available for the analysis and associated with the same gene.
DM CpGs associated with muscle-growth related genes
Two hypomethylated exonic CpGs were associated with dusp28, which encodes a protein from a family of dual-specificity phosphatase (DUSP) that can activate the p38 mitogen-activated protein kinase (MAPK) signalling pathway [Citation22] and therefore regulate muscle stem cells [Citation23]. Three hypomethylated DM CpG sites were linked to acvr2a, encoding Activin receptor type 2a that is linked to myostatin activity in muscle growth [Citation24]. Another hypomethylated CpG site was associated with sirt3, a gene involved in metabolic flexibility of skeletal muscle (i.e., ability to switch between glucose and lipid oxidation) by impacting downstream signalling through peroxisome proliferator–activated receptor (PPAR)γ coactivator-1α [Citation25]. The activity of sirt3 is regulated by nutrient signals and contractile activity of the muscle [Citation26] and has been linked to the fast growing fish of gilthead sea bream species [Citation27]. Hypermethylated CpG sites in the domestic group were present in pdzrn3, encoding a ubiquitin ligase regulating myoblast differentiation [Citation28] and in shkbp1, encoding a SHK3BP1 binding protein involved in development of skeletal muscle fibres [Citation29]. Moreover, hypermethylation was also found in an exonic CpG site related to egr1, an early growth response gene. This transcription factor regulates myoG gene expression and promotes differentiation of muscle satellite cells [Citation30], but is also affected by high intensity exercise [Citation31]. Egr1 also inhibits another transcription factor, MEF2, important in muscle differentiation [Citation32]. Importantly, gene body methylation of egr1 was associated with intramuscular fat in pigs at different developmental levels [Citation33], its expression is up-regulated by myoD [Citation34] and downregulated by Transforming growth factor-β1 TGF-β1 [Citation35]. Two hypomethylated intronic CpG sites were also associated with clcn1, coding for Chloride channel protein 1, which, although not involved in muscle growth specifically, plays a role in muscle physiology through membrane repolarization after muscular contraction [Citation36].
DM CpG associated with both muscle growth and immunity-related genes
Several DM CpG associated genes had relevant functions not only in muscle growth but also in the immune response (). For example, stub1 was associated with the highest number of DM CpGs, all hypermethylated in domestic fish and situated in exons. There was a clear difference in methylation levels between wild and domestic fish in individual CpG sites associated with stub1 (Figure S2), which are present in two close clusters in a volcano plot (). Stub1-associated DM CpG were also detected by DMR analysis (Table S5). This gene codes for E3 Ubiquitin ligase CHIP (carboxyl terminus of Hsc70-interacting protein), which is involved in ubiquitin-mediated protein degradation and regulation of TGF-β1 [Citation37]. TGF-β kinase has a well-known function in inflammation and wound healing [Citation38], and is important for muscle regeneration after damage or exercise [Citation39]. TGF-β also induces skeletal muscle atrophy [Citation40] by delaying myoblast differentiation while increasing cellular proliferation [Citation41]. Another gene associated with hypermethylated CpG was ltbp1, encoding the Latent transforming growth factor beta binding protein 1, which is critical for the assembly of TGF- β1 [Citation42], a cytokine important in muscle regeneration [Citation43]. Interestingly, a significant correlation between gene expression of ltbp1 and the methylation level of its CpG sites was found in sheep [Citation44]. Another gene known as a key regulator of TGF-β1 signalling pathway and associated hypermethylated CpG sites was smad4. This gene is also affected by diet in grass carp and is associated with muscle hardness through regulation of type 1 collagen expression [Citation45]. Smad4 is also involved in skeletal muscle regeneration by promoting the expansion of satellite cell derived progenitors [Citation46]. Besides the muscle growth-related function, this transcription factor regulates the activity of PPARγ via TGF-β1 [Citation47], but also plays a direct role in the immune response [Citation48]. Nine hypermethylated exonic CpG sites were related to dgkz encoding diacylglycerol kinase zeta (DGKZ), which is involved in cardiomyocyte hypertrophy through the PPAR (peroxisome proliferator-activated receptor)-DGK pathway [Citation49]. This gene was associated with feed conversion ratio in broilers by GWAS study [Citation50] and its methylation was suggested as a biomarker of prostate cancer [Citation51]. Importantly, roles of dgkz in myogenic differentiation [Citation52], in muscle hypertrophy and myoblast fusion through mTOR-dependent signalling [Citation53,Citation54] and in T cell activation [Citation55] were experimentally validated, and multiple studies of its function in modulating lipid metabolism has been reviewed [Citation56]. Two hypermethylated exonic CpGs were associated with the gene itb2 encoding Integrin beta-2, with known functions in skeletal muscle hypertrophy [Citation57] and immune response [Citation58]. Another DM CpG-related gene was nfkbia, which codes for an inhibitor of the transcription factor NF-kappa-B, the latter being important in muscle atrophy [Citation59] and inflammation [Citation60].
