
ABSTRACT
Research on m6A-associated SNPs (m6A-SNPs) has emerged recently due to their possible critical roles in many key biological processes. In this sense, several investigations have identified m6A-SNPs in different diseases. In order to gain a more complete understanding of the role that m6A-SNPs can play in breast cancer, we performed an in silico analysis to identify the m6A-SNPs associated with breast cancer and to evaluate their possible effects. For this purpose, we downloaded SNPs related to breast cancer and a list of m6A-SNPs from public databases in order to identify which ones appear in both. Subsequently, we assessed the identified m6A-SNPs in silico by expression quantitative trait loci (eQTL) analysis and differential gene expression analysis. We genotyped the m6A-SNPs found in the in silico analysis in 35 patients with breast cancer, and we carried out a gene expression analysis experimentally on those that showed differences. Our results identified 981 m6A-SNPs related to breast cancer. Four m6A-SNPs showed an eQTL effect and only three were in genes that presented an altered gene expression. When the three m6A-SNPs were evaluated in the tissue sample of our breast cancer patients, only the m6A-SNP rs76563149 located in ZNF354A gene presented differences in allele frequencies and a low gene expression in breast cancer tissues, especially in luminal B HER2+ subtype. Future investigations of these m6A-SNPs should expand the study in different ethnic groups and increase the sample sizes to test their association with breast cancer and elucidate their molecular function.
Introduction
Breast cancer (BC) is a heterogeneous disease, both biologically and clinically. Worldwide, BC is the most commonly diagnosed cancer among women [Citation1]. Its incidence, mortality, and survival rates vary considerably between different geographical areas, which could be due to many factors such as population structure, lifestyle, genetic factors, and environment [Citation2]. Genetic factors play an important role in the aetiology of BC. Investigations carried out by genome-wide association studies (GWAS) have successfully identified many genetic markers like Single Nucleotide Polymorphisms (SNPs) that are associated with BC risk [Citation3]. Moreover, epigenetic changes have recently emerged as markers since they might be involved in cancer initiation and progression. Consequently, they might be useful for early detection, prognosis, and targeted therapy of cancer [Citation4].
Epitranscriptomics is still an unexplored area that, like epigenomics, may be of great interest to respond to the relationship between environmental factors and cancer, since it involves modifications in the RNAs that modulate gene expression in response to external factors. In this sense, N6-methyladenosine (m6A) is the most common post-transcriptional modification in eukaryotic messenger RNA (mRNA) [Citation5]. m6A is a reversible modification that occurs throughout the entire transcriptome into a consensus motif known as DRA*CH (D = G, A, or U; R = G, or A; A* = A methylatable, and H = A, C, or U) [Citation6,Citation7] mainly by the action of methyltransferases METTL3, METTL14, and WTAP (‘writers’), and can be reversed by the enzymes FTO and ALKBH5 (‘erasers’). In addition, multiple proteins, known as ‘readers,’ specifically bind to the methylated m6A position. The m6A plays a critical role in controlling mRNA structure, stability, splicing, export, localization, and translation [Citation5, Citation8–10].
Recent works indicate that SNPs located within or near m6A motifs may affect m6A methylation and subsequent related biological processes by altering the RNA sequence of the target site or key flanking nucleotides [Citation11,Citation12]. These genetic variations are known as m6A-associated SNPs (m6A-SNPs). In this context, these m6A-SNPs could have an eQTL (expression Quantitative Trait Loci) effect. The cis-eQTL effect on the expression of local genes may result from multiple causes, such as altering the level of m6A methylation, the binding of transcription factors and proteins, the mRNA stability, nuclear export, transcription elongation, splicing, or modifying the secondary structure of mRNA [Citation11, Citation13–15] and, consequently, influence the BC development and progression.
To date, several investigations have identified the role of m6A-SNPs in rheumatoid arthritis [Citation16], osteoporosis [Citation17,Citation18], coronary artery disease [Citation19], periodontitis [Citation13], development of sepsis [Citation14], adiposity [Citation20], lipid levels (cholesterol and triglycerides) [Citation21], Parkinson’s disease [Citation22], inflammatory bowel disease [Citation23], colorectal cancer [Citation24], type 2 diabetes [Citation25], and thyroid cancer [Citation26]. Recently, Xuan et al. (2021) [Citation27] have found eight m6A-SNPs that could have an effect on BC. In order to have a more complete understanding of the role those m6A-SNPs could play in BC, the present study aimed to carry out an independent parallel in silico analysis using publicly available data to identify new m6A-SNPs associated with this disease and evaluate their possible effects on gene expression.
Materials and methods
Determination of m6A-SNPs related to breast cancer
In order to determine the effect of the m6A-SNPs on BC, first, we had to identify the SNPs related to this disease. We searched for these SNPs through the NCI Genomic Data Commons (GDC, https://portal.gdc.cancer.gov/) [Citation28], the GWAS Catalogue (https://www.ebi.ac.uk/gwas/) [Citation29], and the SNPedia (https://www.snpedia.com/) [Citation30]. Additionally, we also searched the SNPs present in the microRNAs (miRNAs) related to BC in the following databases: the miRdSNP (http://mirdsnp.ccr.buffalo.edu/index.php) [Citation31], the miRNASNP (http://bioinfo.life.hust.edu.cn/miRNASNP/#!/) [Citation32], the MiRNA SNP Disease Database (MSDD, http://bio-bigdata.hrbmu.edu.cn/msdd/home.jsp) [Citation33], and the SomamiR (http://compbio.uthsc.edu/SomamiR/) [Citation34]. Subsequently, a list of m6A-SNPs and the associated data were downloaded from the m6Avar database (http://m6avar.renlab.org/help.html) [Citation12]. This database presents human m6A-SNPs with high, medium, and low confidence levels. Finally, we compared the list of the SNPs associated with BC and the m6A-SNPs with the purpose of identifying which ones appear in both.
