
ABSTRACT
Critically ill children requiring intensive care suffer from impaired physical/neurocognitive development 2 y later, partially preventable by omitting early use of parenteral nutrition (early-PN) in the paediatric intensive-care-unit (PICU). Altered methylation of DNA from peripheral blood during PICU-stay provided a molecular basis hereof. Whether DNA-methylation of former PICU patients, assessed 2 y after critical illness, is different from that of healthy children remained unknown. In a pre-planned secondary analysis of the PEPaNIC-RCT (clinicaltrials.gov-NCT01536275) 2-year follow-up, we assessed buccal-mucosal DNA-methylation (Infinium-HumanMethylation-EPIC-BeadChip) of former PICU-patients (N = 406 early-PN; N = 414 late-PN) and matched healthy children (N = 392). CpG-sites differentially methylated between groups were identified with multivariable linear regression and differentially methylated DNA-regions via clustering of differentially methylated CpG-sites using kernel-estimates. Analyses were adjusted for technical variation and baseline risk factors, and corrected for multiple testing (false-discovery-rate <0.05). Differentially methylated genes were functionally annotated (KEGG-pathway database), and allocated to three classes depending on involvement in physical/neurocognitive development, critical illness and intensive medical care, or pre-PICU-admission disorders. As compared with matched healthy children, former PICU-patients showed significantly different DNA-methylation at 4047 CpG-sites (2186 genes) and 494 DNA-regions (468 genes), with most CpG-sites being hypomethylated (90.3%) and with an average absolute 2% effect-size, irrespective of timing of PN initiation. Of the differentially methylated KEGG-pathways, 41.2% were related to physical/neurocognitive development, 32.8% to critical illness and intensive medical care and 26.0% to pre-PICU-admission disorders. Two years after critical illness in children, buccal-mucosal DNA showed abnormal methylation of CpG-sites and DNA-regions located in pathways known to be important for physical/neurocognitive development.
Introduction
Children who have been critically ill requiring life support in a paediatric intensive care unit (PICU) suffer from impaired physical and neurocognitive development documented years later [Citation1–6]. Part of this adverse legacy of paediatric critical illness was found to be modifiable by altering the intensive medical care, as revealed by the PEPaNIC multicentre randomized controlled trial (RCT). The PEPaNIC-RCT assessed the short- and long-term impact of withholding supplemental parenteral nutrition in the first week in the PICU (late-PN) as compared with early supplementation of insufficient enteral nutrition with parenteral nutrition within 24 hours (early-PN), in comparison with matched healthy children [Citation4,Citation6,Citation7]. Two and four years after PICU admission, these former patients revealed broad abnormalities in physical and neurocognitive development, and in emotional and behavioural functioning [Citation4,Citation6]. As compared with the children who had received early-PN in the PICU, those in the late-PN group performed better in the long term with regard to executive functioning, visual-motor integration, emotion and behaviour [Citation4,Citation6].
To investigate epigenetic changes as an underlying mechanism, a genome-wide DNA-methylation analysis has been performed on DNA extracted from peripheral blood samples collected from healthy children and from PEPaNIC patients. After excluding all DNA-methylation differences already present upon PICU admission, including those that could be related to altered white blood cell (WBC) composition, 159 CpG-sites were identified as de novo differentially methylated (also known as Differentially Methylated Positions, DMPs) during the stay in the PICU, with 23% of these attributable to the use of early-PN [Citation8]. Several of these de novo differentially methylated CpG-sites were located in genes important for physical and neurocognitive developmental processes or in genes known to be involved in neurodegenerative and psychiatric diseases and were found to statistically explain part of the long-term developmental impairments, suggesting indeed a plausible molecular basis hereof [Citation4,Citation6,Citation8].
Aberrant changes in DNA-methylation are considered to be quite stable, though they can also be restored [Citation9–11]. It is thus unclear whether the DNA methylation status of former PICU patients, assessed 2 y after critical illness and randomization to late-PN or early-PN, is different from that of healthy children and whether this could be relevant for the long-term developmental impairments. To address this question, we extracted DNA from buccal mucosal swabs collected from healthy children and from former PEPaNIC patients 2 y after PICU admission, ensuring that the children were out of the acute illness phase and not sick when approached for follow-up, and performed a genome-wide DNA-methylation analysis to identify any differentially methylated positions and regions, followed by a functional annotation analysis.