Immunity-related genes associated with DM CpG
Thirteen hypo- and hypermethylated exonic CpG sites in domestic group were associated with cst7 encoding Cystatin F, a cysteine protease inhibitor involved in eosinophil survival [Citation61] and in immune response [Citation62]. Another gene associated with a hypermethylated exonic CpG was the gene cic encoding Capicua, a transcriptional repressor that maintains peripheral immune tolerance via T-cell homoeostasis [Citation63] and whose methylation is inversely correlated with its transcription level in pigs [Citation64].
Genes associated with DM CpG and related to epigenetic mechanisms
Several DM CpG sites were associated with genes that have important roles in histone binding, RNA degradation and RNA methylation. For example, ubr7 encoding E3 Ubiquitin ligase UBR7, is associated with histone marked segments of the genome [Citation65], interacts with histone H3 [Citation66] and has been suggested to function as histone E3 monoubiquitin ligase [Citation67]. Two hypomethylated exonic CpGs were associated with the gene cdyl encoding the chromodomain protein CDYL, which can bind to H3K27 histone and interact with Polycomb Repressive Complex 2 [Citation68], a histone methyltransferase that is also involved in regulation of muscle cell differentiation [Citation69]. Another DM CpG-related gene was dcp1a coding for an mRNA decapping enzyme involved in RNA degradation and translation regulation [Citation70]. Interestingly, the closely related gene dcp2 is also involved in epigenetic regulation of autophagy [Citation71]. Finally, a gene alkbh5 associated with hypermethylated intronic CpG encodes for a m6A RNA demethylase with epigenetic activity linked to autophagy [Citation72] and to the transcription factor EB, involved in metabolic flexibility [Citation73]. Moreover, alkbh5 expression can be affected by diet supplementation [Citation74]. These last two genes were not only related to epigenetic processes but also to autophagy.
Genes associated with DM CpG and related to autophagy and proteolysis
Hypomethylated CpG sites in domestic fish were related to fbx7, atg2a and capn2. Fbxl7 encodes for F-box and leucine rich repeat protein 7 that modulates mitochondrial functions such as proteasomal degradation by negatively regulating an anti-apoptotic protein survivin [Citation75]. Atg2a, autophagy related gene 2, is involved in glucose starvation related autophagy with a suggested function in controlling the extent of autophagosome membrane formation [Citation76,Citation77]. Capn2, encoding Calpain 2, is involved in autophagosome formation and protein degradation [Citation78] and was associated with meat quality in birds [Citation79]. Interestingly, the expression of this protease is positively regulated by testosterone in fish [Citation80]. One hypomethylated CpG was related to the gene capn1 encoding Calpain 1, which plays a crucial role in autophagy via autophagy-related genes [Citation81] and is associated with nutritional state in halibut [Citation82] and gilthead sea bream [Citation83]. The activity of this protease was also induced by a moderate and sustained exercise in the latter [Citation84].
Genes associated with DM CpG and related to obesity and high fat diet
Several hypermethylated CpG sites were associated with genes related to obesity and fat composition, such as adcy3, mlkl, irx3, elovl6 and acot1. The expression of adcy3 increases in skeletal muscle in response to fat overfeeding [Citation85] and its related protein is associated with diet-induced obesity [Citation86]. Mlkl codes for mixed lineage kinase domain-like protein, also involved in obesity-induced metabolic complications [Citation87]. Three exonic DM CpG sites were associated with irx3, encoding for Iroquois homeobox 3, strongly related to obese phenotype and obesity-associated gene fto [Citation88]. The gene elovl6 encodes a Fatty acid elongase and is associated with a QTL effect on fatty composition [Citation89] and obesity-induced insulin resistance in vertebrates [Citation90]. Acot1 codes for Acyl-coenzyme A thioesterase 1, which is upregulated in response to high fat overload in animals fed with high-fat diet [Citation91].
Discussion
In this study, we report the reduced representation profiling of methylated cytosines in fast muscle between wild females and their hatchery-born and reared female offspring, i.e., subjected to one generation of domestication. The overall mortality values were relatively low and within the range expected for Nile tilapia (Table S1), indicating that the effect of artificial selection was negligible. Therefore, our study gives insight into epigenetic patterns influenced by the early stages of domestication. Little is known on the genetic differences between the fish, however, the experimental design and analysis (same sampling site for the wild fish, parent-offspring relation of each pair of fish used as covariate) allowed to rule out the influence of genetic background in the study, if any. Moreover, a study performed in coho salmon has shown that hatchery conditions have stronger impact on the epigenome than fish origin and that no significant genetic differentiation arose just after one generation of domestication process [Citation92]. A recent study in sea bass under early stages of domestication has shown that domestication-related methylation patterns not only could explain the onset of domestic traits, such as lower jaw malformations, but also that differentially methylated sites correspond to genetic polymorphisms observed after long periods of selective breeding and coincide with genes under positive selection during domestication process [Citation93].