Data on allele and genotype frequencies for the identified m6A-SNPs were obtained from Ensembl Genome Browser (https://www.ensembl.org/index.html) [Citation35].
Expression quantitative trait loci analysis for the m6A-SNPs related to breast cancer
To find out if the identified m6A-SNPs change the expression of local gene, we carried out a search of cis-eQTL analyses using public data from the GTEx Portal (https://gtexportal.org/home/) and the HaploReg browser (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php) [Citation36]. For that, we introduced the m6A-SNPs identified in the databases and observed if they presented an cis-eQTL effect in the breast tissue when we used the data from GTEx Portal and in different human tissues and cells when we used HaploReg browser. Moreover, we also searched for potential functions in gene expression regulation, such as altering protein binding or the regulation motif in HaploReg browser.
Differential expression analysis
For the m6A-SNPs that showed cis-eQTL effect, we investigated if their genes presented a differential expression in BC using the expression profile data available in GEO database (http://www.ncbi.nlm.nih.gov/geo) [Citation37]. For this purpose, we downloaded two datasets that contained the normalized gene expression signals for breast tissue (normal and tumour). The dataset GSE5764 contained data on gene expression levels of 10 individuals with BC (five with invasive ductal and five with invasive lobular breast carcinoma) and 20 corresponding to normal tissue (10 to normal ductal and 10 to normal lobular tissue) [Citation38]. Dataset GSE21422 contained data from five healthy tissue samples, nine mammary ductal carcinomas in situ, and five invasive ductal carcinomas [Citation39]. We analysed the differences in gene expression levels using the Mann–Whitney U-test through IBM SPSS Statistics 25.0 (SPSS Inc., IBM Corporation, New York, USA). We considered differential expression genes if they had p-values <0.05 and if they had been detected in at least one of the two studies. In the case of the m6A-SNPs in the miRNA that presented cis-eQTL effect, data about the number of samples, gene expression mean, and p-values were obtained from the ENCORI for RNA Interactomes database (http://starbase.sysu.edu.cn/panMirDiffExp.php) [Citation40].
Validation of the m6A-SNPs in patients with breast cancer
Patients
In order to validate the m6A-SNPs that showed a cis-eQTL effect and differences in gene expression after in silico analysis, we analysed breast tissues from BC patients. Samples and associated clinicopathologic data were provided by the Basque Biobank after approval by the Basque Clinical Research Ethics Committee (PI2018044). Frozen breast tumour and corresponding adjacent normal-appearing tissues (located at least 3 cm away from the site at which the tumour was sampled) were collected from 35 patients. All patients presented invasive ductal BC, a mean age of 59.77 ± 8.63, and 68.57% of them were menopausal. The tumour samples were classified into five molecular subtypes: seven samples belonged to luminal A, seven to luminal B HER2-, seven to triple-negative breast cancer (TNBC), eight samples to luminal B HER2+, and six to HER2+ subtype. Tissue histology, as well as molecular subtype classification, was confirmed by two experienced pathologists.
Total RNA was extracted from the tissue samples using TRIzol® Reagent (Invitrogen, Carlsbad, USA) and PureLink® RNA Mini Kit (Invitrogen) following the manufacturer’s instructions. The cDNA synthesis was performed through SuperScript® VILO™ Synthesis kit (Invitrogen) following the manufacturer’s instructions.
As a control group of gene frequencies, data of Iberian populations in Spain (IBS) for the m6A-SNPs were downloaded from the 1000 Genomes database (https://www.internationalgenome.org/home) [Citation41]. Only data from females were selected for this analysis.
Genotype determination
The genotypes for each m6A-SNP identified in the in silico analysis were determined for our patient samples by sequencing. The primers for each m6A-SNPs were designed using the PerlPrimer v1.1.21 program [Citation42]. All primer pairs were verified using NCBI’s Primer-BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and synthesized by Metabion (Planegg, Germany). The sequences of the primers, as well as the size of the amplified, and the annealing temperature can be seen in Table S1. We performed Real-time PCR (qRT-PCR) amplification on a C1000 Thermal Cycler coupled to a CFX96 Real-Time PCR Detection System (Bio-Rad, California, USA). Reactions were carried out in a final volume of 5 μL and contained: 0.50 μL of each forward and reverse primers (1 μM), 2.50 μL of SsoFast EvaGreen Supermix (Bio-Rad), 1.00 μL of cDNA template at 2.50 ng/μL, and 0.50 μL of milliQ water (Merck Millipore, Germany). The qRT-PCR conditions were as follows: an initial denaturalization at 98°C for 2 min, followed by 35 cycles at 98°C for 10 s and the annealing temperature for each set of primers for 30 s. Subsequently, the amplicons were purified with ExoSAP-ITTM Express (Applied Biosystems, Waltham, USA). We performed sequencing reactions with BigDye® TerminatorTM v1.1 Cycle Sequencing Kit (Applied Biosystems) and we purified sequencing products with BigDye® XTerminatorTM Purification Kit (Applied Biosystems), following the manufacturer’s instructions. We performed capillary electrophoresis on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems). We analysed sequencing reaction products with the Sequencing Analysis software v.5.2 (Applied Biosystems) and we determined the genotype of the patients for each m6A-SNPs by ChromasPro software v.2.6.5 (Technelysium Pty Ltd, Brisbane, Australia).
Gene expression analysis
We carried out a gene expression analysis for the m6A-SNPs that presented significant differences in terms of genotype and/or allele frequencies. We conducted qRT-PCR of gene expression described in the previous section. All qRT-PCR analyses were performed in triplicate for each sample. The relative gene expression levels were estimated through the 2−ΔΔCt method [Citation43] using as reference genes GAPDH, β-ACTIN, and GUSB. We choose these reference genes since they are the most commonly used in this type of studies. However, recently, some authors reported variability between different tissues and conditions for these genes [Citation44], which would make them not optimal as reference genes. Therefore, as other authors did to get over this drawback, we used an average of their Ct values [Citation45,Citation46]. Table S1 shows information about the sequence, annealing temperature, amplicon size, and efficiency of the reference genes primers.
