
ABSTRACT
DNA methylation on CpGs regulates transcription in mammals, both by decreasing the binding of methylation-repelled factors and by increasing the binding of methylation-attracted factors. Among the latter, zinc finger proteins have the potential to bind methylated CpGs in a sequence-specific context. The protein ZBTB38 is unique in that it has two independent sets of zinc fingers, which recognize two different methylated consensus sequences in vitro. Here, we identify the binding sites of ZBTB38 in a human cell line, and show that they contain the two methylated consensus sequences identified in vitro. In addition, we show that the distribution of ZBTB38 sites is highly unusual: while 10% of the ZBTB38 sites are also bound by CTCF, the other 90% of sites reside in closed chromatin and are not bound by any of the other factors mapped in our model cell line. Finally, a third of ZBTB38 sites are found upstream of long and active CpG islands. Our work therefore validates ZBTB38 as a methyl-DNA binder in vivo and identifies its unique distribution in the genome.
Introduction
DNA methylation plays a central role in the control of gene expression and cell fate in mammals [Citation1–3]. Methylation of CpGs can profoundly alter the binding specificity and affinity of transcription factors (TFs) to their cognate binding sites in promoter and cis-regulatory regions of genes, altering in consequence chromatin organization and gene expression at target loci [Citation1,Citation4–6]. The consequences of DNA methylation on TF binding are actually quite variable depending on the presence of CpG in cognate binding sites, the density of CpGs, and the relative level of methylation of these CpGs [Citation7–9]. For instance, methyl-DNA binding proteins MeCP2, MBD1, MBD2, and MBD4 bind within chromatin domains rich in methylated CpG dinucleotides, independently of the surrounding DNA sequence, and they contribute to gene silencing [Citation10]. In contrast, many transcription factors are repealed by CpG methylation in their cognate binding site, while others have stronger or similar affinity for CpG-methylated consensus sequences than for the same unmethylated sequences [Citation11–18]. For instance, zinc finger (ZNF) protein ZFP57 contributes to the silencing of a limited number of genes in mice embryonic stem cells, known as imprinted genes, owing to its strong affinity for a defined consensus sequence containing a central methylated CpG present at certain imprinted regions [Citation19]. The transcription factors EGR-1, KLF4, ZBTB4, and KAISO/ZBTB33 recognize DNA sequences that contain a central methyl-CpG in a defined nucleotide context in vitro, and can either activate or repress gene expression in vivo [Citation20–24]. Yet, in many cases, in vitro data remain conflicting with in vivo data and/or do not fully predict the binding specificity of a TF in vivo. For instance, the targets of KAISO/ZBTB33 in the genome seem to be mostly unmethylated in ENCODE cell lines [Citation23,Citation25–27]. Similarly, less than 5% of EGR-1 and KLF4 binding sites in human cells actually contain fully methylated CpG sites [Citation20,Citation21,Citation28]. The characterization of TFs that bind to methylated DNA, their mode of interaction with the methyl-CpGs, and the cellular and molecular consequences of such interactions are still far from being fully understood.
ZBTB38 is a ZNF transcription factor considered a methyl-CpG binding protein due to its ability to bind with high-affinity methylated sequences in vitro [Citation24,Citation29–31]. Two clusters of ZNFs exhibit methyl-CpG binding activity in the protein. A central KAISO-like ZNFs cluster was first described for its ability to bind methyl-CpG sequences [Citation24,Citation29]. A second C-terminal ZNFs cluster recognizes a SELEX-enriched ATmeCGGmeCG sequence (also called mCZ38BS) in vitro and in cells [Citation30–32]. Several studies have revealed that ZBTB38 (Cibz/Zbtb38 in the mouse) regulates cell proliferation, growth, and differentiation [Citation24,Citation29,Citation30,Citation33–41]. The function of ZBTB38 in cell growth is complex as its depletion (or genetic inactivation) can either promote, reduce, or not affect cell proliferation according to cell types, which echoes its potential function as an oncogene or tumour suppressor in cancers [Citation32,Citation33,Citation35,Citation36,Citation38,Citation42–45]. Further highlighting the pleiotropic, context-specific, and tissue-specific effects of ZBTB38, many single-nucleotide polymorphisms (SNPs) either within or in close proximity to ZBTB38 have been associated with adult height and many diseases including idiopathic short stature, atopic dermatitis, macular degeneration, osteoporosis, prion disease, and prostate cancer [Citation42,Citation46–56].
To shed light on the functions of ZBTB38 and its relationship with DNA methylation, we investigated the genome-wide distribution of ZBTB38 binding sites in the human genome. We demonstrate that ZBTB38 binds a large number of regions in the genome in a DNA methylation- and sequence-dependent manner, including a transcriptional program involved in the response to doxorubicin.
Results
Unbiased genome-wide identification of ZBTB38 binding sites in the human genome
To elucidate the regulatory functions of ZBTB38 in human cells and its relationship to DNA methylation, we utilized a previously published stable HeLa-S3 cell line stably expressing HA-Flag-ZBTB38 protein [Citation34]; the expression level of the tagged protein is only about 20% of that of endogenous ZBTB38, to guard against overexpression artefacts. We then performed chromatin immunoprecipitation followed by deep sequencing with antibodies directed against the tags (). We sequenced two independent ChIP-seq replicates (and their matching input DNAs) and observed a strong correlation between average tag density across the genome between the two ChIPs and significant enrichment in ChIP signal vs Input ( and S1A-C). Using the CisGenome software with stringent criteria (see online ‘Material and Methods’), we identified 3032 regions unambiguously bound by ZBTB38. We confirmed ZBTB38 binding at several of these regions by ChIP-qPCR on independent biological replicates (Figure S1D) [Citation30].
Figure 1. Genomic landscape of ZBTB38 binding sites determined by ChIP-sequencing in human cells. (a) Genomic tracks display ZBTB38 ChIP-sequencing data (replicates 1 and 2) and input data (replicates 1 and 2) on a representative 5-Mb region of chromosome 11. (b) Average tag intensity of ZBTB38 ChIP-sequencing and matched Input samples at 3032 called ZBTB38 binding sites. (c) Heatmap representing binding intensities of ZBTB38 ChIP-sequencing and input samples at ZBTB38 binding sites. (d) Genomic distribution of ZBTB38 binding regions across the 10 chromatin states defined by a Hidden Markov Model using multiple histone modifications and genomic features. The asterisk indicates a P-value <10−3. (e) Venn diagram showing the overlap between ZBTB38 binding regions, transcription start sites (TSS), and CpG islands. (f) Venn diagram showing the overlay between ZBTB38 binding sites and CGI, CGI-shores, and CGI-shelves. (g) Box plot representing the relative size of CpG islands associated with ZBTB38 binding. P-value was calculated by Mann–Whitney/U-test.
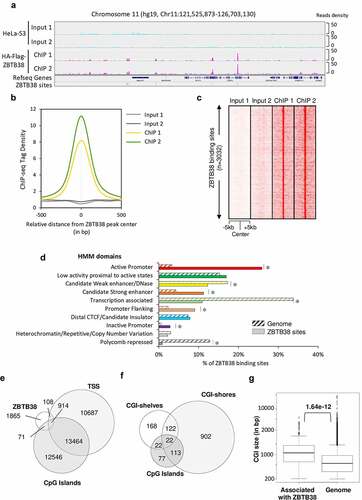
ZBTB38 binding is enriched at a subset of repetitive sequences, CpG island shores, and enhancers
To further understand the function of ZBTB38 in gene expression regulation, we functionally annotated the 3032 regions bound by ZBTB38. Correlation with chromatin domains, defined using integration of ChIP-seq, FAIRE-seq, and DNAse-seq experiments from six different cell types [Citation57], indicates that ZBTB38 binding is enriched at enhancers, promoters, and regions close to active transcription states, as is typical for TFs (). These regulatory regions are defined by co-binding of many TFs [Citation58]. We observed that ZBTB38 binding is enriched in TF binding active regions (BAR) of the genome that are often proximal and distal regulatory modules (PRM and DRM) and high-occupancy transcription-related factor (HOT) regions, which are the regions with the highest co-occurrence of transcription factors in the genome (Figure S1E). These data indicate that ZBTB38 binds regions involved in gene expression control.
A close look at promoters associated with ZBTB38 binding shows that most of them (89.4%) are CpG-rich islands according to annotation in the UCSC genome browser (). This prompted us to investigate the relationship with CpG islands (CGI) size, CGI-shores (the regions ±2 kb from CGI), and CGI-shelves (regions between 2 and 4 kb from CGI) [Citation59–61]. We found that most ZBTB38 binding sites are located in CGI-shores and prevalent at large CGI ().
We also observed that ZBTB38 binding sites are enriched for certain repetitive DNA sequences (Figure S1F). At the genome scale, more than one-third of ZBTB38 binding sites encompass an Alu DNA repeat (Figure S1F). We thus re-analysed ZBTB38 ChIP-seq reads on a reconstituted pseudo-genome, where ChIP-seq reads abundance is calculated on each family of DNA repeats based on RepeatMasker database. Using this approach, the enrichment of ZBTB38 ChIP-tags at Alu sequences was indistinguishable from the background (i.e., input DNA), indicating that ZBTB38 binds only a subset of Alu sequences in the genome (Figure S1G).
The analysis of ZBTB38 binding sites distribution shows an enrichment upstream of TSS and at putative enhancers, suggesting that ZBTB38 might contribute to transduce regulatory information to the promoter.
ZBTB38 target genes are highly expressed and important for metabolic processes
We investigated the relationship between ZBTB38 binding and gene expression. We observed that ZBTB38-associated TSS present very high levels of polymerase II Serine 5 phosphorylation compared to other TSS (). Consistent with this observation, at these ZBTB38-associated promoters, we also observed higher levels of transcription, defined by CAGE experiment (); higher levels of histone marks associated with open chromatin and gene activation such as histone H3 lysine 27 acetylation, H3 lysine 4 di/tri-methylation, H3 lysine 79 dimethylation, and H3 lysine 9 acetylation () and higher levels of DNAse I accessibility (). The data indicate that TSS targeted by ZBTB38 are transcribed at higher levels compared to the bulk genome.
