
ABSTRACT
Epigenetic regulation is a crucial factor controlling gene expression. Here, we report our CRISPR/dCas9-based modular epigenetic toolkit that enables gene-specific modulation of epigenetic architecture. By modifying the SunTag framework of dCas9 tagged with five GCN4 moieties, each epigenetic writer is bound to scFv and target-specific sgRNA, and this system is able to modify multiple epigenetic marks in a target-specific manner. We successfully demonstrated that this system is efficient in modifying individual histone post-translational modifications. We display its utility as a tool to understand the contributions of specific histone marks on gene expression by screening a large promoter region and identifying differential outcomes with high base-pair resolution. This epigenetic toolkit can be easily altered with a large variety of epigenetic effectors and is a useful tool for researchers to use in understanding gene-specific epigenetic changes and their relation to gene expression.
Introduction
Utilization of the CRISPR/Cas9 system is now well established for precise genome editing [Citation1–12]. Taking advantage of the genomic locus-specific targeting abilities with catalytically dead Cas9 [Citation13] (dCas9), this system has also been employed to modulate the focal epigenetic architecture and gene expression [Citation14]. Early transcription modulation systems using dCas9 fused to synthetic activators such as VP64, SAM, and VPR have been shown to alter a specific gene expression. However, this effect is transient [Citation15], and they ignore the endogenous epigenetic environment.
Each gene’s distinct regulatory regions such as enhancers or promoters have unique combinations of different histone posttranslational modifications (PTM) that modulate expression. To understand the precise epigenetic regulation of a gene, we need a system that can systematically edit and explore the effect of histone PTM on gene expression with a high resolution. To address this gap, we developed a modular epigenetic toolkit by modifying the previously developed SunTag framework, originally introduced by Tanenbaum et al., in 2014 [Citation16]. SunTag, a trans-activator system consisting of a repeating polypeptide array of 24 GCN4 moieties, recruits multiple anti-GCN4 scFv-fused effector to a targeted location inside the cell. Later, other groups utilized this system to recruit DNMT3A or TET1 to edit the epigenome [Citation17–19]. We further enhanced the SunTag framework by utilizing 22 amino acid spaces as suggested by Morita et al. [Citation17], 5x GCN4 repeats instead of the original 24, and added convenient restriction sites to allow multiple effectors to be quickly and easily incorporated into the system as needed by the researcher. We generated a broad array of constructs with endogenous epigenetic effectors. Here, we demonstrate this toolkit with the writers JMJD3, EZH2, PRDM9, p300, and JARID1A. We show that this switchable modular system efficiently worked in precisely modifying each histone PTM of several target genes, with resolution limited only by the availability of candidate sgRNA sites and allows for combinations of effectors to be easily used at the same locus. This CRISPR/dCas9-based modular epigenetic toolkit paves a new avenue to study the epigenetic architectures of an individual gene with great precision.
Materials and methods
For full details, see Additional File 1.
sgRNA design sgRNA was designed with zero or one off-target matches using (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. For histone mark
analysis in A549 cells, sequences were selected from regions of histone mark enrichment (Additional File 2, Fig. S2b). For SNCA, only sgRNA in the targeted region were selected for analysis (Additional File 4).
Cell Culture All cells were maintained in humidified 5% CO2 chambers with DMEM +10%FBS. To generate stable dCas9-5xGCN4 HEK293 and A549 cell lines, cells were transduced with dCas9-5xGCN4 lentivirus and selected for blasticidin resistance. A549 cells were further transduced with sgRNA-expressing lentiviral particles and selected for puromycin resistance.
ChIP A549 cells expressing dCas9-5xGCN4 and gene-specific RNA were transfected with scFv-expressing editors or controls using Helix-IN (Oz Biosciences). After 7 2 hours, cells were fixed with 1% formaldehyde, GFP Sorted, and sheared DNA was incubated with antibody against the relevant histone mark, reverse crosslinked, and isolated before PCR (Additional File 5).