Statistical analysis
Allele and genotype frequencies were determined for each m6A-SNP. Differences between the group of patients with BC and the control group from the IBS population were analysed using Pearson’s chi-square (χ2) test. On the other hand, the differences in gene expression between the tumour breast tissue of the patients and their corresponding adjacent normal breast tissues were analysed using the Mann–Whitney U-test. The variation between different tumour subtypes was determined using the Kruskal–Wallis H test. We used IBM SPSS Statistics 25.0 software (SPSS Inc., IBM Corporation, NY, USA) for all tests. We took values of p < 0.05 as statistically significant except in the Kruskal–Wallis H test where a Bonferroni correction was applied (with a statistically significant cut-off value of p < 0.005).
Results
m6A-SNPs related to breast cancer
In total, we identified 981 BC-related m6A-SNPs after comparing 69,713 SNPs related to BC (65,331 from GDC, 1,775 from GWAS Catalogue, 71 from SNPedia, 1,864 from miRdSNP, 42 from miRNASNP, 43 from MSDD, and 587 from SomamiR) with 301,875 m6A-SNPs in RNA (301,529 m6A-SNPs present in mRNA and 346 in miRNA) (). From these 981 m6A-SNPs, there were 26 that belonged to the high confidence category, 147 belonged to the medium confidence category, and 808 to the low confidence category. These different confidence categories relate to the identification method for the m6A-SNPs [Citation12]. PA-m6A-seq (photo-crosslinking-assisted m6A-seq) and miCLIP (m6A individual-nucleotide-resolution crosslinking and immunoprecipitation) methods provide high confidence data. On the other hand, MeRIP-seq (m6A-specific methylated RNA immunoprecipitation with next-generation sequencing) provides medium-confidence. Finally, a genome-wide prediction based on the Random Forest algorithm provides low confidence.
Figure 1. Flow chart of study design and the main results. IBS: Iberian Population in Spain from 1000 Genomes Project.
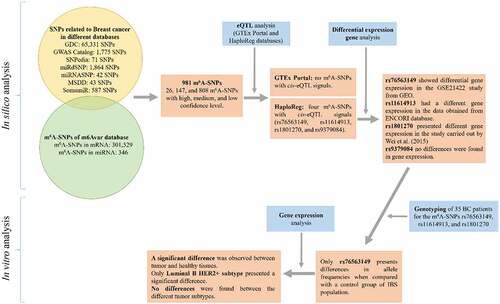
Of the 981 m6A-SNPs, all but one were in mRNA. The other one was in miRNA. It is well known that most m6A methylated sites are in the 5’ untranslated region (UTR), in long exons, near the stop codon, in the 3’-UTR, and non-coding RNAs (ncRNA) [Citation8, Citation47–49]. In our study, we found that most of the m6A-SNPs, i.e., SNPs located within or near m6A motifs, are distributed in the coding sequence (90.62%), 6.63% are in the 3’-UTR, 2.65% in the 5’-UTR, and only 0.10% in the miRNA.
Cis-eQTL analysis
To further explore the potential effect of the 981 m6A-SNPs in BC, we investigated whether they present cis-eQTL effect. Considering the data from the GTEx Portal for BC samples, none of these m6A-SNPs presented a cis-eQTL effect. However, when we searched in the HaploReg database, four m6A-SNPs (rs1801270, rs9379084, rs76563149, and rs11614913) showed a cis-eQTL effect in different cells or tissues ().
The m6A-SNP rs1801270 (C/A) presents cis-eQTL effect in fibroblasts [Citation50]. This m6A-SNP is a missense variant localized in the Cyclin-Dependent Kinase Inhibitor 1A gene (CDKN1A, gene ID: 1026) on chromosome 6 (). Variant A of this m6A-SNP is found in an incomplete DRACH motif (AGA*CG, where the A* is the methylated adenosine). Moreover, this m6A-SNP is near one complete DRACH motif and one incomplete one (at 16 nucleotides upstream and at five nucleotides downstream in the RNA, respectively). The frequency of the A allele varies depending on the population, between 0.067 and 0.488 for European (EUR) and East Asian (EAS) populations, respectively (). On the other hand, according to the ENCODE transcription factor ChIP-seq datasets [Citation51] obtained in HaploReg database, this m6A-SNP could also be involved in gene expression regulation by altering the regulatory motif, i.e., a binding site of transcription factors, and changing the binding site of proteins POL2 and ZBTB33.
Table 1. List of identified m6A-SNPs associated with BC that presented cis-eQTL effect.
Table 2. Alleles and genotypes frequencies for the four m6A-SNPs in different populations. AFR: African; AMR: American; EAS: East Asian; EUR: European; and SAS: South Asian.
The other m6A-SNP is rs9379084 (G/A) that presented a cis-eQTL effect in whole blood. This m6A-SNP is a missense variant found in the Ras Responsive Element Binding Protein 1 gene (RREB1, gene ID: 6239) on chromosome 6 (). This m6A-SNP is part of an incomplete DRACH motif (TGA*AC). Near this m6A-SNP, a complete DRACH motif can also be observed at seven nucleotides downstream and two incomplete DRACH motifs localized at 14 and 25 nucleotides upstream in the RNA. shows the occurrence for allele A, with a maximum frequency of 0.159 in EAS populations and the minimum frequency of 0.006 in African (AFR) populations. Moreover, this m6A-SNP alters a VDR motif, which could result in altered regulation of gene expression ().