Figure 2. ZBTB38 binds upstream of CpG island of actively transcribed genes. (a) Average binding intensity of ZBTB38 ChIP-sequencing, phospho-Serine 5 Polymerase II (S5P) ChIP-sequencing, and input samples at TSS bound by ZBTB38 (left panel) and all TSS (right panel). (b) Average signal reads of CAGE at TSS bound by ZBTB38 (left panel) and all TSS (right panel). (c) Average intensity of histone-sequencing and input samples at TSS bound by ZBTB38 (left panel) and all TSS (right panel). (d) Average intensity of DNAseI-sequencing samples at TSS bound by ZBTB38 (left panel) and all TSS (right panel). (e) Association of ZBTB38 bound genes with transcriptomic studies present the Molecular Signatures Database. CML up-regulated are genes up-regulated in CD34+ cells isolated from bone marrow of CML (chronic myelogenous leukaemia) patients, compared to those from normal donors; HCC up-regulated are genes up-regulated in hepatocellular carcinoma (HCC) compared to normal liver samples; colon carcinoma up-regulated are genes up-regulated genes in colon carcinoma tumours compared to the matched normal mucosa samples; AML (rearranged MLL) up-regulated are genes up-regulated in paediatric AML (acute myeloid leukaemia) with rearranged MLL compared to all AML cases with the intact gene; Housekeeping are Housekeeping genes identified as expressed across 19 normal tissues; NPC down-regulated are genes down-regulated in nasopharyngeal carcinoma (NPC) compared to the normal tissue; MBD Knock-down up-regulated are genes up-regulated in HeLa cells after simultaneous knockdown of all three MBD (methyl-CpG binding domain) proteins MeCP2, MBD1 and MBD2 by RNAi. (f) Association of ZBTB38-bound genes with transcriptomic signatures of chemical alterations listed in the Molecular Signatures Database.
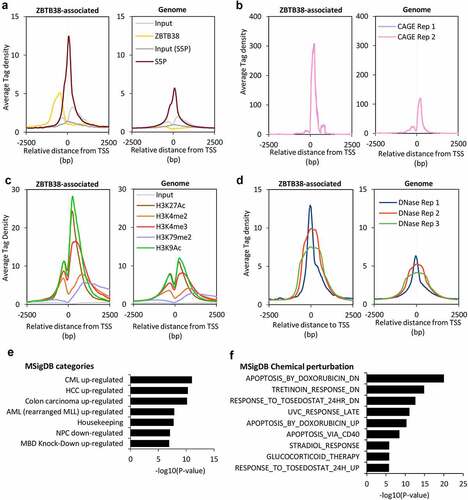
A Gene Ontology analysis shows that ZBTB38 target genes belong to general biological functions including mRNA metabolism, metabolic processes, and cell homoeostasis, and are enriched for genes coding mitochondrion constituents (Table S1). A functional annotation of ZBTB38 target genes using the MSigDB database indicates an association with group of genes deregulated in cancers and with cancer gene set signatures, including CML and AML ( and Table S1). This observation is consistent with previous findings reporting a function of ZBTB38 in AML and CML [Citation35]. Finally, a significant proportion of ZBTB38 targets are either up- or down-regulated upon exposure to doxorubicin, a compound causing DNA damage and reactive oxygen species (ROS) accumulation in cells ( and Table S1). We thus investigated the susceptibility of cancer cells transfected with siRNA against ZBTB38 to doxorubicin. In HeLa, U2OS, and HCT116 cells, depletion of ZBTB38 using three different validated siRNAs enhances the toxicity of doxorubicin (Figure S2).
These data indicate that ZBTB38 primarily associates with active promoters and cis-regulatory elements of transcription of genes important in cancer and for doxorubicin response.
ZBTB38 binding profile is dissimilar to most chromatin remodellers and transcription factors
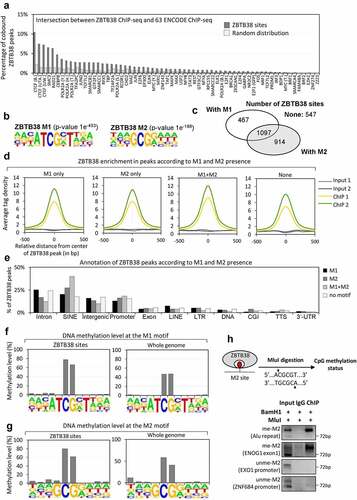
To better understand the binding specificity of ZBTB38, we compared ZBTB38 binding sites with the >60 factors for which ChIP-seq datasets were available in HeLa-S3 cells from the ENCODE portal [Citation62]. We found very little overlap between ZBTB38 and other transcription factors (). CTCF, SMC3, and RAD21 were in the top 3 factors overlapping with ZBTB38, and the overlaps were of 8.65%, 6.59%, and 6.39% of ZBTB38 sites, respectively (). These factors play a concerted role at insulators and chromosome contact points [Citation63,Citation64]. Accordingly, we observed that ZBTB38 co-bounds with the three other factors at a few hundred sites in the genome (Figure S3A and S3B). The shared sites are mostly intronic and intergenic regions (Figure S3C). For the remaining ~94% of its sites, ZBTB38 distribution is distinct from the previously characterized transcription factor and chromatin remodeller profiles. This could indicate either that ZBTB38 associates with a large number of different partners at different sites and/or that ZBTB38 binds a new class of cis-regulatory elements with yet to be characterized transcription factor and chromatin remodelling partners. Consistent with a function of ZBTB38 in various transcription factor networks, we previously observed that ZBTB38 co-purifies with different TFs in vivo (for which ChIP-seq data in HeLa-S3 are not available) [Citation34,Citation44]. This observation suggests a mode of interaction with the chromatin different from many other studied TFs in HeLa-S3 cells.
Figure 3. ZBTB38 binding sites contain a methylated CpG consensus. (a) Overlap between ZBTB38 binding regions and 63 chromatin binding factors in HeLa-S3 cells (ENCODE data). (b) De novo DNA motifs discovery in ZBTB38 binding regions using HOMER tools identify two sequence motifs: M1 (P-value of 1.0 × 10−453) and M2 (P-value of 1.0 × 10−198). The logo and enrichment statistics for M1 and M2 are presented. (c) Venn diagram indicating the proportion of ZBTB38 binding regions containing M1, M2, M1 + M2, or none of these motifs. (d) Average tag intensity of ZBTB38 ChIP-sequencing and Input samples in ZBTB38-bound regions containing M1, M2, M1 + M2, or none. (e) Genomic distribution of ZBTB38 binding regions containing M1, M2, M1 + M2, or none determined using HOMER tools. (f) Methylation level of the M1 motif at ZBTB38 binding regions (left panel) and at whole genome (right panel). (g) Methylation level of the M2 motif at ZBTB38 binding regions (left panel) and at whole genome (right panel). (h) Input DNAs, ZBTB38 ChIPed DNAs, and control IgG-bound DNAs were digested with BamHI and MluI prior to analysis on a 2% agarose gel stained with ethidium bromide. A control 72 base pair fragment without MluI and BamHI site was used as a control ChIP specificity.
ZBTB38 binds two different DNA consensus sequences containing a methylated CpG motif
We further analysed enriched DNA motifs at ZTB38 binding sites (). We identified several known DNA motifs including the CTCF consensus binding site (P-value of 1.0 × 10−38) (Table S2). However, the vast majority of ZBTB38 sites overlap with two uncharacterized motifs that we called motif ‘M1’ (P-value of 1 × 10−453) and ‘M2’ (P-value of 1 × 10−198). We identified 1564 ZBTB38 binding regions containing M1, 2011 regions containing M2, 1097 (one-third) containing M1 + M2, and 547 regions without motif in the list of 3032 ZBTB38 binding regions (). When we examined the binding intensity, we observed that ZBTB38 binds both M1 and M2 with very similar affinity (based on the average tag-enrichment values) and that the presence of the two motifs slightly increases the affinity of ZBTB38 onto the DNA (). A similar observation is made if we compute DNA binding score from the CisGenome analysis (data not shown). We finally found that the genomic distributions of ZBTB38 regions containing M1, M2, or both were quite similar with the striking exception of SINE elements (and especially Alu repeats) that are over-represented in M1 + M2 containing binding sites ().
Both M1 and M2 contain a conserved CpG site. We thus directly assessed the methylation level of the CpG dinucleotide contained in M1 and M2 using a high-throughput bisulphite sequencing map [Citation65]. In HeLa-S3 cells, very little, if any, oxidized version of DNA methylation (i.e., 5hmC, 5fC, and 5caC) exists, so bisulphite conversion is almost directly indicative of the methylation status [Citation66]. The average methylation level of the CpG in M1 and in M2 is 80% at ZBTB38 binding sites compared to 50–60% at the genome-wide level ( F and g). We experimentally confirmed the methylation at ZBTB38 targets by digesting ZBTB38 immunoprecipitated DNAs with a methylation-sensitive restriction enzyme, MluI, that recognizes a sequence similar to the M2 motif (). We selected four genomic sites containing a M2 motif with perfect match to the MluI restriction site for analysis: two regions in promoter of EXO1 and ZNF684 that are not identified in the ChIP-sequencing analysis and two regions, Alu and ENOG1 (i.e., intronic sequence), that are bound by ZBTB38 in our ChIP analysis. We observed that in ZBTB38-immunoprecipitated DNAs, the two ZBTB38 target sites, Alu and ENOG1, are resistant to MluI digestion and PCR amplified in contrast to the promoters of EXO1 and ZNF684 that are not PCR amplified (). DNAs immunoprecipitated by ZBTB38 are therefore methylated on the central CpG site of the M2 motif, indicating that ZBTB38 binds with high-affinity regions of the chromatin containing a methylated CpG site in cells.