SNCA Promoter Screening In triplicate, HEK293 cells expressing dCas9-5xGCN4 were transfected with sgRNA (Additional File 4) and epigenetic effectors or empty control. Seventy-two hours after transfection, we performed FACS, extracted RNA, and ran qPCR for SNCA and β- Actin to determine relative expression levels (ΔΔCt) (Additional File 5). All five sgRNA sites that induced significant expression reduction using either JARID1A or EZH2 were further analysed with ChIP. To screen the promoter region, 13 PCR primer pairs were selected to cover the entire length of the SNCA promoter (Average PCR bin size: 274 base pairs, Additional File 5) and provide a map of histone mark enrichment in these cells. ChIP analysis was performed once in technical triplicate.
Statistical Analysis ChIP samples were normalized to input, and differences between empty and treated were assessed using parametric t-test. qPCR samples were normalized to β-Actin and fold change (2^(- ΔΔCt)) from empty was calculated. Statistical analysis of the qPCR data fold change (from 1) was performed via the bootstrap resampling method using an open web portal [Citation20,Citation21].
Results
Recently, we reported that the epigenetic mark H3K4me3 was enriched on the promoter of a Parkinson’s disease-related gene, SNCA, and we were able to modulate its expression by target-specifically altering its epigenetic code [Citation22]. We envisioned a broader system where highly specific changes to individual genes can be achieved using an expanded suite of epigenetic modifiers that adjusts the endogenous structure while minimally disturbing other loci. To this end, we took advantage of the previously developed SunTag system [Citation16]. To recruit multiple functional domains of epigenetic writers, we used dCas9 with 5xGCN4 moieties (five GCN4 moieties spaced by 22 amino acids [Citation17]). This allows us to recruit multiple anti-GCN4 antibody (scFv)-fused epigenetic effectors to the sgRNA-directed loci and directly modulate the local histone PMTs (). We modified the original plasmids to include a small RsrII-based cloning site that allowed us to easily generate constructs to target a wide variety of epigenetic marks. We generated five plasmid constructs with scFv and sfGFP fused into different key epigenetic effectors (, Additional File 2: Fig. S1a,b, Additional File 1). Histone acetyltransferase p300 catalyses the acetylation at the 27th lysine residue on histone H3 (H3K27ac), although it can also target other lysine residues as well [Citation23–26]. JMJD3 (Jumonji domain-containing protein D3) and EZH2 (Enhancer of zeste homolog 2) decreases and increases H3K27 tri-methylation (H3K27me3), respectively [Citation27–31]. We also generated constructs to decrease or increase H3K4 tri-methylation (H3K4me3) using JARID1A (Jumonji, AT-rich interactive domain 1) and PRDM9 (PR/SET Domain 9), respectively [Citation32,Citation33]. Previously, it was shown that only using the catalytic core of an enzyme with dCas9 is more effective [Citation4], so we subcloned the catalytic core of each epigenetic writer into the SunTag-scFv system (Additional File 2: Fig. S1c) to develop our modular epigenetic toolkit.
Figure 1. Toolkit of Epigenetic Editors. a Schematic of epigenetic modulator design based on SunTag framework. 5xGCN4 sequences bound to dCas9 recruit epigenetic effectors fused with anti-GCN4 scFv antibody fragments to the sites directed by sgRNA. b Epigenetic modulators used in this study and their effects on histone marks. c Epigenetic editors alter histone marks in a site-specific manner. Each graph is normalized to input and represents biological triplicates.
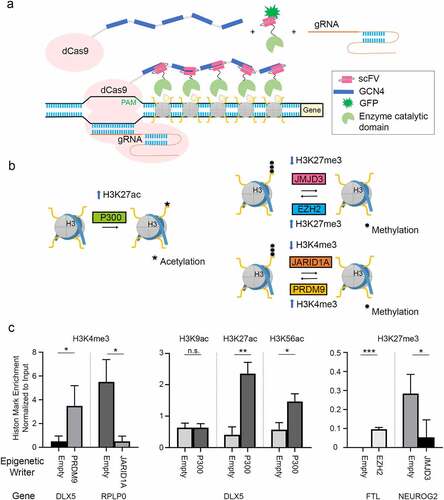
To assess the efficiency of the modular epigenetic toolkit, we analysed relevant epigenetic marks in specific genes in the well-characterized A549 cell line [Citation34,Citation35]. We data mined available epigenetic information around ±2.5 kb of transcriptional start sites, sorted genes for epigenetic marks, and selected four genes with distinctive patterns of histone modifications that would demonstrate the toolkit’s flexibility – RPLP0, FTL, DLX5, and NEUROG2 (Additional File 1–3: Fig. S2a,b). We confirmed a similar enrichment pattern of histone PTM enrichment as in the ENCODE database with only minor differences in our cells (Additional File 2: Fig. S2c).