The m6A-SNP rs76563149 (G/T) showed a cis-eQTL effect in different cells and tissues: fibroblast, adipose tissue, artery, oesophagus, lung, skeletal muscle, tibial nerve, stomach, testis, thyroid, and whole blood. This m6A-SNP appears in the 5’-UTR region of the Zinc Finger Protein 354A gene (ZNF354A, gene ID: 6940) on chromosome 5 (). Since this gene is oriented in the minus strand, the variants of this m6A-SNP in the mRNA will be C/A. Variant A of this m6A-SNP belongs to an incomplete DRACH motif (GAA*GC). In this case, the frequency of allele A is in the range of 0.081 to 0.368 for AFR and South Asian (SAS) populations, respectively (). Moreover, this m6A-SNP could be involved in gene expression regulation by altering a regulatory motif and the binding of 11 proteins ().
Finally, the m6A-SNP rs11614913 (C/T) showed cis-eQTL effect in different cells and tissues: fibroblast, tibial artery and nerve, skin, and whole blood. The rs11614913 is present in the microRNA 196a-2 gene (MIR196A2, gene ID: 406,973) between Hox10 and Hox9 genes on chromosome 12 (). This m6A-SNP is located near two complete DRACH motifs (at 4 and 19 nucleotides upstream in the RNA) and one incomplete DRACH motif (at 11 nucleotides upstream in the RNA), and, therefore, it could influence the m6A modification (). In this case, the frequency of allele C is in the range of 0.458–0.860 for EAS and AFR populations, respectively (). The change from the U to the C variant does not alter the secondary structure of this miRNA (). However, this m6A-SNP could also be involved in gene expression regulation by altering a regulatory motif.
Figure 2. A graphical representation of the miRNA structure obtained from the UNAFold Web Server (http://www.unafold.org/mfold/applications/rna-folding-form.php) [Citation52]. The m6A-SNP rs11614913 (C/U) is found inside the red circle and it may influence the m6A modification of the complete DRACH motif (green boxes) and incomplete one (blue box). (a) Representation of the miRNA structure with the variant C for the m6A-SNP rs11614913. (b) Representation of the miRNA structure with the variant U for the m6A-SNP rs11614913.
![Figure 2. A graphical representation of the miRNA structure obtained from the UNAFold Web Server (http://www.unafold.org/mfold/applications/rna-folding-form.php) [Citation52]. The m6A-SNP rs11614913 (C/U) is found inside the red circle and it may influence the m6A modification of the complete DRACH motif (green boxes) and incomplete one (blue box). (a) Representation of the miRNA structure with the variant C for the m6A-SNP rs11614913. (b) Representation of the miRNA structure with the variant U for the m6A-SNP rs11614913.](/cms/asset/bfceb457-022a-415b-b9b7-3e7fa6b6dcd0/kepi_a_2111137_f0002_oc.jpg)
Differential expression analysis
For the four genes, in which the m6A-SNPs showed cis-eQTL effect, we analysed the gene expression level in breast tissue. In the cases of the m6A-SNPs rs1801270, rs9379084, and rs76563149 that are present in the CDKN1A, RREB1, and ZNF354A genes, respectively, the gene expression levels were obtained from two gene expression microarray data sets from GEO database. In the case of the m6A-SNP rs11614913 present in the MIR196A2 gene, the gene expression level was obtained from the ENCORI database. We found that only ZNF354A and MIR196A2 present a differential gene expression. With respect to ZNF354A, differential expression was obtained in the GSE21422 study (p = 0.019) that analysed five healthy tissue samples and 14 carcinoma samples (9 for ductal carcinomas in situ and 5 for invasive ductal carcinomas). For MIR196A2, a significant difference in gene expression was found when comparing 1,085 samples of breast invasive carcinoma tissue with 104 normal tissues (p = 4.0e-14).
Although CDKN1A did not present significant differences in gene expression in the two datasets analysed, the study of Wei et al. (2015) [Citation53] suggested otherwise. They analysed the gene and protein expression of CDKN1A in 65 breast tumour samples and their corresponding noncancerous adjacent tissues and their results showed that the differences in gene and protein expression of CDKN1A were statistically significant (p < 0.01). For RREB1, no statistical significance was found after analysing the data from GEO database, and we did not find any related study of gene expression.
Validation of the m6A-SNPs in patients with breast cancer
We determined the genotype of 35 BC patients for the three m6A-SNPs (rs1801270, rs76563149, and rs11614913) in order to calculate the genotype and allele frequencies. We compared these frequencies with data available in the 1000 Genomes database for females in the IBS population. In the group of patients with BC, the percentage of individuals with the A allele was 11.40% (AA = 5.70% and AC = 5.70%) for the m6A-SNP rs1801270 and 65.70% (AA = 14.30% and AC = 51.40%) for the m6A-SNP rs76563149 (). In the case of the m6A-SNP rs11614913, the percentage of individuals that presented the C allele, that is the variant associated with BC in different studies [Citation54–58], was 80.00% (CC = 51.40% and CT = 28.60%). On the other hand, in the IBS population, 17.00% (AA = 0% and AC = 17.00%) and 45.30% (AA = 3.80% and AC = 41.50%) showed the A allele for the m6A-SNPs rs1801270 y rs76563149, respectively, while the 83.00% (CC = 43.40% and CT = 39.60%) had the C allele for the m6A-SNP rs11614913 (). No statistically significant differences were detected between the patients with BC and the IBS population when we compared the frequencies of the genotypes for the three m6A-SNPs (). However, when we compared allele frequencies, statistically significant differences were observed in the m6A-SNP rs76563149 (p = 0.029).
Table 3. Alleles and genotypes frequencies and χ2 test for the patients with BC and female from the IBS population. We took values of p < 0.05 as significant (indicated in bold in the table below).