ZBTB38 binds with high affinity to the methylated version of the M2 motif in vitro
We used the resemblance between the M2 motif and the MluI restriction site to further investigate the consequences of changes in ZBTB38 expression on the methylation level at the M2 motif. Indeed, correlative analysis suggest a possible function for ZBTB38 in the regulation of DNA methylation at specific sites [Citation67]. We analysed two loci, ENOG1 and Alu, with perfect match to the MluI consensus. We purified the genomic DNA from cells treated with siRNA against ZBTB38, performed an overnight digestion with MluI, and performed a PCR amplification of MluI/M2 containing regions. Using such an approach, we observed that the M2 motif was methylated at ENOG1 and Alu sites (MluI-resistant) in the control cells as well as in cells treated with siRNA against ZBTB38 for 48 hours (). In the same samples, EXO1 and ZNF684 promoters were not amplified in the PCR reaction and thus unmethylated, as expected (). Similarly, we observed that the level of methylation of M2 at ZBTB38 targets was similar in HeLa-S3 cells expressing HA-Flag-ZBTB38 (and used for the ChIP-sequencing) and isogenic parental cells (). Our analysis indicate that the level of CpG methylation at the M2 motif is not influenced by the depletion of ZBTB38 in HeLa-S3 cells.
Figure 4. ZBTB38 binds the methylated M2 motif in vitro and it does not regulate its methylation in vivo. (a) Analysis of M2 motif methylation at two ZBTB38 binding sites and two control regions using the DNA methylation-sensitive MluI restriction enzyme. Genomic DNAs prepared from HeLa S3 cells transfected with siRNAs against ZBTB38 or control siRNAs were digested overnight by BamHI or BamHI+MluI and analysed by PCR amplification. (b) Analysis of M2 motif methylation at two ZBTB38 binding sites and two control regions using the DNA methylation-sensitive MluI restriction enzyme in HeLa-S3 cells expressing the HA-Flag-ZBTB38 protein and parental cells. (c) In vitro binding assays. GST-fusions of ZBTB38 central zinc fingers and mutated zinc fingers (H491R) or GST alone were incubated with equimolar mix of methylated, unmethylated, and mutated DNA probe containing the M2 motif. Left panel: relative quantification of methyl, unmethyl, and mutated level of DNAs recovered on the beads. Right panel: migration on agarose gel stained with ethidium bromide of total DNAs recovered on the beads prior enzymatic digestion.
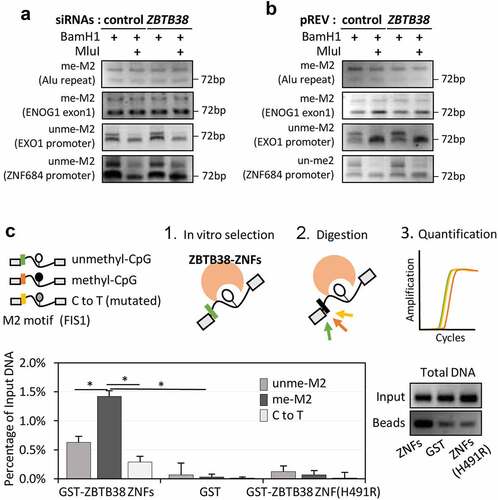
We speculated that ZBTB38 might actually directly interact with the methylated CpG sequence. Consistent with this hypothesis, the M1 motif resembles the mCZ38BS site bound by ZBTB38 C-terminus ZNFs in vitro and in ChIP experiments, and mutation of either ZBTB38 ZNFs or the meCpG in the motif reduces the interaction [Citation30]. To test whether ZBTB38 directly interacts with the methylated M2 CpG sequence, we performed an in vitro binding assay and used the central set of three zinc fingers [Citation24]. A GST-fusion protein of these zinc fingers of ZBTB38 (GST-ZBTB38-ZF) was produced in bacteria, and purified and incubated with oligo-nucleotides containing the M2 sequence from the FIS1 promoter (a validated in vivo target; Figure S1D) either methylated, unmethylated, or with the key cytosine mutated into thymidine. In addition to the M2 sequence, each oligonucleotide contained a specific restriction sites allowing by enzymatic digestion the discrimination between M2-CpG-modifications and on both extremities similar sequences for PCR amplification (; see Materials and Methods). After incubation, digestion with MluI, and quantitative PCR amplification, the level of methyl-, mutated-, and unmethylated-oligonucleotides retained by GST-ZBTB38-ZF was calculated. We observed that DNAs containing the M2 motif are better retained by ZBTB38 zinc fingers than by GST alone or by a mutated version of the zinc fingers (H491R, inactivating the C2H2 motif) that binds DNA poorly (). By qPCR, we could also demonstrate that ZBTB38-ZF binds more strongly the methylated M2-containing sequences than the mutated (fourfold increase) or unmethylated (threefold increase) M2-containing sequences (). These results indicate that the three central zinc fingers of ZBTB38 bind the M2 motif in vitro and that they bind the methylated form with higher affinity than the unmethylated or mutated forms. These results strongly suggest that in vivo ZBTB38 directly recognizes the methylated M2 sequence.
Large-scale analysis of Zinc Finger proteins did not identify other ZNF with high affinity to methylated sequences in vivo
We re-analysed ChIP-sequencing datasets for other ZNF factors to see whether other ZNF factors would show a similar trend as ZBTB38. We used a previously described methodology that counts the level of CpG methylation in a binding site to assess the relationship between CpG methylation and ZNF binding profiles [Citation13]. In the case of ZBTB38, this method indicates that the large majority (92.8%) of ZBTB38 sites have at least one CpG fully methylated (Table S3 and here after).
We first focused our analysis on KAISO/ZBTB33, a protein closely related to ZBTB38 [Citation29]. We found that KAISO binding sites identified in HCT116 cells are for the large majority devoid of CpG methylation as previously observed (Table S3) [Citation26]. We also observed that very few sites are common between KAISO (in HCT116) and ZBTB38 (in HeLa-S3) (n = 16). This indicates that despite very high amino-acid sequence similarity in their respective DNA-binding domains, KAISO/ZBTB33 and ZBTB38 have different DNA-binding properties in vivo, probably due to their divergence in their C-terminus ZNFs.
We then studied other ChIP-sequencing datasets of ZNF factors in HeLa-S3 cells and in 293 T cells [Citation62,Citation68,Citation69]. For most ZNF factors, there is a preference to bind DNA sequences with unmethylated CpG sites (Table S3). Among the few exceptions, we noticed ZFP57 that is already known for its ability to bind methylated-CpG sites in vivo [Citation19]. Additional ZNFs factors, including KRAB-containing zinc finger proteins ZNF284, ZNF287, ZNF445, and ZNF570, show high scores towards CpG-methylated sites in our analysis; still their scores are lower than ZBTB38 and ZFP57 scores (Table S3). This analysis provided further evidence that the genomic distribution and relationship towards DNA methylation shown by ZBTB38 are rare within the ZNF family.
We also observed that a limited number of TFs and ZNF factors exhibit preferential binding in CGI-shores compared to CpG islands (Table S4). Among them are the histone acetyltransferase P300 (or EP300), the transcription factor JUN, and the RNA polymerase III C1 subunit POLR3A (S4 Table). These different factors have been previously involved in the formation of DNA loops and in the bending of the DNA to favour promoter–enhancer communications [Citation62,Citation70,Citation71].
These analyses further support that ZBTB38 interaction with DNA in vivo is distinct from other ZNF TFs, and that it suggests an involvement in promoter–enhancer communications.
The M2 motif resembles an E2F4 consensus sequence but ZBTB38 targets are not cell cycle regulated
The M2 motif resembles the binding site of transcription factor E2F4, a transcriptional regulator of cell cycle genes in mammals, and the MluI cell cycle box, found within the promoters of G1/S activated genes in yeast [Citation72–74] (). We thus analysed the relationship between ZBTB38 and E2F4 ChIP-sequencing data in HeLa-S3 cells. We found no enrichment of E2F4 ChIP-sequencing tags at ZBTB38 sites containing M2 (). Conversely, we found no enrichment of ZBTB38 ChIP-sequencing tags at E2F4 binding sites containing M2 (). Since E2F4 binding to DNA is blocked by DNA methylation [Citation12,Citation75], we thought that methylation of the central CpG could discriminate E2F4 and ZBTB38 binding. We observed that most ZBTB38 but not E2F4 peaks have at least one fully methylated cytosine (). Furthermore, the average methylation level of M2 is 80% when bound by ZBTB38, while it is close to 0% when bound by E2F4 (). Because CpG methylation is often associated with chromatin compaction, we assessed the DNA nuclease I accessibility at ZBTB38 and E2F4 regions associated with M2. We observed that E2F4-bound regions are positive for DNAseI signals, while on the contrary, ZBTB38-bound regions are indistinguishable from the rest of the genome ().
Figure 5. The M2 motif resembles an E2F4 binding consensus site but ZBTB38 targets are not cell cycle regulated. (a) Resemblance between the ZBTB38 M2 consensus and the E2F4 consensus sites derived from ChIP-sequencing analysis. (b) Average tag intensity of ZBTB38 ChIP-sequencing, E2F4 ChIP-sequencing, and input samples at ZBTB38 (left panel) and E2F4 (right panel) binding regions defined by the M2 motif. (c) Proportion of ZBTB38 and E2F4 peaks containing one or more unmethylathylated CpG (0%) or one or more fully methylated CpG (100%). (d) Profile of CpG methylation at the M2 motif in ZBTB38 (left panel) and E2F4 (right panel) binding regions. (e) Average intensity of DNAseI-sequencing reads at ZBTB38-bound (left panel) and E2F4-bound (right panel) regions. (f) Overlay between cyclically expressed genes in HeLa-S3 and targets of ZBTB38 and E2F4. The number of E2F4 and ZBTB38 targets present on the array and the actual number of genes intersecting with the list genes cyclically expressed are indicated as cyclic gene/total gene over the corresponding bars. (g) Overlay between genes activated at the G1/S transition in HeLa-S3 cells and targets of ZBTB38 and E2F4. (h) Overlay between housekeeping genes and targets of ZBTB38 and E2F4. (i) Overlay between tissue-specific genes and targets of ZBTB38 and E2F4.