Methods
Study population, buccal mucosal swab sampling and ethical approval
This study is a pre-planned secondary analysis of the multicentre PEPaNIC-RCT (registered at ClinicalTrials.gov NCT01536275) that included 1440 critically ill children aged 0–17 y admitted to the PICUs of Leuven (Belgium), Rotterdam (The Netherlands) or Edmonton (Canada) and its 2-year follow-up study [Citation4,Citation7]. The patients in this RCT had been randomly assigned to early initiation of parenteral nutrition within 24 hours when enteral nutrition was insufficient (‘early-PN’), or to postponing any supplemental PN to beyond the first week in PICU (‘late-PN’). After one week, for both groups equally, PN could be administered if necessary [Citation7]. The full study protocol has been published [Citation7]. Matched healthy children, either siblings and relatives of the patients or unrelated children from the same geographic area, were included as controls, attempting to adjust as much as possible for genetic and socio-economic/environmental background [Citation4].
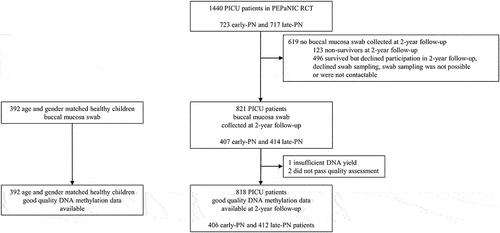
At the 2-year follow-up of the PEPaNIC patients, upon which time also the matched healthy children were assessed, buccal mucosal swabs (Isohelix, Cell Projects, Harrietsham, Kent, England) were collected following a standardized collection procedure after obtaining written informed consent from parents and/or the children. Swabs were stored in a DNA stabilizing solution (DSK kit, Isohelix) at −80°C until further processing. All former patients and healthy children from whom a buccal mucosal swab was available (only taken from participants recruited in Leuven or Rotterdam) were eligible for this DNA-methylation study ().
Figure 1. CONSORT diagram of study participants. Participants included in the epigenetic 2-year follow-up analysis are shown.
The institutional review boards at each participating site approved this follow-up study (Ethische Commissie Onderzoek UZ Leuven/KU Leuven: ML8052; Medische Ethische Toetsingscommissie Erasmus MC: NL49708.078). The study was performed in accordance with the 1964 Declaration of Helsinki and its amendments. Written informed consent was acquired from parents, legal guardians, or the child if 18 y or older.
DNA extraction and DNA-methylation data processing
DNA was extracted from all available buccal mucosal swabs from patients and healthy children (DDK DNA isolation kit, Isohelix). DNA concentrations were quantified with the Qubit® 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA). In total, 200 ng DNA was subsequently subjected to bisulphite-conversion with use of the EZ-96 DNA-methylation-Direct® Kit (Zymo Research, Irvine, CA). Bisulphite-converted DNA was profiled using the Infinium® HumanMethylation EPIC BeadChip (Illumina Inc., San Diego, CA), which interrogates 865,859 CpG-sites. All mandatory laboratory health and safety procedures have been complied within the course of conducting these experiments.
Data were processed using R statistical software version 4.0.2 using the LICMEpigenetics package (version 0.1.0) and the ilm10b4.hg19 annotation file using human genome 19 (hg19) as a reference was used to annotate the CpG sites. This package contains R functions to exclude low-quality samples and probes, normalize the methylation data, adjust for batch effect, and find differentially methylated positions and regions, as described below [Citation12–14].