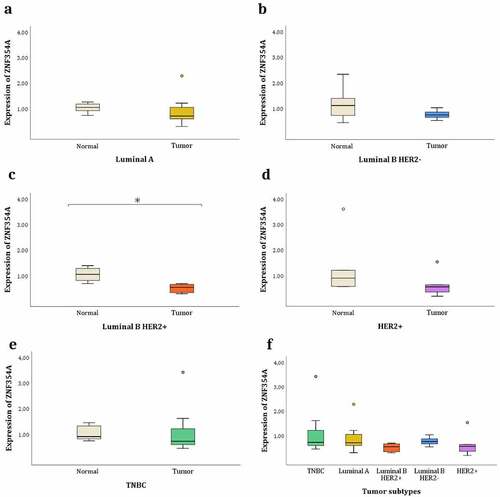
Since the m6A-SNP rs76563149 located in the ZNF354A gene was the only one that presented significant differences, we proceeded to evaluate its expression in the tumour breast tissue and its corresponding adjacent normal tissue for our BC patients. A significant difference was observed between the tumour and normal breast tissue (p < 0.001), with a reduced expression in the tumour samples (0.785 ± 0.613 in tumour breast tissue and 1.095 ± 0.564 in normal breast tissue). Given that BC tumorigenesis/prognosis is affected significantly by menopausal status, we evaluated the ZNF354A expression considering this variable. No significant difference was found when compared the menopausal status of the patients (p = 0.522). Subsequently, we evaluated the ZNF354A expression taking into account the molecular subtypes. In this case, only the luminal B HER2+ subtype presented a significant difference (p = 0.001), with a reduced expression in the tumour breast tissue ( and ). No significant differences were found when comparing the tumour subtypes with each other (p = 0.117) ( and ).
Table 4. Average of relative gene expression for the m6A-SNP rs76563149 localized in ZNF354A gene with its standard deviation (SD) in the tumour breast tissue and its corresponding adjacent normal breast tissue and the p-values obtained for the Mann–Whitney U and Kruskal–Wallis H tests. We took p-values of <0.05 as significant, except in the Kruskal–Wallis H test, where a p-value of <0.005 was considered as significant (indicated in bold in the table below).
Figure 3. Relative expression levels of the ZNF354A gene. The fragment analysed contains the m6A-SNP rs76563149. Gene expression between each tumour subtype and its corresponding adjacent normal tissues: (a) TNBC. (b) Luminal A. (c) Luminal B HER2 +. (d) Luminal B HER2-. (e) HER2 +. (f) Gene expression between the different tumour subtypes. * P-value <0.05. The circles are outlier.
Discussion
Recently, m6A-SNPs have begun to be considered an important class of functional genetic variants that may provide new insights into the onset, development, and progression of diseases. In this regard, m6A-SNPs may be involved through multiple effects. Among others, they can affect the levels of m6A methylation, affect the binding of transcription factors and proteins, modify the secondary structure of mRNA, and affect the production of mature mRNA with correct biological function when they are found at splice sites [Citation13,Citation14]. For this reason, several studies have been carried out in order to identify m6A-SNPs and evaluate the role that they play in different diseases [Citation13, Citation14, Citation16, Citation17, Citation19–27].
In the present study, we found 981 m6A-SNPs after comparing the list of SNPs related to BC with the list of m6A-SNPs. Of these, only four m6A-SNPs presented a cis-eQTL effect: rs1801270 in CDKN1A, rs76563149 in ZNF354A, rs9379084 in REEB1, and rs11614913 in MIR196A2, and all of them, except rs9379084, showed an altered gene expression in breast tissue when using data from GEO and ENCORI databases and Wei et al. (2015) [Citation53]. These m6A-SNPs are different from those found by Xuan et al. (2021) [Citation27], so our study allows us to expand the list of m6A-SNPs that could be involved in BC.
m6A-SNP rs9379084 present in the RREB1 gene did not show differences in gene expression after analysing the data from the GEO database. However, the protein encoded by this gene acts in different biological processes, such as DNA damage repair, cell growth and proliferation, cell differentiation, fat development, glucose balance fasting, zinc transport, and transcriptional regulation [Citation59], which may play a role in cancer. In fact, RREB1 has been involved in the development of several cancers and other diseases [Citation59], indicating that it may also be related to the development of BC although the effects of the methylation of this m6A-SNP would be involved in processes subsequent to gene transcription.
To understand the potential role of these three m6A-SNPs identified in the present study and their corresponding genes in BC, we investigated their biological functions. The m6A-SNP rs11614913 is in the MIR196A2 gene. MIR196A2 encodes two different mature miRNAs, miR-196a-5P and miR-196a-3P, from the same stem-loop and rs11614913 is in miR-196a-3P. This mature miRNA regulates the expression of multiple target genes, mainly by pairing to the 3′-UTR region of mRNA, and affecting either stability, translational inhibition, or degradation of mRNA. Indeed, under different conditions, this miRNA may function as an oncomiR [Citation60]. We genotyped this m6A-SNP in our BC patients and we found no significant difference in relation to the IBS population (). These results are in agreement with those obtained by other authors in Caucasian populations [Citation54, Citation61, Citation62]. However, analysis of Asian populations shows significant associations where variant C increases the BC risk [Citation54–58, Citation60]. Therefore, we believe that it is important to evaluate the role of this m6A-SNP in different ethnic groups. On the other hand, as shown in , the presence of the C or U variant does not alter the secondary structure of this miRNA, and since this m6A-SNP is near four m6A positions, future work should address how this m6A-SNP may affect the m6A modification and its influence on BC.