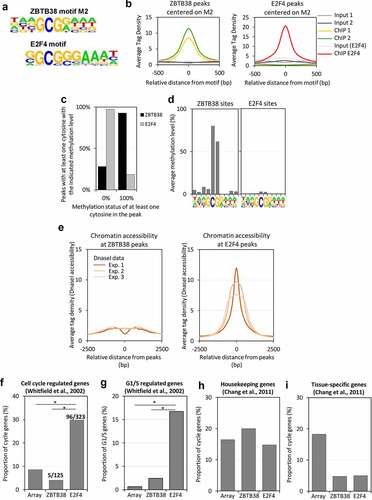
We next investigated whether ZBTB38 binding and M2 methylation show any overlap with genes that are transcriptionally regulated during the cell cycle. We overlapped the lists of ZBTB38 and E2F4 target genes with experimentally validated cell cycle regulated genes in HeLa cells [Citation76]. We observed a minimal overlap between cell cycle regulated genes and ZBTB38 targets (). On the contrary, E2F4 target genes are enriched for cell cycle genes in Gene Ontology analyses, which we could confirm by showing that, at least, one-third of cell-cycle regulated genes are bound by E2F4 in HeLa cells (). In addition, if we restrict our analysis to G1/S activated genes, we observed that only 2.8% of ZBTB38 targets are activated at the G1/S transition compared to 16.3% of E2F4 target genes (). We thus conclude that methylation of the M2 motif (and ZBTB38 binding) is correlated with the mode of expression of genes during the cell cycle. We then intersected the lists of ZBTB38 and E2F4 targets with a curated list of housekeeping and tissue-specific genes [Citation77]. We found that the proportions of housekeeping and tissue-specific genes associated with E2F4 and ZBTB38 binding at promoter are roughly similar (). This indicates that ZBTB38 binding and the methylation of M2 do not discriminate universally expressed genes and tissue-specific genes. On the contrary, ZBTB38 and E2F4 targets are different regarding how and when they are expressed during the cell cycle: ZBTB38 targets are mostly expressed throughout the cell cycle, while E2F4 targets are induced at the G1/S transition.
Discussion
ZBTB38 binds a consensus methylated DNA sequence in vitro and in vivo
CpG methylation controls, both positively and negatively, the affinity and the selectivity of hundreds of transcription factors for specific DNA sequences [Citation8,Citation13]. Herein, we characterize the binding profile of the human transcription factor ZBTB38 and demonstrate that two-thirds of ZBTB38 sites are dictated by the presence of a methyl-CpG site in a consensus sequence. However, in contrast to ZFP57, which binds a limited number of loci to maintain the mono-allelic expression of imprinted loci, ZBTB38 binds thousands of genomic sites in human cancer cells, and those are mostly located in the vicinity of active sites of transcription.
We identified two consensus motifs, M1 and M2, that often coincide at these ZBTB38 target sites. The M1 motif is very similar to a motif bound by the C-terminal ZNFs of ZBTB38 in vitro and in ChIP experiments [Citation30]. M2 is similar to an E2F binding site and an MluI restriction site. We provide evidence that the central zinc fingers of ZBTB38 recognize the M2 motif in vitro, with stronger affinity for the methylated form. We note that, even though ZBTB38 (XENON in rat) was initially found to bind a TpG-containing motif, we did not recover this motif in our analyses [Citation78].
The recognition of two motifs is not unprecedented in the family of ZNF factors [Citation79–81]. However, ZBTB38 does appear unique in the fact that it uses two different sets of ZF, located in the central part and in the C-terminal part of the protein, to selectively read out two distinct methylated DNA motifs. The presence of two methyl-CpG binding modules in ZBTB38 could easily explain the presence of the two motifs at most sites. It is also possible that protein–protein interactions with partners or homodimerization capabilities of ZBTB38 mediate binding specificities and recruitment onto the chromatin. Partners of ZBTB38 may dictate DNA binding preference. In vitro, E2F6 and E2F3 can bind specific methylated E2F-like sequences [Citation12,Citation16]. Importantly, E2F6 lacks the sequences involved in gene transactivation and in retinoblastoma (Rb) protein binding, and hence its transcriptional activity is not cell-cycle regulated [Citation82]. The methylation of the E2F/M2 motif might thus direct the co-recruitment of E2F family members that are not sensitive to Rb and cell-cycle regulation and that may cooperate with ZBTB38 to control expression at highly expressed loci. In , it is noteworthy to observe that E2F6 is the E2F factors that best overlap with ZBTB38, although it colocalizes with only 2% of ZBTB38 sites in the genome.
In vivo data show that the CpG sites are methylated at ZBTB38 binding sites (containing M1 and/or M2), and in vitro data show that ZBTB38 binds preferentially the methylated form over an unmethylated form of M2 (this study) and binds to the methylated form of M1 [Citation30,Citation31]. Yet, we could not manipulate DNA methylation using DNMT inhibitors to further document the role of CpG methylation in vivo. We observed that such inhibitors cause the degradation of ZBTB38 in a proteasome-dependent manner [Citation35].
ZBTB38 and DNA methylation controls gene expression during the cell cycle
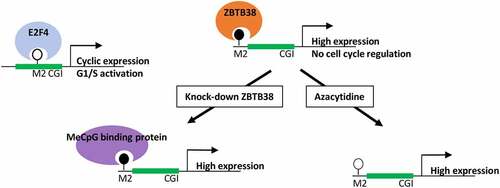
We demonstrated that ZBTB38 binding sites were enriched upstream of constitutively expressed genes, with general functions, including metabolism of RNAs, proteins, and DNAs. At these ZBTB38 sites, the DNA is either organized as nucleosomes or enriched for stably bound nuclear factors which may explain the low concordance of ZBTB38 binding with other transcription factors and the lack of E2F4 binding in these regions. We thus speculate that the methylation of the M2 motif, as well as its relative location to the transcription start site, may contribute to the discrimination between active expression at ZBTB38-associated genes and cell-cycle-regulated expression at E2F4 targets (). This is reminiscent of a previous study showing that an ultrastable methylated CpG site in CGI-shores is associated with genes exhibiting housekeeping functions [Citation83] or the role of methyl-CpG site to engage in promoter–enhancer communications [Citation28] In addition, in mice, CGI-shores associated with unmethylated CGI and highly transcribed genes actually present high level of DNA methyltransferase 3a, Dnmt3a, activity [Citation84]. It is thus likely that the methylation of the M2 motif and ZBTB38 binding defines a new promoter-proximal cis-regulatory module to protect genes from cell-cycle regulation by the Rb/E2F factors. Intriguingly, findings in plants have shown that methyl-DNA binding factors convey positive signal to promoters and that KLF4 activates transcription by binding to a methyl-CpG sites [Citation16,Citation28,Citation85].
Figure 6. Model of gene expression regulation by ZBTB38 and M2 motif methylation. We find that ZBTB38 binds many genomic sites that are methylated, contain an M2 motif, and are located outside of CpG islands. E2F4, in contrast, binds sites containing an unmethylated M2 motif, proximal to genes that are cell-cycle regulated.
Studies have shown various effect of ZBTB38 depletion on gene expression in different human cell lines. ZBTB38 knock-down barely alters gene expression in a human lymphoblastoid cell line [Citation86]. Only two genes, GDF15 and MMP7, were up-regulated upon ZBTB38 depletion and none were down-regulated at 48 hours [Citation86]. This is likely due to a redundancy with other methyl-CpG binding proteins or alternative mechanisms of gene expression regulation at play in the absence of ZBTB38 (). In contrast, in a neuroblastoma cell line and in a prostate cancer cell line, ZBTB38 depletion causes a more pronounced change in gene expression, although it is not known whether these genes are direct or indirect targets of ZBTB38 [Citation36,Citation44]. In a Zbtb38 mice knock-out model, the changes in gene expression pattern were also very modest, with 27 genes misregulated in B-cells lacking ZBTB38 [Citation87]. Our Gene Ontology functional analysis indicates that several targets of ZBTB38 are also regulated by MBD family members in HeLa cells, notably MeCP2. Intriguingly, MeCP2 binds DNA sequences containing a methylated E2F site in vitro and it exhibits transcription activating activity by promoting long-range interaction at active genes [Citation88,Citation89]. These observations provide further support that ZBTB38, and multiple methyl-CpG factors, might co-regulate ZBTB38 target genes expression by conveying information towards the promoter. It is not uncommon that paralog recognizes similar DNA binding targets, with more pronounced differences in specific chromatin and genomic context or the use of multiple ZNF domains [Citation90–92]. As such, ZBTB38 colocalizes at a subset of sites with CTCF and Cohesin complex factors that are known for modulating gene expression at a distance. Our work also indicates that ZBTB38 binds the M2 motif in its unmethylated version in vitro. This may have important biological and pathological consequences. We reported that ZBTB38 is an unstable protein with an estimated half-life of 4 hours in HeLa cells [Citation33]. Hence, the depletion of E3 ligase RBBP6 causes an accumulation of ZBTB38 protein and consequently defects in DNA replication. We, and others, provided evidence that ZBTB38 accumulation causes the repression of the cell-cycle gene MCM10, a well-characterized E2F target with both M1 and M2 containing sequences [Citation30,Citation33]. It is thus tempting to speculate that fluctuation of ZBTB38 protein abundance in different cell growth conditions or subsequently to abnormal DNA methylation changes will fine tune cell-cycle gene expression through the low affinity of ZBTB38 for the unmethylated E2F-motif. Alternatively, ZBTB38 binding at methylated and unmethylated sites may differentially alter DNA bending or chromatin organization [Citation92]. Multiple genetic variants in the ZBTB38 locus are associated with human stature, an increased risk of prostate cancer, myopia, and prion disease, and some of these features and pathologies are associated with increased expression of ZBTB38 mRNA in tissues [Citation46,Citation47,Citation53,Citation93,Citation94]. Whether these phenotypes rely on the control of cell cycle-gene expression is an attractive possibility that will deserve further investigation. A limitation of our work is that it was carried out in a single cancer cell line. Additional analyses in other cancer backgrounds, as well as in normal cells, will help shed light on the functions of ZBTB38 in physiological and pathological contexts. In addition, while our data are compatible with ZBTB38 repressing some of its target genes, they do not rule out the possibility that ZBTB38 activates other targets.