For each gene, we chose a sgRNA at the gene locus enriched for the relevant epigenetic target (Additional File 4). We generated A549 cell lines stably expressing dCas9-5xGCN4 and the desired sgRNA (Additional File 1) and confirmed the presence of dCas9-5xGCN4 at the sgRNA target site. (Additional File 2: Fig. S3a). The appropriate dCas9-5xGCN4/sgRNA cell lines were transfected with the scFv-fused epigenetic writers (p300, JMJD3, EZH3, JARID1A, or PRDM9) or empty scFv, FACS sorted followed by ChIP analysis, and analysed changes in histone PTM (, Additional File 2: Fig. S3b). PRDM9 increased DLX5 H3K4me3 enrichment 6.9-fold, conversely, JARID1A decreased RPLP0 H3K4me3 enrichment by 11-fold. p300 led to a 5.8-fold increase in DLX5 H3K27ac as well as a 1.79-fold increase in H3K56ac, although it did not affect H3K9ac enrichment at that locus. EZH2 increased H3K27me3 in the FTL regulatory region from below detection limit, while JMJD3 reduced Neurog2 H3K27me3 by 5.3-fold (1c). These data show that our modular epigenetic toolkit system target-specifically modulates endogenous histone marks. Surprisingly, despite these epigenetic changes, we found little effect on gene expression in these cells (Data not shown). We theorized that to specifically affect gene activity, we would need a more in-depth exploration of how the sgRNA site selection affects the native epigenetic architecture, and how this may drive gene expression. With this, we may be able to reveal new insights into endogenous gene regulation and inform future decisions about where to target epigenetic writers to achieve maximum effect. Additionally, there is always a possibility that multiple epigenetic factors regulate a gene expression simultaneously in combination with some cis-regulatory elements. In those cases, removing or adding one single epigenetic modification might result in partial activation/inactivation.
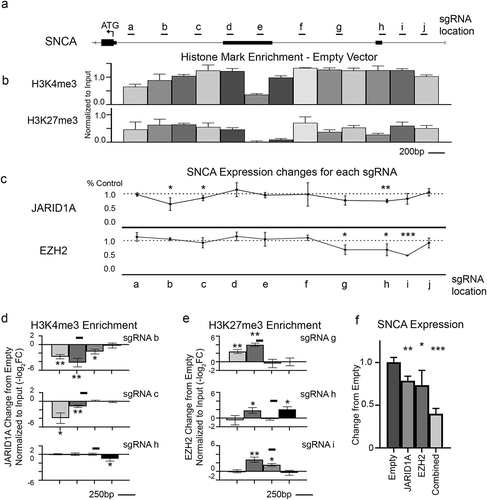
To test this idea, we turned to a gene we have previously explored the epigenetic landscape of SNCA – which encodes α-synuclein [Citation22]. The promoter region of SNCA spans over 3200 base pairs; to thoroughly investigate this area, we selected 10 sgRNA targets spaced every 200–400 base pairs, based on the availability of high-quality sgRNA sites (, Additional File 2: Fig S4, Additional File 4). We hypothesized that targeting our modular epigenetic toolkit at the sites of histone mark enrichment would allow us to modulate SNCA expression and provide insight into the functional importance of epigenetic architecture with a hundred-base resolution. We previously found JARID1A and EZH2 to be effective at modulating SNCA expression levels, so we elected to screen the promoter with these two editors. We transfected sgRNA with empty control constructs or our epigenetic editors into HEK293 cells that stably express dCas9-5xGCN4. After 72 hours, we FACS-sorted cells and performed ChIP analysis of H3K4me3 and H3K27me3 for the entire ~3200 base-pair region () to establish a baseline of the endogenous epigenetic landscape in our cells. We then analysed the expression of α-synuclein by qRT-PCR in control (empty-scFv) to JARID1A-scFv and EZH2-scFv targeted at each sgRNA site. Interestingly, we noted that each epigenetic effector was able to successfully modulate SNCA expression, but that sgRNAs in different promoter regions had differential results (). In particular, the removal of permissive H3K4me3 was effective at one distal site (sgRNA h), and at two sites near the SNCA start codon. In contrast, the addition of the inhibitory H3K27me3 was effective near the more distal transcriptional start site but had no effect on the more proximal region of the gene. ChIP analysis of nearby chromatin showed that