The m6A-SNP rs1801270 is present in the CDKN1A gene. The expression of CDKN1A is controlled by the tumour suppressor protein p53 and thus it links DNA damage to cell cycle arrest. The protein encoded by this gene is involved in multiple functions. On the one hand, the CDKN1A protein binds and inhibits the activity of the cyclin-dependent kinases, functioning as a regulator of cell cycle progression at G1 when the DNA is damaged [Citation63,Citation64]. On the other hand, this protein interacts with proliferating cell nuclear antigen (PCNA) and plays a regulatory role in S phase DNA replication and DNA damage repair. Moreover, it also plays a role in apoptosis [Citation65]. Given the involvement of CDKN1A in these cellular processes, different works have studied its role in the development, progression, and therapy of cancer [Citation66,Citation67]. In this sense, in comparison with their corresponding noncancerous adjacent tissues, BC samples show an increased mRNA and protein expression of CDKN1A [Citation53]. On the other hand, several studies have associated the m6A-SNP rs1801270 with different types of cancer and reported that the presence of the A allele is a risk factor [Citation68–73]. However, the studies carried out on BC in the European population [Citation74,Citation75] did not confirm its association with variant A, whose frequency in this population is the lowest (). This corresponds to the experimental results obtained in the present study for patients with BC. In our patients, the presence of the A allele did not present significant differences with the IBS population. Therefore, this m6A-SNP should be evaluated in other populations where its frequency is higher, e.g. in the EAS, AFR, or AMR populations, in order to determine its involvement in BC. Despite not finding differences, we hypothesize that since m6A-SNP rs1801270 is a missense variant found in the coding region, this m6A-SNP could not only have an amino acid change but could also affect the RNA processing by altering the stability, nuclear export, transcription elongation, or splicing of the CDKN1A mRNA in BC [Citation11, Citation13–15]. Moreover, this m6A-SNP could regulate the translation by reducing the kinetics of codon-anticodon pairing [Citation15] or resolving secondary structures of mRNA [Citation15,Citation76].
The m6A-SNP rs76563149, located in the ZNF354A gene, belongs to the Krüppel‐associated box domain zinc finger proteins (KRAB-ZNF), one of the most abundant families of epigenetic repressors [Citation77]. KRAB-ZFPs silence transcription by the deposition of repressive chromatin marks [Citation77]. Moreover, KRAB-ZFPs may also facilitate DNA methylation adjacent to their binding motifs, especially in cells with stem cell phenotype [Citation77]. To date, the role of rs76563149 remains unexplored. This m6A-SNP is part of a DRACH motif and, consequently, its presence could influence m6A methylation. We genotyped this m6A-SNP in our BC patient samples and our results showed a higher frequency of allele A in the patients than in the females of IBS population (). As a result, we carried out a gene expression analysis and found low levels of expression in tumour breast tissues when compared with their corresponding adjacent normal breast tissues. When we carried out the gene expression analysis taking into account the molecular subtypes, the luminal B HER2+ subtype was the only one that presented significant differences. These results suggest that m6A-SNP rs76563149 could alter the m6A methylation and it may affect the expression of ZNF354A. In this way, it could affect the splicing, nuclear export, and stability of mRNA, as well as the binding of reader proteins [Citation76]. Indeed, it has been observed that this m6A-SNP could alter the binding site of transcription factors and affect the binding of 11 proteins (). Moreover, a decrease of ZNF354A expression in tumour BC tissues could influence cell proliferation, migration, invasion, and apoptosis, like other best-studied members of KRAB-ZNF family [Citation78]. Further studies are needed to evaluate the role that ZNF354A may play in luminal B HER2+ subtype because no data was found in the literature. However, since this molecular subtype is characterized as oestrogen receptor-positive (ER+), we hypothesize that ZNF354A could act as a transcriptional regulatory factor that modulates the ER pathway, like ZNF496 [Citation79,Citation80]. This member of KRAB-ZNF family interacts with the DBD domain of ER type α (ER-α), inhibiting the binding of ER-α to the oestrogen response element, which selectively reduces the expression of ER-α target genes and inhibits the growth of ER-α-positive BC cells [Citation79,Citation80]. However, in the case of ZN354A the low expression would produce an opposite effect, increasing the growth of ER-α-positive BC cells. In addition, the effect of ZNF354A could be enhanced when the tumour is not only ER+ but also human epidermal growth factor receptor 2-positive (HER2+), as is the case of luminal B HER2 +.
While in this work we have focused on the m6A-SNPs that can influence gene expression and are statistically significant, the remaining m6A-SNPs that did not present these characteristics should not be ruled out. The m6A methylation could have more subtle effects that imply a fine modulation of gene expression and, in this case, the influence of methylation could not be observed by the analysis of eQTL. For this reason, more experimental studies should be carried out in order to confirm whether these 978 m6A-SNPs are involved in BC.
In conclusion, our in silico analysis identified three m6A-SNPs that may be associated with BC. These m6A-SNPs, rs11614913, rs1801270, and rs76563149 are in the MIR196A2, CDKN1A, and ZNF354A genes, respectively. After in vitro analysis, only the m6A-SNP rs76563149 shows significant differences. Future investigations of these m6A-SNPs should expand the study in different ethnic groups and increase the sample sizes to test their association with BC and elucidate their molecular function.
Supplemental Material
Download MS Word (12.2 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors, MMP and FO, upon request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2022.2111137
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Momenimovahed Z, Salehiniya H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer (Dove Med Press). 2019;11:151–164.
- Lilyquist J, Ruddy KJ, Vachon CM, et al. Common genetic variation and breast cancer risk - Past, present, and future. Cancer Epidemiol Biomarkers Prev. 2018;27(4):380–394.
- Bhat SA, Majid S, Wani HA, et al. Diagnostic utility of epigenetics in breast cancer – a review. Cancer Treat Res Commun. 2019;19:100125.
- Maity A, Das B. N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. 2016;283(9):1607–1630.
- Wang Y, Zhao JC. Update: mechanisms underlying N6-methyladenosine modification of eukaryotic mRNA. Trends Genet. 2016;32(12):763–773.
- Linder B, Grozhik AV, Olarerin-George AO, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–772.
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15(5):313–326.
- Cao G, Li HB, Yin Z, et al. Recent advances in dynamic m6A RNA modification. Open Biol. 2016;6(4):160003.