Other zinc finger proteins sense DNA methylation and bind in CGI shores
Initially described as a mark of transcriptional silencing memory, CpG methylation is emerging as a more complex mark. Herein, we identified a methylated E2F motif associated with several genes highly expressed and with metabolic functions that do not undergo cell cycle regulation. The CGIs associated with the promoter of these genes tend to be larger than regular CGI. In addition, these promoters exhibit intermediate levels of tissue specificity which typically require complex gene regulation mechanisms.
Our reanalysis of datasets of ZNF ChIP-sequencing produced by different labs identified a small number of other ZNFs that present binding features similar to ZBTB38. Among these, human ZFP57 shows a bias to methylated CpG sequences compared to unmethylated sequences consistent with previous work in mice [Citation19]. More intriguingly, ZNF284 and ZNF287 bind CGI-shore with higher frequency than CGI in 293 T cells and show preference for methylated CpG sequences. Applying similar search criteria as ZBTB38, we thus identify two additional candidate factors that may help convey information from proximal promoter regions. Study of these factors may help better comprehend the role of DNA methylation and the different layers of proteins involved in CpG methylation function.
Materials and methods
Cell lines
The HeLa S3 (XLP) cells stably expressing HA-Flag-ZBTB38, and its control Flag-HA cell line, were previously published. The HeLa-S3-HA-Flag-ZBTB38 cell line expressed the HA-Flag-ZBTB38 protein at lower level than the endogenous protein [Citation34]. Cell lines were maintained in DMEM (GlutaMAX, Glucose 4.5 g/L, and pyruvate) supplemented with 10% foetal bovine serum and penicillin/streptomycin. U2OS and HCT-116 cells were maintained in DMEM (GlutaMAX, Glucose 4.5 g/L, and pyruvate) and McCoy’s 5A Medium, respectively, supplemented with 10% foetal bovine serum and penicillin/streptomycin. Cells were mycoplasma-free, and their status was checked regularly with the VenorGeM kit according to the manufacturer’s protocol (Minerva Biolabs, Germany).
Chromatin immunoprecipitation
Cell were harvested, washed twice in PBS, and fixed in PBS – 1% formaldehyde for 10 minutes at room temperature. Formaldehyde was neutralized by adding glycine at 125 mM final concentration for 2 minutes. Fixed cells were extensively washed in cold PBS.
Nuclei were isolated by resuspending cells into cell lysis buffer (Hepes pH 7.8 25 mM, MgCl2 1.5 mM, KCl 10 mM, DTT 1 mM, NP-40 0.1%) and incubated for 10 minutes on ice, followed by centrifugation (5 minutes, 2000 rpm) and supernatant removal. Nuclei were resuspended in nuclear lysis buffer (Hepes pH 7.9 50 mM, NaCl 140 mM, EDTA 1 mM, Triton X100 1%, Sodium deoxycholate 0.1%, SDS 0.5%) and sonicated on the Bioruptor system (Diagenode, Belgium) in cold water to obtain a homogeneous population of 150–300 base pairs DNA fragments. Following centrifugation (10 min, 13,000 rpm), the supernatant was used for immunoprecipitation.
Immunoprecipitation was performed by preparing antibodies–beads complexes prior to mixing with the chromatin. Protein-A/G beads (Thermo Fisher Scientific, 88802) were incubated at 4°C for 4 hours with 2.5 µg antibodies against HA (Abcam, ab9110) plus 2.5 µg of antibodies against Flag (Sigma-Aldrich, F1804) or 2.5 µg of mouse IgG plus 2.5 µg of rabbit IgG (Thermo Fisher Scientific, 31903 and 10500C). Antibody–beads complexes were then incubated overnight with 60–80 µg of chromatin in IP buffer (Hepes pH 7.9 50 mM, NaCl 140 mM, EDTA 1 mM, Triton X100 1%).
Immunoprecipitates were washed three times with IP buffer: once with wash buffer (Tris pH 8.0 20 mM, LiCl 250 mM, EDTA 1 mM, NP-40 0.5%, Na-deoxycholate 0.5%) and twice with elution buffer (Tris pH 8.0 20 mM, EDTA 1 mM). Then, immunoprecipated chromatin was eluted by incubating beads with extraction buffer supplemented with 1% SDS at 65°C.
Chromatin was then reverse cross-linked by adding NaCl (200 mM) and Ribonuclease A (0.3 µg/µL) (Sigma-Aldrich, R4642) and incubating overnight at 65°C. The proteins were digested by adding Proteinase K (0.2 µg/µL) (Sigma-Aldrich, 3115844001) and incubating for 4 hours at 37°C. Finally, DNA was purified by phenol-chloroform extraction and salt precipitation.
Validation of ChIP-sequencing results by qPCR
We validated the ChIP data by real-time qPCR on a number of target sites on different biological samples that were not the one sequenced. The primers used are as follows: control negative region 5’-TGA CAG GTT ACT GCC TCT AGT TGA-3’ and 5’-AAG GAA CCA GGC TAT GAC TAA GAA-3’; DNM1L (ZBTB38 site) 5’-AGG AAG TCA CAC ACT TGC TCA CG-3’ and 5’-ACT ACA GGC ACC CAC CAC TA-3’; FIS1 (ZBTB38 site) 5’-ATG GGG AGC TTA GCA GTG AG-3’ and 5’-GCA AGG TAA TGC TCT GCC CT-3’; DNM1L downstream 5’-TTG AGC TGG GAG TTC GAG AC-3’ and 5’-CCC AGC TCT TCC CCT GTA A-3’; DNM1L upstream 5’-TCG CAG ACC AAG GAA ATG T-3’ and 5’-GGA GCG GTT TCC CCA TCA TT-3’; RBX1 (ZBTB38 site) 5’-CTG CAG ATG GGT CGG TTT CA-3’ and 5’-CCA CTC CGC ATT CCT CAG TT-3’; PRRC2C (ZBTB38 site) 5’-TAG TGG GGA GGG AGG TGT TC-3’ and 5’-GGT CTG AAC GAT CTT CCC CG-3’; PHYH (ZBTB38 site) 5’-TCG AAA GCA GCC TGG GTA AC-3’ and 5’-TAC ACC GTT CTC CTG CCT CA-3’; CTSD (ZBTB38 site) 5’-GGT CGA GGT GGG CAG ATT AC-3’ and 5’-CCA CCT CAG CCT CTC CAG TA-3’; PDPR (ZBTB38 site) 5’-TGA AAG CCA GAG GTG AGG TT-3’ and 5’-AAT TTT GTC ACT CCG GCT GG-3’; VHL (ZBTB38 site) 5’-AGC GTG ATG ATT GGG TGT TC-3’ and 5’-CTT GGC CTC TCA AAG TGC AG-3’; YTHDC1 (ZBTB38 site) 5’-GCT CTG TCG CTA GGT TGG AG-3’ and 5’-AGT CCC AGC TAC TCA GGA GG-3’; Intergenic region 1 (ZBTB38 site) 5’-CGT TTG TGT CTT TCC GCC AG-3’ and 5’-AGC GGG TAG TGT TAG GGG AA-3’; Intergenic region 2 (ZBTB38 site) 5’-CAC TAG CTC CTG GAT CTG TGC-3’ and 5’-GCA CAC TCA CTC AGC GTT CT-3’; Intergenic region 3 (ZBTB38 site) 5’-GCC CCA GAT AAT GAA GAC GC-3’ and 5’-CAC CAA CGT TCT TCC TTG CA-3’; Intergenic region 4 (ZBTB38 site) 5’-ATT AGA CGG GTG TGT GGC AT-3’ and 5’-CTT GGC TCA CTA CAA CAT CCG-3’; Intergenic region 5 (ZBTB38 site) 5’-CCA AGG GAC GGC TAG ATG AT-3’ and 5’-TGG TTA AAC GCG CAT AGG TG-3’; Intergenic region 6 (ZBTB38 site) 5’-CGC TGA ACT GCT CTG TTG TT-3’ and 5’-GGG AGG GGA CAT CAG AGA AG-3’; Intergenic region 7 (ZBTB38 site) 5’-GCC AGG CAC CTT TTC TTC TT-3’ and 5’-GGA ACA CAC CCA AAG CAG TT-3’; Intergenic region 8 (ZBTB38 site) 5’-CTT CTG CTT GAG GTT CGT TGA-3’ and 5’-ACG CCT GAA ACT TGT GCA AT-3’.
Libraries preparation and deep sequencing
Two validated ChIP samples and two input samples (matching the ChIP samples) were analysed by deep sequencing. The libraries were prepared by the sequencing platform of the ‘Centre National de Génotypage’ (CEA, Evry, France) according to the manufacturer’s recommendation using the NextFlex™ ChIP-sequencing kit (Bioo Scientific, 5143-01) and adaptors NextFlex™ ChIP-seq barcodes (Bioo Scientific, 514120). Twenty nanograms of ChIP and Input DNA was used as starting material, DNA extremities were repaired, and the resulting DNA PCR was amplified (11–12 cycles). DNA materials were then size-selected and purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, A63881) prior to analysis on a Illumina HiSeq 2000.
Quality control and analysis of the ChIP-sequencing data
We retrieved the ChIP-sequencing and Input-sequencing data in fasta format from the sequencing platform. Sequencing quality was assessed with fastqc tool (www.bioinformatics.babraham.ac.uk).
Mapping was performed using bowtie 1.1.2 [Citation95] on the human reference genome (hg19), allowing two mismatches per read and not keeping reads with multiple alignments. Duplicated reads were removed with SAMtools tool [Citation96]. The number of mapped reads considered for further analysis for the four samples ranged from 5.38 to 34.90 millions reads.
Reproducibility assessment was performed by comparison of reads mapped on a 500 base pair sliding window along chromosome 11 for the two HA-Flag ChIP replicates and by calculating the Pearson correlation coefficient.