To the best of our knowledge, this is the first study providing a cytosine resolution methylation profiling in Nile tilapia between generations in the context of early domestication. Previous studies have shown cytosine methylation differences in fast muscle associated with Nile tilapia growth and sex in a single generation of hatchery conditions [Citation94], regional cytosine methylation differences in fast muscle associated with sex in a Nile tilapia hybrid [Citation95] or associated with sex in gonads [Citation96]. A limitation of these studies and indeed the present paper is the use of complex tissues composed by multiple cell types. Differences in tissue composition can result in the identification of additional differentially methylated genes between groups, namely immune-related genes due to the presence of blood or neuronal markers due to variations in innervation. Recent advances in single-nucleus methylome sequencing will make it possible to address this issue in the near future.
Non-tilapiine methylome profiling within the context of domestication has been performed in a few other species, namely in fast muscle of coho salmon [Citation92] and in the steelhead fin tissue [Citation97], sperm and red blood cells [Citation18]. We compared our results with the closest tissue- and context-specific studies [Citation92,Citation94] and found a few common genes associated with differentially methylated cytosines or regions. Genes from a family encoding dual specificity phosphatases (DUSP) and protocadherins (CADH) were associated with cytosine hypomethylation of domesticated groups in both our and the previous study [Citation92], and those encoding for serine/threonine phosphatase kinases (SGK) and syntaxins (STX) were associated with cytosine hypomethylation of domesticated fish in our study as opposed to hypermethylation in the above mentioned study. However, it is difficult to interpret since none of the gene families concerned strictly the same gene or species, and the genes were associated with large differentially methylated regions in coho salmon, in contrast to our single cytosine-specific association. More common genes were found when compared to the single cytosine methylome analysis between fast- and slow growing Nile tilapia [Citation94]. For example, within the family of autophagy-related genes, where atg14 was recently shown as associated with the highest number of growth- and sex-related DM CpG sites. It strengthens the idea that autophagy might be an epigenetically regulated and both growth-specific and sex-dependent mechanism in Nile tilapia.
Although the average methylation level of the Nile tilapia genome is similar to that reported in previous studies, the proportion and distribution of DM CpG sites throughout the genome suggests a non-random pattern of methylation associated with domestication. The systematic analysis of functional ontology of genes associated with these patterns has highlighted the GO terms related to ribosomal RNA modifications, and specifically rRNA methylation. Such rRNA modification can have various consequences on the phenotype, since ribosomes are a general and essential element of protein synthesis. DNA methylation patterns involved in rRNA methylation make a link between pre- and post-transcriptional epigenetic marks and add an extra layer of complexity of epigenetic mechanisms involved in domestication. Moreover, although more processes were not detected as significantly enriched by systematic analyses, we found many DM CpG-associated genes related to growth, immunity, autophagy, diet or epigenetic mechanisms () that can help explaining the changes occurring during the early stages of domestication. One of the main differences between any wild and captive population is the access to food, restrained in the first and often ad libitum in the second group. Farmed fish receive more food at regular times and without wasting energy for foraging, and the food is nutritionally optimized to yield high growth rates. Thus, such important dietary changes can be reflected in gene regulation related to diet components, which in our case were associated particularly with high fat nutritional profiles. The growth-promoting farming conditions were reflected by several DM CpG associated genes, many of which, intriguingly, were also implicated in immunity-related processes. This growth-immunity relationship is in line with the life-history theory predicting trade-offs between physiologically costly functions and it has been shown that, in case of growth-selected fish [Citation98] and other farmed animals [Citation99], energy investment in growth is associated with decreased energy allocation in immunity-related functions. It is difficult to predict the consequences of methylation of these genes in our study, however, it is important to highlight that even when energy intake is not restrained by resource availability, the pleiotropic nature of the epigenetically affected genes can lead to important changes in both traits simultaneously. We also do not rule out the hypothesis that in highly controlled farm conditions, the relaxed pathogen pressures lead first to changes in immune profiles of fish, which in consequence will also affect the growth rate. Several selected genes were also associated with proteolysis and autophagy. Protein lysis might be linked to catabolism, i.e., muscle degradation in order to extract energy when food is scarce. In a context of unrestrained food availability together with regular and frequent feeding periods in farm conditions, methylation of these genes could act as a protective mechanism against catabolism, thus promoting once again muscle growth. Moreover, autophagy related genes have been already identified in this species as growth and sex-related in the context of early domestication [Citation94]. Finally, several differentially methylated genes were involved in other epigenetic mechanisms, i.e., mRNA degradation, histone modifications or RNA methylation, suggesting that early domestication involves changes at different levels of gene-phenotype axis.
Figure 7. Schematic representation of genes that are associated with differentially methylated CpGs (domestic VS wild females) and whose functions are related muscle growth, autophagy, immunity, diet or epigenetic regulation processes.