- Chen XY, Zhang J, Zhu JS. The role of m6A RNA methylation in human cancer. Mol Cancer. 2019;18(1):103.
- Jiang S, Xie Y, He Z, et al. m6ASNP: a tool for annotating genetic variants by m6A function. Gigascience. 2018;7(5):giy035.
- Zheng Y, Nie P, Peng D, et al. M6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 2018;46(D1):D139–145.
- Lin W, Xu H, Wu Y, et al. In silico genome-wide identification of m6A-associated SNPs as potential functional variants for periodontitis. J Cell Physiol. 2020;235(2):900–908.
- Sun X, Dai Y, Tan G, et al. Integration analysis of m6A-SNPs and eQTLs associated with sepsis reveals platelet degranulation and Staphylococcus aureus infection are mediated by m6A mRNA methylation. Front Genet. 2020;11:7.
- Slobodin B, Dikstein R. So close, no matter how far: multiple paths connecting transcription to mRNA translation in eukaryotes. EMBO Rep. 2020;21(9):e50799.
- Mo XB, Zhang YH, Lei SF. Genome-wide identification of N6-methyladenosine (m6A) SNPs associated with rheumatoid arthritis. Front Genet. 2018;9:299.
- Mo XB, Zhang YH, Lei SF. Genome-wide identification of m6A-associated SNPs as potential functional variants for bone mineral density. Osteoporos Int. 2018;29(9):2029–2039.
- Hu Y, Zhao X. Role of m6A in osteoporosis, arthritis and osteosarcoma (Review). Exp Ther Med. 2021;22:926.
- Mo XB, Lei SF, Zhang YH, et al. Detection of m6A-associated SNPs as potential functional variants for coronary artery disease. Epigenomics. 2018;10(10):1279–1287.
- Lin W, Xu H, Yuan Q, et al. Integrative genomic analysis predicts regulatory role of N6-methyladenosine-associated SNPs for adiposity. Front Cell Dev Biol. 2020;8:551.
- Mo X, Lei S, Zhang Y, et al. Genome-wide enrichment of m6A-associated single-nucleotide polymorphisms in the lipid loci. Pharmacogenomics J. 2019;19(4):347–357.
- Qiu X, He H, Huang Y, et al. Genome-wide identification of m6A-associated single-nucleotide polymorphisms in Parkinson’s disease. Neurosci Lett. 2020;737:135315.
- Sebastian-delaCruz M, Olazagoitia-Garmendia A, Gonzalez-Moro I, et al. Implication of m6A mRNA methylation in susceptibility to iflammatory bowel disease. Epigenomes. 2020;4(3):16.
- Zhao H, Jiang J, Wang M, et al. Genome-wide identification of m6A-associated single-nucleotide polymorphisms in colorectal cancer. Pharmgenomics Pers Med. 2021;14:887–892.
- Chen M, Lin W, Yi J, et al. Exploring the epigenetic regulatory role of m6A-associated SNPs in type 2 diabetes pathogenesis. Pharmgenomics Pers Med. 2021;14:1369–1378.
- Ruan X, Tian M, Kang N, et al. Genome-wide identification of m6A-associated functional SNPs as potential functional variants for thyroid cancer. Am J Cancer Res. 2021;11(11):5402–5414.
- Xuan Z, Zhang Y, Jiang J, et al. Integrative genomic analysis of N6-methyladenosine-single nucleotide polymorphisms (m6A-SNPs) associated with breast cancer. Bioengineered. 2021;12(1):2389–2397.
- Grossman RL, Heath AP, Ferretti V, et al. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375(12):1109–1112.
- Buniello A, Macarthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012.
- Cariaso M, Lennon G. SNPedia: a wiki supporting personal genome annotation, interpretation and analysis. Nucleic Acids Res. 2012;40( Database issue):D1308–1312.
- Bruno AE, Li L, Kalabus JL, et al. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3′UTRs of human genes. BMC Genomics. 2012;13:44.
- Liu C-J, Fu X, Xia M, et al. Guo A-Y. miRNASNP-v3: a comprehensive database for SNPs and disease-related variations in miRNAs and miRNA targets. Nucleic Acids Res. 2021;49(D1):D1276–1281.
- Yue M, Zhou D, Zhi H, et al. MSDD: a manually curated database of experimentally supported associations among miRNAs, SNPs and human diseases. Nucleic Acids Res. 2018;46(D1):D181–D185.
- Bhattacharya A, Ziebarth JD, Cui Y. SomamiR: a database for somatic mutations impacting microRNA function in cancer. Nucleic Acids Res. 2013;41(DI):D977–982.
- Howe KL, Achuthan P, Allen J, et al. Ensembl 2021. Nucleic Acids Res. 2021;49(DI):D884–891.
- Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(D1):D930–934.
- Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets - Update. Nucleic Acids Res. 2013;41(D1):D991–995.
- Turashvili G, Bouchal J, Baumforth K, et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer. 2007;7(1):55.
- Kretschmer C, Sterner-Kock A, Siedentopf F, et al. Identification of early molecular markers for breast cancer. Mol Cancer. 2011;10(1):15.
- Li JH, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(DI):D92–97.
- Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
- Marshall OJ. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics. 2004;20(15):2471–2472.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408.
- Tan SC. Use of arbitrary reference genes may lead to misleading conclusions. Gynecol Obstet Invest. 2019;84(5):519–520.
- Schmid H, Cohen CD, Henger A, et al. Validation of endogenous controls for gene expression analysis in microdissected human renal biopsies. Kidney Int. 2003;64(1):356–360.
- Yin WZ, Yang QW, Niu K, et al. Validation of reference genes for the normalization of RT-qPCR expression studies on human laryngeal cancer and hypopharyngeal cancer. Eur Rev Med Pharmacol Sci. 2019;23(10):4199–4209.