A joint peak-calling analysis of the two ChIP replicates and the corresponding inputs was performed with CisGenome tool [Citation97], with a stringent cut-off at 5 and a maximal P-value set-up at 10−40 (except for Figure S1A for which default cut-off has been used and multiple P-values were tested). Coverage files (BedGraph format) were generated using the HOMER suite and visualized using the Integrated Genomics Viewer [Citation98,Citation99]. Histograms of average tag density were generated using the HOMER suite, and heatmaps were generated using SeqMINER [Citation100].
Genomic annotation of ZBTB38 binding regions and Gene Ontology analysis
Annotation of ZBTB38-bound regions was performed using the HOMER suite based on the default HOMER annotation database. The coordinates of transcription start sites (TSS) and tRNA loci from reference genome hg19 were obtained from HOMER database. Gene Ontology analysis of ZBTB38 target genes was performed using the PANTHER classification system and the molecular signatures database [Citation101,Citation102].
Bound DNA repeats identification
DNA repeats coordinates were downloaded from RepeatMasker on UCSC website [Citation103]. Genome coordinates of DNA repeats bound by ZBTB38 have been identified using a homemade code available at GitHub (https://github.com/ClaireMarchal). Percentage of repeats has been estimated by calculating the ratio of total length of DNA repeats bound by ZBTB38 on the total length of chromatin bound by ZBTB38. Venn diagrams have been generated by HOMER suite.
A pseudogenome has been generated with human DNA repeats sequences from RepeatMasker. Reads from fastq have been mapped on this pseudogenome, using bowtie 1.1.2 allowing two mismatches and without keeping reads mapped at more than one site, as previously performed [Citation104]. Duplicated reads have been removed using SAMtools, and total reads mapped on each DNA repeats have been calculated using SAMtools. The total number of reads mapped on each repeat has been normalized on the number of reads sequenced for each samples.
Transfection of siRNA molecules and doxorubicin treatment
Control scrambled siRNAs and siRNAs directed against ZBTB38 were purchased from Thermo Fisher Scientific and were validated in previous studies [Citation33–35]. siRNAs were transfected using the Neon® transfection system using the kit MAK10096 and the following optimized parameters given from the constructors for HeLa-S3, U2OS, and HCT-116 cells (Thermo Fisher Scientific). Cells were then seeded in six-well plate and treated for 6 hours with doxorubicin (Sigma-Aldrich, D1515) at final concentration of up to 1 μM and cell death measured by trypan blue scoring 18 hours later.
ChIP-sequencing datasets used in the study
ChIP-sequencing data analysed in this study were previously published, publicly available though GSE accession numbers, and in the case of ENCODE data the embargo release date has passed. All the ENCODE datasets were directly retrieved from the dedicated website (either as Fasta or BED format). Histone marks datasets generated by Broad Institute and RNA-sequencing datasets generated by Caltech Institute were downloaded from the UCSC website. ChromHMM and DNA binding domain datasets were downloaded from the UCSC website and from metatracks.encodenets.gersteinlab.org, respectively [Citation57,Citation58].
Other accession numbers for publicly available datasets are the following: phospho-Serine 5 Polymerase II (GSE71848) [Citation105], CAGE experiment (GSM849330) [Citation106], BRF1 and Polymerase III (GSE20309) [Citation107], HSF1 (GSE43579) [Citation108], DNaseI sensitivity [Citation62], MNase sensitivity (SRR029442) [Citation109], and Zinc Finger transcription factors profiling in HEK293 cells (GSE58341 and GSE78099) [Citation68,Citation69]. Of note, in the case of GSE58341 and GSE78099 solely, ZNF factors with more than 100 binding sites in the peak calling analysis were further considered.
Intersection between datasets peaks coordinates was considered positive, and highlighted as co-bound, when distance from centres of respective peaks is ≤100 bp.
De novo DNA motif discovery and analysis
Discovery and analysis of DNA motifs at ZBTB38-bound regions were performed using HOMER tool. We performed the analysis on DNA segments of 400 base pair centred on the centre of the ZBTB38 peak. We conducted a de novo motif discovery analysis and identified DNA motifs enriched at ZBTB38 sites compared to the rest of the genome by HOMER tool. We then compared the list of motifs with known motifs contained in the HOMER database of DNA motifs and obtained two lists: motif with known associated transcription factor and new motifs. Coordinates of peaks centred on motif have been generated using HOMER tool. Coordinates of genomic sequences with the motifs have been generated using FIMO tool [Citation110].
Methylation level correlation
Methyl-cytosine position on the whole genome was obtained from Bis-sequencing BedGraph: GSM949621 for HeLa cells [Citation65], GSM1465024 for HCT116 cells [Citation26], and GSE58341 for HEK293 [Citation68]. The average methylation at a given position in peaks centred on motif has been calculated with HOMER tool. The percentage of peaks with at least one cytosine at a given methylation status was obtained by merging the peaks with the list of cytosine coordinates of the given status.
MluI digestion test
Genomic DNAs from cells treated with siRNA molecules or constitutively expressing HA-Flag-ZBTB38 were prepared by adding SDS and proteinase K (Roche; 03115844001) directly to cell pellets overnight at 56°C prior to phenol/chloroform extraction and ethanol precipitation. Similar amount of genomic DNA (1 µg) from the different conditions was further digested overnight with BamHI (R3136S, New England BioLabs) and MluI (R3198S, New England BioLabs). Resulting digested DNA was PCR amplified using the One Taq Hot Start Quick-Load 2X master mix according to the manufacturer’s recommendation (New England Biolabs; M0481L) and run on a MasterCycler Nexus machine (Eppendorf). PCR was analysed on a 2% agarose gel and visualized using ethidium bromide.
DNAs from ChIP samples were analysed using a similar strategy. Following the ChIP protocol, purified DNAs were subjected to overnight MluI and BamHI digestion prior to PCR analysis.
The primer pairs used for these analyses are as follows: ZBTB38_Alu 5’-TCA GGA GTT CAC AAC CAG CC-3’ and 5’-TGG AGT GCA GTG GTG TGA TC-3’; ZBTB38_ENOG1 (exon1) 5’-GCG AGT CGT ACG TGC TGT-3’ and 5’-GGT AGT CGG CGT TGG TGG-3’; ZNF684 (promoter) 5’-GAG CTC CAC TGG CCT TAT GG-3’ and 5’-GCG GCG CTG ATT TGA AGA TT-3’; EXO1 (promoter) 5’-CCT ATG AGT TGG AAG CCG CA-3’ and 5’-TGA CCT TTC AAT TTG CGC GG-3’; Control (72 base pair amplicon; with no BamHI and MluI restriction site) 5’-CCC CCA TGA TTC ATT ACC TC-3’ and 5’-CCC ACC CAA ATC TCA TCT TG-3’.
GST-fusion purification and interaction with methylated DNA in vitro
GST and GST-fusion proteins were produced in E. coli cells. After centrifugation, cells were resuspended in lysis buffer (Hepes pH 7.5 25 mM, KCl 20 mM, EDTA 2.5 mM, DTT 1 mM, and Triton X-100 1%) and completed with PMSF and protease inhibitors (Roche mini-tablet EDTA-free). Cells resuspended in lysis buffer were sonicated in a Bioruptor (Diagenode) for a round of three times for 30 seconds. Lysates were kept on ice and 1/10e NaCl 5 M was added for 15 minutes of incubation. Lysates were centrifuged for 15 minutes at 4°C at 12,000 rpm. Supernatant containing GST-fusion proteins was incubated with glutathione-sepharose beads (Thermo Fisher Scientific; 16100) for an hour at 4°C on a wheel. After extensive washing of the beads with Tris HCl pH 8.0 20 mM, the presence of GST-fusion was monitored by western blot and protein levels were quantified.
On the next day, sepharose beads with GST, GST-ZBTB38-ZF, or GST-ZBTB38-ZF(H491R) were incubated with oligonucleotides as previously described [Citation111]. Briefly, a mix containing equimolar levels of methylated, mutated, and unmethylated oligonucleotides was freshly prepared. An aliquot from this mix was saved as the input, and the remaining mix was incubated with the proteins on the beads in the following binding buffer (Hepes pH 7.9 20 mM; KCl 50 mM, MgCl2 2 mM, EDTA 0.5 mM, Glycerol 10%, BSA 0.1 mg/ml, and DTT 2 mM). The mix was incubated for 5 minutes on the wheel at 4°C and washed 10 times for 5 minutes with the binding buffer solution. Bound DNAs were then digested by either EcoRI (New England Biolabs; R0101S), BamHI-HF (New England Biolabs; R3136S), or Kpn1 (Thermo Fisher Scientific; ER0521) in appropriate buffers. Digested DNAs and Input DNAs were then amplified by qPCR, and the relative amount of methylated, mutated, and unmethylated DNAs bound to each protein was determined using the Ct methodology.