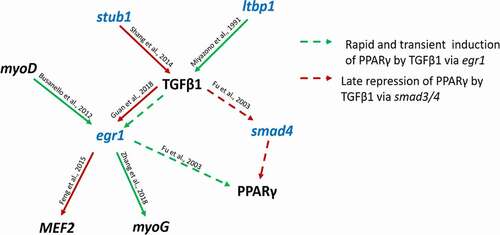
Several DM CpG associated genes were related to the Transforming growth factor beta 1 (TGF-β1) signalling pathway and regulation (). For example, ltbp1 and stub1 are known to promote or inhibit TGF-β1, respectively, while egr1 and smad4 can be repressed or indirectly involved in repression and induction of other genes via TGF-β1. Downstream regulated genes such as mef2, myoG and ppargy, are known for their key role in myogenesis. Even though this kinase was not detected as associated with differentially methylated cytosines, it seems to be affected by upstream regulators and to involve downstream components that are also associated with DM CpGs. We suggest that in the context of early fish domestication, TGF-β1 signalling pathway is one of the candidates that should receive further attention in functional studies.
Figure 8. Schematic representation of a gene network associated with differentially methylated CpGs (in blue) and whose functions are related to transforming growth factor beta 1 or to other genes relevant to the process of muscle growth (in black). Red/green and continuous/dashed arrows indicate positive/negative and direct/indirect regulation, respectively.
Taken together, our study shows the importance of the epigenetic changes that occur during the early stages of domestication. The differentially methylated genes observed between the wild and first-generation farmed fish could explain, to some extent, the onset of a ‘domestic phenotype’ in aquaculture. Myogenic and immune functions were common to many pleiotropic genes, while other genes are known to be affected by diet and fat in particular. Differential methylation in autophagy- and proteolysis-related genes can potentially decrease catabolism in an environment of regular feeding and unlimited food availability. We suggest that the modulation of phenotypic traits related to growth and immunity might occur already within the first generation of farmed fish, i.e., very early in domestication, and in the absence of artificial selection, this epigenetic mechanism plays a crucial role in early domestication.
Materials and methods
Ethics statement
This study was approved by the Nord University (Bodø, Norway) ethics board and all procedures involving animals were performed according to the instructions of the Norwegian Animal Research Authority (FOTS ID 1042).
Collection of wild specimens, fish husbandry and sampling
Fertilized eggs were collected from six wild mouthbrooding females in the river Nile, Egypt (location GPS: 25°39'56”N, 32°37'07”). They were then euthanized as detailed below and measured. Their fast muscle was sampled, while their fertilized eggs were transported to our research station at Nord University (Norway), where they hatched and were reared in separate tanks in a recirculating aquaculture system (pH = 7.6, temperature = 28°C, 11 h dark/13 h light cycle, density = 27 fish/ m3, food: Amber Neptun Skretting® feed 0.15–0.8 mm) during 5 months. Then one female offspring corresponding to each wild female was euthanized and its fast muscle sampled.
The wild and domestic fish were euthanized and sampled following the same procedure. In short, they were euthanized with clove oil (Sigma Aldrich, USA) using a 1:10 mix of 15 mL clove oil with 95% ethanol diluted in 10 L of water, the fast muscle was carefully excised just prior to snap-freezing and then stored at −80°C until DNA extraction. DNA was extracted with the DNEasy Blood & Tissue kit (Qiagen, Germany) according to the manufacturer’s recommendations. DNA quality check was performed by NanoDrop and Tape Station (Agilent, USA) Assays, while DNA quantification was performed with Qubit (Thermofisher Scientific, USA).
Preparation of reduced representation bisulphite sequencing (RRBS) libraries
Library preparation for RRBS was performed with the NuGen ovation RRBS methyl-seq system 1–16 (Tecan Genomics, Inc, Redwood City, USA) following the producer’s instructions. Genomic DNA was digested with MspI at 37°C for 1 hr, followed by adapter ligation and final repair. For the bisulphite conversion of adapter ligated libraries and subsequent cleaning, the EpiTect fast bisulphite conversion kit (Qiagen, Hilden, Germany) was used following the manufacturer’s protocol. The resulting bisulphite converted libraries were amplified for 12 PCR cycles and later purified using reagents and recommendations of the NuGen RRBS kit. Quality and quantity of the RRBS libraries were assessed using the TapeStation. Single-end 75 bp sequencing was performed for a pool of 12 libraries on an Illumina NextSeq instrument (San Diego, USA) with 4% PhiX DNA (Illumina) as an internal control, following the instructions for RRBS sequencing from NuGen. Sequencing data are publicly available at NCBI under the Bioproject PRJNA661533 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA661533).