all significant changes in expression coincided with significant changes in histone modifications in the sgRNA neighbourhood (). Interestingly, the epigenetic alterations were significantly altered within 500 base pairs of the sgRNA () and we saw no significant changes outside of this region, suggesting that the epigenetic modulators exert only a local effect. We further confirmed the ability of the toolkit to enact combinatorial changes at a single locus by co-transfection of EZH2 and JARID1A in the 5’ end of the SNCA gene (sgRNA h). The synergistic effect resulted in a reduction of SNCA expression by 60.1%, significantly lower than either JARID1A or EZH2 alone (22.5% and 26.8% respectively) (). The ability to alter multiple histone post-translational modifications in one step highlights the ease and potential of this modular system. The action of JARID1A but not EZH2 at sgRNAb and sgRNAc underscores the highly selective nature of these modifications in gene expression. Notably, the same effectors exhibited dramatically different expression effects when the sgRNAs were only 200 bp apart (sgRNA h,i,j), highlighting the ability of our system to enact changes at high resolution determined only by the availability of specific sgRNA sites. Each cell type has a specific epigenetic landscape for any given gene, for example, SNCA’s epigenetic landscape is different between neuronal and non-neuronal cells, and this may influence the extent of effect by modular epigenetic toolkit as well as selection of optimal targeting sgRNA.
Figure 2. Screening of single gene promoter. a Scale diagram of promoter region of SNCA gene showing two alternative non-coding Exon 1 and Exon 2 with a start codon (ATG) as well as locations of sgRNA used. b Histone mark enrichment from our HEK293 dCas9 cell line for the indicated marks. (Peak locations are scaled to diagram shown in a. Average PCR bin size = 275 ± 25 base pairs). c qPCR assay was carried out to determine the change in α-synuclein expression levels relative to empty scFv control (Set to 1) for JARID1A and EZH2 targeted to specified loci by sgRNA. (Locations are scaled to diagram shown in a). Each point is normalized to empty control and represents biological triplicates. d,e Histone mark changes after transfection with indicated sgRNA and d JARID1A-scFv or e EZH2-scFv. Black lines indicate location of the sgRNA targeting site on the chromosome. Average PCR bin size = 275 ± 25 base pairs. f qPCR assay shows the change in α-synuclein expression levels relative to empty scFv control for JARID1A and EZH2 targeted separately or together to a single sgRNA target site shows that combined targeting is more effective than either alone. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Discussion
This epigenetic toolkit has the potential to make highly specific changes to the epigenetic architecture of any gene targetable by dCas9, and focused targeting of specific endogenous epigenetic modifiers may prove to be an effective strategy for persistently altering pathologic transcriptional activity. Recently, there has been increased focus on using specific gene targeting as a therapeutic avenue for genetic disease. This system offers a strong tool to dissect and understand the underlying epigenetic architecture and opens potential new avenues for therapeutic strategies for various disease conditions.
Authors’ contributions
SGT and YSK conceived the study and designed all the experiments. SGT created all modular epigenetic toolkit system. LA, SGT, IHJ, AS, and MBF performed the experiments. MC and HNH performed bioinformatics analysis. LA SGT, HNH, AS, MBF, and YSK wrote the manuscript. All authors read and approved the final manuscript. YSK supervised the entire project.