The list of oligonucleotides is provided below. For each sequences, we indicated the location of the restriction site in bold; the cytosine, either mutated or methylated (5med), into brackets; and in italics the common sequences for PCR amplification. The sequences are as follows: Fis1_BamH1_meC (sense) 5’-TCC CAG ATC CCG TCA GTC TAG GAT CCA GCC CAA CAA CTT GGG AGG CCG [5MedC]GG [5MedC]GG GAA GAT CGC TGG AGG CCA GGA GTT CAA GAC CAG CCT GAG CAA CAT ACC CCT ACC TGA CTC TCC TCA-3’; Fis1_BamH1_meC (antisense) 5’-TGA GGA GAG TCA GGT AGG GGT ATG TTG CTC AGG CTG GTC TTG AAC TCC TGG CCT CCA GCG ATC TTC C[5MedC]G C[5MedC]G CGG CCT CCC AAG TTG TTG GGC TGG ATC CTA GAC TGA CGG GAT CTG GGA-3’; Fis1_EcoR1_C (sense) 5’-TCC CAG ATC CCG TCA GTC TA G AAT TC A GCC CAA CAA CTT GGG AGG CCG [C]GG [C]GG GAA GAT CGC TGG AGG CCA GGA GTT CAA GAC CAG CCT GAG CAA CAT ACC CCT ACC TGA CTC TCC TCA-3’; Fis1_EcoR1_C (sense) 5’-TGA GGA GAG TCA GGT AGG GGT ATG TTG CTC AGG CTG GTC TTG AAC TCC TGG CCT CCA GCG ATC TTC C[C]G C[C]G CGG CCT CCC AAG TTG TTG GGC TGA ATT CTA GAC TGA CGG GAT CTG GGA-3’; Fis1_Kpn1_T (sense) 5’-TCC CAG ATC CCG TCA GTC TAG GTA CCA GCC CAA CAA CTT GGG AGG CCG [T]GG [T]GG GAA GAT CGC TGG AGG CCA GGA GTT CAA GAC CAG CCT GAG CAA CAT ACC CCT ACC TGA CTC TCC TCA-3’ and Fis1_Kpn1_T (antisense) 5’-TGA GGA GAG TCA GGT AGG GGT ATG TTG CTC AGG CTG GTC TTG AAC TCC TGG CCT CCA GCG ATC TTC CC[A] CC[A] CGG CCT CCC AAG TTG TTG GGC TGG TAC CTA GAC TGA CGG GAT CTG GGA-3’.
Cell Cycle genes and housekeeping gene annotation
Periodically expressed genes in HeLa-S3 cells were retrieved from the web portal: http://genome-www.stanford.edu/human-cellcycle/hela/data.shtml [Citation76]. This study reports 1134 cell-cycle-regulated genes using a home-made microarray covering around 13,000 genes. We manually re-annotated and curated the list of genes to fit with human genome hg19 annotation. We then intersected these lists of genes with ZBTB38 and E2F4 target genes (defined by ZBTB38 and E2F4 binding at the TSS). The significance of the concordance was then determined using a hypergeometric test. A similar analysis was conducted using a curated list of housekeeping and tissue-specific genes based on the analysis of 1737 microarrays studies in 26 tissues [Citation77].
Graphics generation
Boxplots and corresponding statistical analysis have been generated using R (R Core Team, 2015). Scatter plots and Pearson correlation coefficients have been generated using R.
Accession codes
ZBTB38 ChIP-sequencing and Input samples have been submitted to the Gene Expression Omnibus repository (GSE108618).
Grant support
Laboratories of B.M. and P.A.D. are partners of Labex ‘Who am I?’ (ANR-11-LABX-0071 and ANR-11-IDEX-005-02). This work was supported by Electricité de France (RB2018-14), by Ligue contre le cancer Comité de Paris (RS11/75-8; RS12/75/95-21; RS13/75-59), by a FP7 Marie Curie action grant (PIRG07-GA-2010-268,448), and by Association pour la Recherche sur les Tumeurs de Prostate (ARTP, RAK22004KKA). Work of B.M. is supported by Fondation pour la Recherche Médicale (AJE20151234749), INCa-Plan Cancer (ASC15018KSA), and INSERM. Work of P.A.D. is supported by Agence Nationale de la Recherche (PRCI INTEGER ANR-19-CE12-0030-01), LabEx ‘Who Am I?’ (ANR-11-LABX-0071), Université de Paris IdEx (ANR-18-IDEX-0001) funded by the French Government through its ‘Investments for the Future’ program, Fondation pour la Recherche Médicale, and Fondation ARC (Programme Labellisé PGA1/RF20180206807).
Author contributions
B.M. and P.A.D. designed the experiments. C.M. and B.M. performed the experiments and analyzed the data. B.M., C.M., and P.-A.D. wrote the manuscript.
Supplemental Material
Download Zip (1.6 MB)Acknowledgments
We would like to thank Dr Jorg Tost, Dr Aurélie Bousard, and the sequencing facility at ‘Centre National de Génotypage’ (CEA, France) for handling the sequencing procedure. We thank Dr Slimane Ait-Si-Ali and Dr Laurianne Fritsch (UMR7216, Paris, France) for their help in establishing the cell line. We thank Alexey Ruzov (University of Nottingham, United Kingdom), Didier Trouche (Centre de Biologie Intégrative, Toulouse, France), and colleagues at UMR7216 ‘Epigenetics and Cell Fate’ and at Institut Cochin (INSERM U1016) for helpful comments and suggestions during the conduct of this study. C.M. was the recipient of a MESNT PhD fellowship and a Fondation pour la Recherche Médicale fellowship (FDT20150532354).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
ChIP-sequencing data are available on NCBI GEO with accession number GSE108618. Software and pipelines used for analysis are listed in Materials and Methods section, and references are cited accordingly. Homemade analysis are available on https://github.com/ClaireMarchal.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2022.2111135
Additional information
Funding
Related Research Data
References
- Tirado-Magallanes R, Rebbani K, Lim R, et al. Whole genome DNA methylation: beyond genes silencing. Oncotarget. 2017;8:5629–5637.
- Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607.
- Yamaguchi K, Chen X, Oji A, et al. Large-scale chromatin rearrangements in cancer. Cancers (Basel). 2022;14:2384.
- Baubec T, Defossez P-A. Reading DNA modifications. J Mol Biol. 2020;S0022-2836(20):30096.
- Fournier A, Sasai N, Nakao M, et al. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief Funct Genomics. 2012;11:251–264.
- Buck-Koehntop BA, Defossez P-A. On how mammalian transcription factors recognize methylated DNA. Epigenetics. 2013;8:131–137.
- Héberlé É, Bardet AF. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019;63:727–741.
- Xuan Lin QX, Sian S, An O, et al. MethMotif: an integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res. 2019;47:D145–D154.
- Wang G, Luo X, Wang J, et al. MeDReaders: a database for transcription factors that bind to methylated DNA. Nucleic Acids Res. 2018;46:D146–D151.
- Baubec T, Ivánek R, Lienert F, et al. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. 2013;153:480–492.
- Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159.
- Bartke T, Vermeulen M, Xhemalce B, et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484.
- Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–565.
- Kribelbauer JF, Laptenko O, Chen S, et al. Quantitative analysis of the DNA methylation sensitivity of transcription factor complexes. Cell Rep. 2017;19:2383–2395.
- Yin Y, Morgunova E, Jolma A, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356. DOI:10.1126/science.aaj2239.
- Hu S, Wan J, Su Y, et al. DNA methylation presents distinct binding sites for human transcription factors. eLife. 2013;2:e00726.
- Luo X, Zhang T, Zhai Y, et al. Effects of DNA methylation on TFs in human embryonic stem cells. Front Genet. 2021;12:639461.
- Zuo Z, Roy B, Chang YK, et al. Measuring quantitative effects of methylation on transcription factor-DNA binding affinity. Sci Adv. 2017;3:eaao1799.
- Quenneville S, Verde G, Corsinotti A, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–372.
- Liu Y, Olanrewaju YO, Zheng Y, et al. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014;42:4859–4867.
- Zandarashvili L, White MA, Esadze A, et al. Structural impact of complete CpG methylation within target DNA on specific complex formation of the inducible transcription factor Egr-1. FEBS Lett. 2015;589:1748–1753.
- Prokhortchouk A, Hendrich B, Jørgensen H, et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–1618.
- Buck-Koehntop BA, Stanfield RL, Ekiert DC, et al. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc Natl Acad Sci U S A. 2012;109:15229–15234.
- Sasai N, Nakao M, Defossez P-A. Sequence-specific recognition of methylated DNA by human zinc-finger proteins. Nucleic Acids Res. 2010;38:5015–5022.
- Ruzov A, Savitskaya E, Hackett JA, et al. The non-methylated DNA-binding function of Kaiso is not required in early Xenopus laevis development. Dev Camb Engl. 2009;136:729–738.
- Blattler A, Yao L, Wang Y, et al. ZBTB33 binds unmethylated regions of the genome associated with actively expressed genes. Epigenetics Chromatin. 2013;6:13.
- Qin S, Zhang B, Tian W, et al. Kaiso mainly locates in the nucleus in vivo and binds to methylated, but not hydroxymethylated DNA. Chin J Cancer Res Chung-Kuo Yen Cheng Yen Chiu. 2015;27:148–155.
- Oyinlade O, Wei S, Kammers K, et al. Analysis of KLF4 regulated genes in cancer cells reveals a role of DNA methylation in promoter- enhancer interactions. Epigenetics. 2018;13:751–768.
- Filion GJP, Zhenilo S, Salozhin S, et al. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol. 2006;26:169–181.
- Pozner A, Hudson NO, Trewhella J, et al. The C-terminal zinc fingers of ZBTB38 are novel selective readers of DNA methylation. J Mol Biol. 2017;430:258–271.
- Hudson NO, Whitby FG, Buck-Koehntop BA. Structural insights into methylated DNA recognition by the C-terminal zinc fingers of the DNA reader protein ZBTB38. J Biol Chem. 2018;293:19835–19843.
- de Dieuleveult M, Miotto B. DNA methylation and chromatin: role(s) of methyl-CpG-binding protein ZBTB38. Epigenetics Insights. 2018;11:2516865718811117.
- Miotto B, Chibi M, Xie P, et al. The RBBP6/ZBTB38/MCM10 axis regulates DNA replication and common fragile site stability. Cell Rep. 2014;7:575–587.
- Miotto B, Marchal C, Adelmant G, et al. Stabilization of the methyl-CpG binding protein ZBTB38 by the deubiquitinase USP9X limits the occurrence and toxicity of oxidative stress in human cells. Nucleic Acids Res. 2018;46:4392–4404.
- Marchal C, de Dieuleveult M, Saint-Ruf C, et al. Depletion of ZBTB38 potentiates the effects of DNA demethylating agents in cancer cells via CDKN1C mRNA up-regulation. Oncogenesis. 2018;7:82.
- Chen J, Xing C, Yan L, et al. Transcriptome profiling reveals the role of ZBTB38 knock-down in human neuroblastoma. PeerJ. 2019;7:e6352.