Bioinformatics and statistical analyses
Raw reads were adapter-trimmed with the Nugen-provided script and Cutadapt (Babraham Bioinformatics) [Citation100], and aligned to the latest reference genome O_niloticus_UMD_NMBU (https://www.ncbi.nlm.nih.gov/assembly/GCF_001858045.2/), using the Bismark v0.19.1 pipeline (Babraham Bioinformatics) [Citation101]. The two strands of oreNil3 were modified in silico (conversion of all C’s to T’s) and indexed according to Bowtie2 [Citation102] requirements. Reads were mapped to the original and in silico modified genome using Bismark (with parameters: -q – p – N1 – bowtie2). The resulting CpG coverage files were used as input in MethylKit package [Citation103] in R to calculate the coverage, methylation levels (i.e., number of methylated counts over a total count at each site) of each sample and differences in methylation among samples. The statistical method used to detect differentially methylated cytosines (DMC) and regions (DMR) between groups was logistic regression (wild group as control, groups as dependent variable, methylation as independent variable), and the significance threshold were FDR < 0.01 and minimum difference in methylation level of 25%. For DMC, only cytosines covered at least 10 times were considered, and for DMR tiling windows were set at 1000bp, initial per base coverage of 3 and a minimum number of 10 cytosines per region. Circos [Citation104] was used to represent and locate all differentially methylated CpG sites. The Oreochromis niloticus annotation release 104 was used as input in Homer [Citation105] to locate CpG sites within their genomic context (introns, exons, promoters, intergenic regions) and to assign the gene reference in RefSeq format with the closest TSS (transcription start site) to each differentially methylated cytosine. Gene references were then converted to gene symbols and full gene names using the NCBI browser and manually associated with function using the GeneCards browser [Citation106] and the literature. In addition, Functional Enrichment Analysis was performed with DAVID [Citation107] and the top 10 pathways were reported, using a custom background gene list corresponding to all the CpG sites analysed in the RRBS dataset, and specific to each comparison.
Acknowledgments
We are thankful to Hilde Ribe, Øivind Torslett, Steinar Johnsen and Kaspar Klaudiussen (Nord University, Norway) for their assistance in fish husbandry and commitment to the welfare of the fish. This study has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 683210) and from the Research Council of Norway under the Toppforsk programme(grant agreement no 250548/F20).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Price EO. Domestication defined Price, EO. In: Animal domestication and behavior. New York: CABI Publishing; 2002. p. 10–12. ISBN: 0-85199-597-7.
- de Mestral LG, Herbinger CM. Reduction in antipredator response detected between first and second generations of endangered juvenile Atlantic salmon Salmo salar in a captive breeding and rearing programme. J Fish Biol. 2013;83(5):1268–1286.
- Trut L, Oskina I, Kharlamova A. Animal evolution during domestication: the domesticated fox as a model. BioEssays. 2009;31(3):349–360.
- Agnvall B, Bélteky J, Katajamaa R, et al. Is evolution of domestication driven by tameness? A selective review with focus on chickens. Appl Anim Behav Sci. 2018;205:227–233.
- Wilkins AS, Wrangham RW, Fitch WT. The “domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics. Genetics. 2014;197(3):795–808.
- Chen X, Wang J, Qian L, et al. Domestication drive the changes of immune and digestive system of Eurasian perch (Perca fluviatilis). PLoS One. 2017;12(3):e0172903–e0172903.
- Rauw WM, Kanis E, Noordhuizen-Stassen EN, et al. Undesirable side effects of selection for high production efficiency in farm animals: a review. Livest Prod Sci. 1998;56(1):15–33.
- Selechnik D, Richardson, MF, Shine, R et al, et al. Bottleneck revisited: increased adaptive variation despite reduced overall genetic diversity in a rapidly adapting invader. Front Genet. 2019;10:1221.
- Christie MR, Marine ML, Fox SE, et al. A single generation of domestication heritably alters the expression of hundreds of genes. Nat Commun. 2016;7:10676.
- Christie MR, Marine ML, French RA, et al. Genetic adaptation to captivity can occur in a single generation. Proc Natl Acad Sci USA. 2012;109(1):238–242.
- Schmitz RJ, Schultz MD, Lewsey MG, et al. Transgenerational epigenetic instability is a source of novel methylation variants. Science. 2011;334(6054):369–373.
- Hu J, Barrett RDH. Epigenetics in natural animal populations. J Evol Biol. 2017;30(9):1612–1632.
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97.
- Tian F, Zhan F, VanderKraats ND, et al. DNMT gene expression and methylome in Marek’s disease resistant and susceptible chickens prior to and following infection by MDV. Epigenetics. 2013;8(4):431–444.
- Hu Y, Xu H, Li Z, et al. Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers. PLoS One. 2013;8(2):e56411.
- Adam A-C, Lie KK, Whatmore P, et al. Profiling DNA methylation patterns of zebrafish liver associated with parental high dietary arachidonic acid. PLoS One. 2019;14(8):e0220934.
- Pértille F, Brantsæter M, Nordgreen J, et al. DNA methylation profiles in red blood cells of adult hens correlate with their rearing conditions. J Exp Biol. 2017;220(19):3579.
- Gavery MR, Nichols KM, Goetz GW, et al. Characterization of genetic and epigenetic variation in sperm and red blood cells from adult hatchery and natural-origin steelhead. Oncorhynchus mykiss G3 (Bethesda). 2018;8(11):3723–3736.
- Gavery MR, Roberts SB. Epigenetic considerations in aquaculture. PeerJ. 2017;12(5):e4147.