Availability of data and materials
All supporting data are included in the manuscript and supplementary files. All vectors described will be deposited at Addgene and will be available under a Uniform Materials Transfer Agreement.
Supplemental Material
Download Zip (4.3 MB)Acknowledgments
Authors would like to acknowledge the efforts by Ms. Siyona Mishra and Ms. Annlisa Simon for their help in maintaining eukaryotic cells and bacterial cultures. The authors also thank Dr. Sambuddha Basu and Dr. Goun Je for their valuable input in study design.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2022.2106646
Additional information
Funding
Related Research Data
References
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823.
- Chavez A, Scheiman J, Vora S, et al. Highly efficient Cas9- mediated transcriptional programming. Nat Methods. 2015;12(4):326–328.
- Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–451.
- Hilton IB, D’Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33(5):510–517.
- Holtzman L, Gersbach CA. Editing the epigenome: reshaping the genomic landscape. Annu Rev Genomics Hum Genet. 2018;19:43–71.
- Joung J, Konermann S, Gootenberg JS, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12(4):828–863.
- Klann TS, Black JB, Chellappan M, et al. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol. 2017;35(6):561–568.
- Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168(1–2):20–36.
- Park M, Keung AJ, Khalil AS. The epigenome: the next substrate for engineering. Genome Biol. 2016;17(1):183.
- Pulecio J, Verma N, Mejia-Ramirez E, et al. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell. 2017;21(4):431–447.
- Thakore PI, Black JB, Hilton IB, et al. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods. 2016;13(2):127–137.
- Thakore PI, D’Ippolito AM, Song L, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12(12):1143–1149.
- Brocken DJW, Tark-Dame M, Dame RT. dCas9: a Versatile Tool for Epigenome Editing. Curr Issues Mol Biol. 2018;26:15–32.
- Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911.
- O’Geen H, Ren C, Nicolet CM, et al. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017;45(17):9901–9916.
- Tanenbaum ME, Gilbert LA, Qi LS, et al. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159(3):46–635.
- Morita S, Noguchi H, Horii T, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34(10):1060–1065.
- Huang YH, Su J, Lei Y, et al. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017;18:176.
- Pflueger C, Tan D, Swain T, et al. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018 Aug;28(8):1193–1206. Epub 2018 Jun 15.
- Cleries R, Galvez J, Espino M, et al. BootstRatio: a web- based statistical analysis of fold-change in qPCR and RT-qPCR data using resampling methods. Comput Biol Med. 2012;42(4):438–445.
- Disease registry statistical tools - bootstratio. Accessed1/4/2020. Available from: http://pdo.iconcologia.net/stats/br/index.html.
- Guhathakurta S, Kim J, Adams L, et al. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol Med. 2021;13(2). DOI:10.15252/emmm.202012188
- Eckner R, Ewen ME, Newsome D, et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein(p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8(8):84–869.
- Wang L, Tang Y, Cole PA, et al. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol. 2008;18(6):741–747.
- Vv O, RL S, Russanova V, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953–959.
- Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. 2006;23(3):289–296.
- Koyanagi M, Baguet A, Martens J, et al. EZH2 and histone 3 trimethyl lysine 27 associated with Il4 and Il13 gene silencing in Th1 cells. J Biol Chem. 2005;280(36):31470–31477.
- Vire E, Brenner C, Deplus R, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–874.
- Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–1043.
- Zhang X, Liu L, Yuan X, et al. JMJD3 in the regulation of human diseases. Protein Cell. 2019;10(12):864–882.
- Burchfield JS, Li Q, Wang HY, et al. JMJD3 as an epigenetic regulator in development and disease. Int J Biochem Cell Biol. 2015;67:148–157.
- DiTacchio L, Le HD, Vollmers C, et al. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333(6051):1881–1885.
- Paigen K, Petkov PM. PRDM9 and its role in genetic recombination. Trends Genet. 2018;34(4):291–300.
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74.
- Davis CA, Hitz BC, Sloan CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46(D1):D794–D801.