- de Dieuleveult M, Marchal C, Jouinot A, et al. Molecular and clinical relevance of ZBTB38 expression levels in prostate cancer. Cancers (Basel). 2020;12:E1106.
- Nishii T, Oikawa Y, Ishida Y, et al. CtBP-interacting BTB zinc finger protein (CIBZ) promotes proliferation and G1/S transition in embryonic stem cells via Nanog. J Biol Chem. 2012;287:12417–12424.
- Oikawa Y, Omori R, Nishii T, et al. The methyl-CpG-binding protein CIBZ suppresses myogenic differentiation by directly inhibiting myogenin expression. Cell Res. 2011;21:1578–1590.
- Kotoku T, Kosaka K, Nishio M, et al. CIBZ regulates mesodermal and cardiac differentiation of by Suppressing T and Mesp1 expression in mouse embryonic stem cells. Sci Rep. 2016;6:34188.
- Nishio M, Matsuura T, Hibi S, et al. Heterozygous loss of Zbtb38 leads to early embryonic lethality via the suppression of Nanog and Sox2 expression. Cell Prolif. 2022;55:e13215.
- Parsons S, Stevens A, Whatmore A, et al. Role of ZBTB38 genotype and expression in growth and response to recombinant human growth hormone treatment. J Endocr Soc. 2022;6:bvac006.
- Jing J, Liu J, Wang Y, et al. The role of ZBTB38 in promoting migration and invasive growth of bladder cancer cells. Oncol Rep. 2019;41:1980–1990.
- Ding G, Lu W, Zhang Q, et al. ZBTB38 suppresses prostate cancer cell proliferation and migration via directly promoting DKK1 expression. Cell Death Dis. 2021;12:998.
- Naciri I, Laisné M, Ferry L, et al. Genetic screens reveal mechanisms for the transcriptional regulation of tissue-specific genes in normal cells and tumors. Nucleic Acids Res. 2019;47:3407–3421.
- Clayton P, Bonnemaire M, Dutailly P, et al. Characterizing short stature by insulin-like growth factor axis status and genetic associations: results from the prospective, cross-sectional, epidemiogenetic EPIGROW study. J Clin Endocrinol Metab. 2013;98:E1122–1130.
- Gudbjartsson DF, Walters GB, Thorleifsson G, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–615.
- Kim -J-J, Lee H-I, Park T, et al. Identification of 15 loci influencing height in a Korean population. J Hum Genet. 2010;55:27–31.
- Wang Y, Wang Z, Teng Y, et al. An SNP of the ZBTB38 gene is associated with idiopathic short stature in the Chinese Han population. Clin Endocrinol (Oxf). 2013;79:402–408.
- Cho YS, Go MJ, Kim YJ, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–534.
- Okada Y, Kamatani Y, Takahashi A, et al. A genome-wide association study in 19 633 Japanese subjects identified LHX3-QSOX2 and IGF1 as adult height loci. Hum Mol Genet. 2010;19:2303–2312.
- Tanaka N, Koido M, Suzuki A, et al. Eight novel susceptibility loci and putative causal variants in atopic dermatitis. J Allergy Clin Immunol. 2021;148:1293–1306.
- Mead S, Uphill J, Beck J, et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet. 2012;21:1897–1906.
- Han X, Gharahkhani P, Mitchell P, et al. Genome-wide meta-analysis identifies novel loci associated with age-related macular degeneration. J Hum Genet. 2020;65:657–665.
- Mullin BH, Tickner J, Zhu K, et al. Characterisation of genetic regulatory effects for osteoporosis risk variants in human osteoclasts. Genome Biol. 2020;21:80.
- Kote-Jarai Z, Olama AAA, Giles GG, et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet. 2011;43:785–791.
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–216.
- Yip KY, Cheng C, Bhardwaj N, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48.
- Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186.
- Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295.
- Grandi FC, Rosser JM, Newkirk SJ, et al. Retrotransposition creates sloping shores: a graded influence of hypomethylated CpG islands on flanking CpG sites. Genome Res. 2015;25:1135–1146.
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
- Parelho V, Hadjur S, Spivakov M, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433.
- Zuin J, Dixon JR, van der Reijden MIJA, et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111:996–1001.
- Varshney D, Vavrova-Anderson J, Oler AJ, et al. SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation. Nat Commun. 2015;6:6569.
- Mariani CJ, Vasanthakumar A, Madzo J, et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014;7:1343–1352.
- Vural S, Palmisano A, Reinhold WC, et al. Association of expression of epigenetic molecular factors with DNA methylation and sensitivity to chemotherapeutic agents in cancer cell lines. Clin Epigenetics. 2021;13:49.
- Najafabadi HS, Mnaimneh S, Schmitges FW, et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol. 2015;33:555–562.
- Imbeault M, Helleboid P-Y, Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature. 2017;543:550–554.
- Fang F, Xu Y, Chew -K-K, et al. Coactivators p300 and CBP maintain the identity of mouse embryonic stem cells by mediating long-range chromatin structure. Stem Cells Dayt Ohio. 2014;32:1805–1816.
- Kerppola TK, Curran T. The transcription activation domains of Fos and Jun induce DNA bending through electrostatic interactions. EMBO J. 1997;16:2907–2916.
- Koch C, Moll T, Neuberg M, et al. A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science. 1993;261:1551–1557.
- Lowndes NF, Johnson AL, Breeden L, et al. SWI6 protein is required for transcription of the periodically expressed DNA synthesis genes in budding yeast. Nature. 1992;357:505–508.
- Grant GD, Brooks L, Zhang X, et al. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol Biol Cell. 2013;24:3634–3650.
- Campanero MR, Armstrong MI, Flemington EK. CpG methylation as a mechanism for the regulation of E2F activity. Proc Natl Acad Sci U S A. 2000;97:6481–6486.
- Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000.
- Chang C-W, Cheng W-C, Chen C-R, et al. Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PloS One. 2011;6:e22859.
- Kiefer H, Chatail-Hermitte F, Ravassard P, et al. ZENON, a novel POZ Kruppel-like DNA binding protein associated with differentiation and/or survival of late postmitotic neurons. Mol Cell Biol. 2005;25:1713–1729.
- Gordân R, Murphy KF, McCord RP, et al. Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biol. 2011;12:R125.
- Siggers T, Reddy J, Barron B, et al. Diversification of transcription factor paralogs via noncanonical modularity in C2H2 zinc finger DNA binding. Mol Cell. 2014;55:640–648.
- Badis G, Berger MF, Philippakis AA, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723.
- Trimarchi JM, Fairchild B, Verona R, et al. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc Natl Acad Sci U S A. 1998;95:2850–2855.
- Edgar R, Tan PPC, Portales-Casamar E, et al. Meta-analysis of human methylomes reveals stably methylated sequences surrounding CpG islands associated with high gene expression. Epigenetics Chromatin. 2014;7:28.
- Wu H, Coskun V, Tao J, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448.
- Harris CJ, Scheibe M, Wongpalee SP, et al. A DNA methylation reader complex that enhances gene transcription. Science. 2018;362:1182–1186.
- Cusanovich DA, Pavlovic B, Pritchard JK, et al. The functional consequences of variation in transcription factor binding. PLoS Genet. 2014;10:e1004226.
- Wong R, Bhattacharya D, Richard Y. ZBTB38 is dispensable for antibody responses. PloS One. 2020;15:e0235183.
- Yasui DH, Peddada S, Bieda MC, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421.
- Di Fiore B, Palena A, Felsani A, et al. Cytosine methylation transforms an E2F site in the retinoblastoma gene promoter into a binding site for the general repressor methylcytosine-binding protein 2 (MeCP2). Nucleic Acids Res. 1999;27:2852–2859.
- Patel A, Yang P, Tinkham M, et al. DNA conformation induces adaptable binding by Tandem zinc finger proteins. Cell. 2018;173:221–233.e12.
- Shen N, Zhao J, Schipper JL, et al. Divergence in DNA specificity among paralogous transcription factors contributes to their differential in vivo binding. Cell Syst. 2018;6:470–483.e8.
- Samee MAH, Bruneau BG, Pollard KS. A De Novo shape motif discovery algorithm reveals preferences of transcription factors for DNA shape beyond sequence motifs. Cell Syst. 2019;8:27–42.e6.
- Bonder MJ, Luijk R, Zhernakova DV, et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat Genet. 2017;49:131–138.
- Chandra A, Mitry D, Wright A, et al. Genome-wide association studies: applications and insights gained in ophthalmology. Eye Lond Engl. 2014;28:1066–1079.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinforma Oxf Engl. 2009;25:2078–2079.
- Ji H, Jiang H, Ma W, et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300.
- Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589.
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192.
- Ye T, Krebs AR, Choukrallah M-A, et al. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35.
- Mi H, Muruganujan A, Casagrande JT, et al. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566.
- Liberzon A, Subramanian A, Pinchback R, et al. Molecular signatures database (MSigDB) 3.0. Bioinforma Oxf Engl. 2011;27:1739–1740.
- Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinforma. 2009; Chapter 4: Unit 4.10.;. DOI:10.1002/0471250953.bi0410s25. 25(1).
- Deplus R, Blanchon L, Rajavelu A, et al. Regulation of DNA methylation patterns by CK2-mediated phosphorylation of Dnmt3a. Cell Rep. 2014;8:743–753.
- Liang K, Woodfin AR, Slaughter BD, et al. Mitotic transcriptional activation: clearance of actively engaged Pol II via transcriptional elongation control in mitosis. Mol Cell. 2015;60:435–445.
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108.
- Oler AJ, Alla RK, Roberts DN, et al. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol. 2010;17:620–628.
- Vihervaara A, Sergelius C, Vasara J, et al. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci U S A. 2013;110:E3388–3397.
- Auerbach RK, Euskirchen G, Rozowsky J, et al. Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci U S A. 2009;106:14926–14931.
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinforma Oxf Engl. 2011;27:1017–1018.
- Pierrou S, Enerbäck S, Carlsson P. Selection of high-affinity binding sites for sequence-specific, DNA binding proteins from random sequence oligonucleotides. Anal Biochem. 1995;229:99–105.