- Diana JS. Aquaculture production and biodiversity conservation. BioScience. 2009;59(1):27–38.
- Teletchea F, Fontaine P. Levels of domestication in fish: implications for the sustainable future of aquaculture. Fish Fish. 2014;15(2):181–195.
- Wang D, Han S, Peng R, et al. DUSP28 contributes to human hepatocellular carcinoma via regulation of the p38 MAPK signaling. Int J Oncol. 2014;45(6):2596–2604.
- Segalés J, Perdiguero E, Muñoz-Cánoves P. Regulation of muscle stem cell functions: a focus on the p38 MAPK signaling pathway. Front Cell Dev Biol. 2016;4(91). DOI:10.3389/fcell.2016.00091
- Solomon AM, Bouloux PMG. Modifying muscle mass - the endocrine perspective. J Endocrinol. 2006;191(2):349–360.
- Jing E, O’Neill BT, Rardin MJ, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62(10):3404–3417.
- Palacios OM, Carmona JJ, Michan S, et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging (Albany Ny). 2009;1(9):771–783.
- Simo-Mirabet P, Perera E, Calduch-Giner JA, et al. Co-expression analysis of sirtuins and related metabolic biomarkers in juveniles of gilthead sea bream (Sparus aurata) with differences in growth performance. Front Physiol. 2018;9:608.
- Honda T, Inui M. PDZRN3 regulates differentiation of myoblasts into myotubes through transcriptional and posttranslational control of Id2. J Cell Physiol. 2019;234(3):2963–2972.
- Guiraud A, Couturier N, Buchman V, et al. Sh3kbp1 involvement during skeletal muscle fibers formation: a new candidate for centronuclear myopathies. Neuromuscul Disord. 2017;27:S248.
- Zhang W, Tong H, Zhang Z, et al. Transcription factor EGR1 promotes differentiation of bovine skeletal muscle satellite cells by regulating MyoG gene expression. J Cell Physiol. 2018;233(1):350–362.
- Edgett BA, Foster WS, Hankinson PB, et al. Dissociation of increases in PGC-1α and its regulators from exercise intensity and muscle activation following acute exercise. PLoS One. 2013;8(8):e71623.
- Feng Y, Desjardins CA, Cooper O, et al. EGR1 functions as a potent repressor of MEF2 transcriptional activity. PLoS One. 2015;10(5):e0131619.
- Wang Y, Ma C, Sun Y, et al. Dynamic transcriptome and DNA methylome analyses on longissimus dorsi to identify genes underlying intramuscular fat content in pigs. BMC Genomics. 2017;18(1). DOI:10.1186/s12864-017-4201-9
- Busanello A, Battistelli C, Carbone M, et al. MyoD regulates p57kip2 expression by interacting with a distant cis-element and modifying a higher order chromatin structure. Nucleic Acids Res. 2012;40(17):8266–8275.
- Guan M, Li W, Xu L, et al. Metformin improves epithelial-to-mesenchymal transition induced by TGF-β1 in renal tubular epithelial NRK-52E cells via inhibiting Egr-1. J Diabetes Res. 2018;2018:1–8.
- Aromataris EC, Rychkov GY. ClC-1 chloride channel: matching its properties to a role in skeletal muscle. Clin Exp Pharmacol Physiol. 2006;33(11):1118–1123.
- Shang Y, Xu X, Duan X, et al. Hsp70 and Hsp90 oppositely regulate TGF-β signaling through CHIP/Stub1. Biochem Biophys Res Commun. 2014;446(1):387–392.
- Taylor AW. Review of the activation of TGF-beta in immunity. J Leukoc Biol. 2009;85(1):29–33.
- Kim J, Lee J. Role of transforming growth factor-β in muscle damage and regeneration: focused on eccentric muscle contraction. J Exerc Rehabil. 2017;13(6):621–626.
- Mendias CL, Gumucio JP, Davis ME, et al. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve. 2012;45(1):55–59.
- Schabort EJ, van der Merwe M, Loos B, et al. TGF-beta’s delay skeletal muscle progenitor cell differentiation in an isoform-independent manner. Exp Cell Res. 2009;315(3):373–384.
- Miyazono K, Olofsson A, Colosetti P, et al. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. Embo J. 1991;10(5):1091–1101.
- Delaney K, Kasprzycka P, Ciemerych MA, et al. The role of TGF-β1 during skeletal muscle regeneration. Cell Biol Int. 2017;41(7):706–715.
- Cao J, Wei C, Liu D, et al. DNA methylation landscape of body size variation in sheep. Sci Rep. 2015;5:13950.
- Yu EM, Ma -L-L, Ji H, et al. Smad4-dependent regulation of type I collagen expression in the muscle of grass carp fed with faba bean. Gene. 2019;685:32–41.
- Paris ND, Soroka A, Klose A, et al. Smad4 restricts differentiation to promote expansion of satellite cell derived progenitors during skeletal muscle regeneration. eLife. 2016;5:e19484.