- Meyer KD, Patil DP, Zhou J, et al. 5′ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
- Ke S, Alemu EA, Mertens C, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29(19):2037–2053.
- Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646.
- Ardlie KG, Deluca DS, Segrè AV. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Consortium Gte. Science. 2015;348(6235):648–660.
- Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42(5):2976–2987.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415.
- Wei CY, Tan QX, Zhu X, et al. Expression of CDKN1A/p21 and TGFBR2 in breast cancer and their prognostic significance. Int J Clin Exp Pathol. 2015;8(11):14619–14629.
- Wang F, Ma YL, Zhang P, et al. A genetic variant in microRNA-196a2 is associated with increased cancer risk: a meta-analysis. Mol Biol Rep. 2012;39(1):269–275.
- Wang PY, Gao ZH, Jiang ZH, et al. The associations of single nucleotide polymorphisms in miR-146a, miR-196a and miR-499 with breast cancer susceptibility. PLoS One. 2013;8(9):e70656.
- Zhang H, Su YL, Yu H, et al. Meta-analysis of the association between miR-196a-2 polymorphism and cancer susceptibility. Cancer Biol Med. 2012;9(1):63–72.
- Hu Z, Liang J, Wang Z, et al. Common genetic variants in pre-microRNAs were associated with increased risk of breast cancer in Chinese women. Hum Mutat. 2009;30(1):79–84.
- Zhao H, Xu J, Zhao D, et al. Somatic mutation of the SNP rs11614913 and its association with increased MIR 196A2 expression in breast cancer. DNA Cell Biol. 2016;35(2):81–87.
- Deng YN, Xia Z, Zhang P, et al. Transcription factor RREB1: from target genes towards biological functions. Int J Biol Sci. 2020;16(8):1463–1473.
- Choupani J, Nariman-Saleh-Fam Z, Saadatian Z, et al. Association of mir-196a-2 rs11614913 and mir-149 rs2292832 polymorphisms with risk of cancer: an updated meta-analysis. Front Genet. 2019;10:186.
- Catucci I, Yang R, Verderio P, et al. Evaluation of SNPs in miR-146a, miR196a2 and miR-499 as low-penetrance alleles in German and Italian familial breast cancer cases. Hum Mutat. 2010;31(1):E1052–1057.
- Jedlinski DJ, Gabrovska PN, Weinstein SR, et al. Single nucleotide polymorphism in hsa-mir-196a-2 and breast cancer risk: a case control study. Twin Res Hum Genet. 2011;14(5):417–421.
- Mousses S, Özçelik H, P DL, et al. Two variants of the CIP1/WAF1 gene occur together and are associated with human cancer. Hum Mol Genet. 1995;4(6):1089–1092.
- Bae I, Fan S, Bhatia K, et al. Relationships between G1 arrest and stability of the p53 and p21CiP1/Waf1 proteins following gamma-irradiation of human lymphoma cells. Cancer Res. 1995;55(11):2387–2393.
- Zhang Y, Fujita N, Tsuruo T. Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene. 1999;18(5):1131–1138.
- Abbas T, Dutta A. P21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2010;9(6):400–414.
- Kreis -N-N, Louwen F, Yuan J. The multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation, migration and cancer therapy. Cancers (Basel). 2019;11(9):1220.
- Carvalho INSR, de Oliveira Reis AH, Cabello PH, et al. Polymorphisms of CDKN1A gene and risk of retinoblastoma. Carcinogenesis. 2013;34(12):2774–2777.
- Lotfi Garavand A, Mohammadi M, Mohammadzadeh S. Evaluation of TP53 codon 72, P21 codon 31, and MDM2 SNP309 polymorphisms in Iranian patients with acute lymphocytic leukemia. Reports Biochem Mol Biol. 2020;9(1):26–32.
- Lin YC, Hour TC, Tsai YC, et al. Preliminary evidence of polymorphisms of cell cycle regulatory genes and their roles in urinary tract urothelial cancer susceptibility and prognosis in a Taiwan population. Urol Oncol Semin Orig Investig. 2017;35(9):543.e7–543.e16.
- Själander A, Birgander R, Rannug A, et al. Association between the p21 codon 31 A1 (arg) allele and lung cancer. Hum Hered. 1996;46(4):221–225.
- Li G, Liu Z, Sturgis EM, et al. Genetic polymorphisms of p21 are associated with risk of squamous cell carcinoma of the head and neck. Carcinogenesis. 2005;26(9):1596–1602.
- Barbieri RB, Bufalo NE, Secolin R, et al. Polymorphisms of cell cycle control genes influence the development of sporadic medullary thyroid carcinoma. Eur J Endocrinol. 2014;171(6):761–767.
- Cox A, Dunning AM, Garcia-Closas M, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39(3):352–358.
- Driver KE, Song H, Lesueur F, et al. Association of single-nucleotide polymorphisms in the cell cycle genes with breast cancer in the British population. Carcinogenesis. 2008;29(2):333–341.
- Mao Y, Dong L, Liu X-M, et al. m6A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019;10(1):5332.
- Machnik M, Cylwa R, Kiełczewski K, et al. The expression signature of cancer-associated KRAB-ZNF factors identified in TCGA pan-cancer transcriptomic data. Mol Oncol. 2019;13(4):701–724.
- Sobocińska J, Molenda S, Machnik M, et al. KRAB-ZFP transcriptional regulators acting as oncogenes and tumor suppressors: an overview. Int J Mol Sci. 2021;22(4):2212.
- Sun M, Ju J, Ding Y, et al. The signaling pathways regulated by KRAB zinc-finger proteins in cancer. Biochim Biophys Acta Rev Cancer. 2022;1877(3):188731.
- Wang J, Zhang X, Ling J, et al. KRAB-containing zinc finger protein ZNF496 inhibits breast cancer cell proliferation by selectively repressing ERα activity. Biochim Biophys Acta Gene Regul Mech. 2018;1861(9):841–853.