- Fu M, Zhang J, Lin Y, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor γ (PPARγ) gene expression by transformino growth factor β in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (AP1) and Smads. Biochem J. 2003;370(3):1019–1025.
- Gu AD, Zhang S, Wang Y, et al. A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor β receptor signaling. Immunity. 2015;42(1):68–79.
- Huang Y, Zhang H, Shao Z, et al. Suppression of endothelin-1-induced cardiac myocyte hypertrophy by PPAR agonists: role of diacylglycerol kinase zeta. Cardiovasc Res. 2011;90(2):267–275.
- Shah TM, Patel NV, Patel AB, et al. A genome-wide approach to screen for genetic variants in broilers (Gallus gallus) with divergent feed conversion ratio. Mol Genet Genomics. 2016;291(4):1715–1725.
- Paziewska A, Dabrowska M, Goryca K, et al. DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy. Br J Cancer. 2014;111(4):781–789.
- Evangelisti C, Tazzari PL, Riccio M, et al. Nuclear diacylglycerol kinase-ζ is a negative regulator of cell cycle progression in C2C12 mouse myoblasts. FASEB J. 2007;21(12):3297–3307.
- You JS, Lincoln HC, Kim C-R, et al. The role of diacylglycerol kinase ζ and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J Biol Chem. 2014;289(3):1551–1563.
- Miao Y, Yang J, Xu Z, et al. RNA sequencing identifies upregulated kyphoscoliosis peptidase and phosphatidic acid signaling pathways in muscle hypertrophy generated by transgenic expression of myostatin propeptide. Int J Mol Sci. 2015;16(4):7976–7994.
- Zhong XP, Hainey EA, Olenchock BA, et al. Enhanced T cell responses due to diacylglycerol kinase ζ deficiency. Nat Immunol. 2003;4(9):882–890.
- Rincón E, Gharbi SI, Santos-Mendoza T, et al. Diacylglycerol kinase ζ: at the crossroads of lipid signaling and protein complex organization. Prog Lipid Res. 2012;51(1):1–10.
- Marino JS, Tausch BJ, Dearth CL, et al. β2-Integrins contribute to skeletal muscle hypertrophy in mice. Am J Physiol Cell Physiol. 2008;295(4):C1026–1036.
- Fagerholm SC, Guenther C, Llort Asens M, et al. Beta2-integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front Immunol. 2019;10(254). DOI:10.3389/fimmu.2019.00254
- Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008;86(10):1113–1126.
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651.
- Matthews SP, McMillan SJ, Colbert JD, et al. Cystatin F ensures eosinophil survival by regulating granule biogenesis. Immunity. 2016;44(4):795–806.
- Hamilton G, Colbert JD, Schuettelkopf AW, et al. Cystatin F is a cathepsin C-directed protease inhibitor regulated by proteolysis. EMBO J. 2008;27(3):499–508.
- Park S, Lee S, Lee C-G, et al. Capicua deficiency induces autoimmunity and promotes follicular helper T cell differentiation via derepression of ETV5. Nat Commun. 2017;8:16037.
- Fu Y, Li J, Tang Q, et al. Integrated analysis of methylome, transcriptome and miRNAome of three pig breeds. Epigenomics. 2018;10(5):597–612.
- Ji X, Dadon DB, Abraham BJ, et al. Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc Natl Acad Sci USA. 2015;112(12):3841.
- Kleiner RE, Hang LE, Molloy KR, et al. A chemical proteomics approach to reveal direct protein-protein interactions in living cells. Cell Chem Biol. 2018;25(1):110–120.
- Xiong L, Darwanto A, Sharma S, et al. Mass spectrometric studies on epigenetic interaction networks in cell differentiation. J Biol Chem. 2011;286(15):13657–13668.
- Zhang Y, Yang X, Gui B, et al. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J Biol Chem. 2011;286(49):42414–42425.
- Adhikari A, Davie J. JARID2 and the PRC2 complex regulate skeletal muscle differentiation through regulation of canonical Wnt signaling. Epigenetics Chromatin. 2018;11(1):46.
- Dougherty JD, Reineke LC, Lloyd RE. mRNA decapping enzyme 1a (Dcp1a)-induced translational arrest through protein kinase R (PKR) activation requires the N-terminal enabled vasodilator-stimulated protein homology 1 (EVH1) domain. J Biol Chem. 2014;289(7):3936–3949.
- Hargarten JC, Williamson PR. Epigenetic regulation of autophagy: a path to the control of autoimmunity. Front Immunol. 2018;9:1864.
- Song H, Feng X, Zhang H, et al. METTL3 and ALKBH5 oppositely regulate m6 A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019;15:1419–1437.
- Mansueto G, Armani A, Viscomi C, et al. Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 2017;25(1):182–196.
- Lu N, Li X, Yu J, et al. Curcumin attenuates lipopolysaccharide-induced hepatic lipid metabolism disorder by modification of m6 A RNA methylation in piglets. Lipids. 2018;53(1):53–63.
- Liu Y, Lear T, Iannone O, et al. The pro-apoptotic F-box protein Fbxl7 regulates mitochondrial function by mediating the ubiquitylation and proteasomal degradation of survivin. J Biol Chem. 2015;290:11843–11852.
- Bakula D, Müller, AJ, Zuleger, T et al . WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Commun Biol. 2017;8:15637.
- Tang Z, Takahashi Y, Chen C, et al. Atg2A/B deficiency switches cytoprotective autophagy to non-canonical caspase-8 activation and apoptosis. Cell Death Differ. 2017;24(12):2127–2138.
- Demarchi F, Bertoli C, Copetti T, et al. Calpain as a novel regulator of autophagosome formation. Autophagy. 2007;3(3):235–237.
- Ye M, Xu M, Chen C, et al. Expression analyses of candidate genes related to meat quality traits in squabs from two breeds of meat-type pigeon. J Anim Physiol Anim Nutr. 2018;102(3):727–735.
- Cleveland BM, Weber GM. Effects of sex steroids on indices of protein turnover in rainbow trout (Oncorhynchus mykiss) white muscle. Gen Comp Endocrinol. 2011;174(2):132–142.
- Xia HG, Zhang L, Chen G, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6(1):61–66.
- Macqueen DJ, Meischke L, Manthri S, et al. Characterisation of capn1, capn2-like, capn3 and capn11 genes in Atlantic halibut (Hippoglossus hippoglossus L.): transcriptional regulation across tissues and in skeletal muscle at distinct nutritional states. Gene. 2010;453(1–2):45–58.
- Vélez EJ, Azizi S, Verheyden D, et al. Proteolytic systems’ expression during myogenesis and transcriptional regulation by amino acids in gilthead sea bream cultured muscle cells. PLoS One. 2017;12(12):e0187339.
- Vélez EJ, Azizi S, Lutfi E, et al. Moderate and sustained exercise modulates muscle proteolytic and myogenic markers in gilthead sea bream (Sparus aurata). Am J Physiol Regul Integr Comp Physiol. 2017;312(5):R643–R653.
- Meugnier E, Bossu C, Oliel M, et al. Changes in gene expression in skeletal muscle in response to fat overfeeding in lean men. Obesity. 2007;15(11):2583–2594.
- Tong T, Shen Y, Lee H-W, et al. Adenylyl cyclase 3 haploinsufficiency confers susceptibility to diet-induced obesity and insulin resistance in mice. Sci Rep. 2016;6:34179.
- Xu H, Du X, Liu G, et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab. 2019;23:14–23.
- Schneeberger M. Irx3, a new leader on obesity genetics. EBioMedicine. 2018;39:19–20.
- Corominas J, Ramayo-Caldas Y, Puig-Oliveras A, et al. Polymorphism in the ELOVL6 gene is associated with a major QTL effect on fatty acid composition in pigs. PLoS One. 2013;8(1):e53687–e53687.
- Matsuzaka T, Shimano H, Yahagi N, et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med. 2007;13(10):1193–1202.
- Fujita M, Momose A, Ohtomo T, et al. Upregulation of fatty acyl-CoA thioesterases in the heart and skeletal muscle of rats fed a high-fat diet. Biol Pharm Bull. 2011;34(1):87–91.
- Le Luyer J, Laporte M, Beacham TD, et al. Parallel epigenetic modifications induced by hatchery rearing in a Pacific salmon. Proc Natl Acad Sci USA. 2017;114(49):12964–12969.
- Anastasiadi D, Piferrer F. Epimutations in developmental genes underlie the onset of domestication in farmed european sea bass. Mol Biol Evol. 2019;36(10):2252–2264.
- Podgorniak T, Brockmann S, Konstantinidis I, et al. Differences in the fast muscle methylome provide insight into sex-specific epigenetic regulation of growth in Nile tilapia during early stages of domestication. Epigenetics. 2019;14:818–836.
- Wan ZY, Xia JH, Lin G, et al. Genome-wide methylation analysis identified sexually dimorphic methylated regions in hybrid tilapia. Sci Rep. 2016;6:35903.
- Chen X, Wang Z, Tang S, et al. Genome-wide mapping of DNA methylation in Nile tilapia. Hydrobiologia. 2016;791(1):1–11.
- Blouin MS, Thuillier V, Cooper B, et al. No evidence for large differences in genomic methylation between wild and hatchery steelhead (Oncorhynchus mykiss). Can J Fish Aquat Sci. 2010;67(2):217–224.
- Visse M, Sild E, Kesler M, et al. Do Atlantic salmon parr trade growth against immunity? Mar Freshwater Behav Physiol. 2015;48(4):225–240.
- van der Most PJ, de Jong B, Parmentier HK, et al. Trade-off between growth and immune function: a meta-analysis of selection experiments. Funct Ecol. 2011;25(1):74–80.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359.
- Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87–R87.
- Krzywinski M, Schein J, Birol İ, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645.
- Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589.
- Rebhan M, Chalifa-Caspi V, Prilusky J, et al. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 1997;13(4):163.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.