The methylation data were subjected to several consecutive quality assessments at sample and probe level. First, samples not showing the typical bi-peak curve of the methylation value distribution in the low- and high-end range on the sample histograms (density plots) were excluded. Beta-values (ranging from 0 or no methylation to 1 or full methylation) and corresponding M-values (log2 ratios of the intensities of methylated probes versus unmethylated probes) were then obtained from the raw intensities after background subtraction, colour correction, and functional normalization. To ensure that signals were expressed above the background defined by negative control probes, probes with a detection p-value greater than 0.01 in 50% of the samples were removed (N = 82) [Citation15–17]. Also probes spanning known single nucleotide polymorphisms or located on sex chromosomes (N = 49,733) were removed [Citation15–17]. Finally, a principal component analysis (PCA) on the M-values with p-value heatmap assessed the possible presence of non-biological or technical variation in the data (‘batch effect’), see Supplementary Figure S1 [Citation18]. Such non-biological or technical variation due to experimental conditions was corrected for by including the first 30 principal components (PCs) of the technical control probes located on the Infinium® HumanMethylation EPIC BeadChip, excluding the negative control probes, as covariates in all multivariable linear regression models, according to the method developed by Lehne et al. [Citation19]. To assess which genes were differentially methylated we used the ilm10b4.hg19 annotation file provided by the minfi package to map the DMPs and DMRs onto the genome.
Statistical analyses
Patient demographics and medical characteristics are reported as mean and standard deviation, median and interquartile range, or number and percentage. Group comparisons were performed with the chi-square test for categorical variables, student t-test for normally distributed continuous variables and Wilcoxon signed rank test for non-normally distributed variables.
Differences in DNA-methylation between former PICU patients and healthy children were assessed, correcting for multiple testing by applying a false discovery rate (FDR) of less than 0.05 as determined with the Benjamini-Hochberg procedure [Citation20]. Such differences, if any, are to be considered the sum of differences evoked by the paediatric critical illness and intensive medical care and those that may have been present in the former patients prior to PICU admission.
First, all differentially methylated positions (DMPs) in former PICU patients as compared with the matched healthy children were identified. Therefore, methylation status of every CpG-site was assessed separately for differences between former patients and healthy children, with use of multivariable linear models using the limma framework adjusting for baseline risk factors [age, centre, race, sex, geographical origin, language, history of malignancy and predefined syndrome (see Supplementary Methods S1 for the definition)] and adjusting for technical variation (batch effect) [Citation19,Citation21].
Second, all differentially methylated regions (DMRs), i.e., regions within the DNA where groups of neighbouring CpG-sites are differentially methylated, in former PICU patients as compared with the matched healthy children were identified with the DMRcate package [Citation22]. A stepwise explanation of the DMRcate method with an illustrative example is provided in Supplementary Methods S2.
To investigate to what extent timing of parenteral nutrition (early-PN versus late-PN) had a direct role in bringing about any of the DMPs or DMRs 2 y after PICU discharge, we performed similar analyses as those described above, limiting them to the DMPs and DMRs identified for former patients and healthy children, further adjusting for type and severity of illness [Paediatric Index of Mortality 3 (PIM3) score, Paediatric Logistic Organ Dysfunction (PeLOD) score], and risk of malnutrition [STRONGkids score]) in addition to the above described baseline risk factors and batch effect [Citation19].
To quantify the extent of the methylation change between patients and healthy children, or the Early-PN and Late-PN group, we calculated the absolute change for all differentially methylated CpG-sites. We computed the absolute difference between the average beta values of both groups. The absolute changes per CpG were summarized using the mean, standard deviation, maximum, median, and interquartile range.
Finally, we assessed whether any of the 159 de novo differentially methylated positions at PICU discharge identified in a previous study that used white blood cell DNA [Citation8] were also differentially methylated in the buccal mucosa DNA harvested at 2-year follow-up of former PICU patients. To identify differences in methylation between these groups within this selection of CpG sites, we created multivariable linear models using the limma framework adjusting for baseline risk factors [age, centre, race, sex, geographical origin, language, history of malignancy and predefined syndrome (see Supplementary Methods S1 for the definition)] and adjusting for technical variation (batch effect) [Citation19,Citation21].
Functional annotation analysis
To assess the function of genes that were identified as differentially methylated, we performed a functional annotation analysis with use of the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways database [Citation23,Citation24]. This database groups genes into biological pathways that fulfil a specific biological function, with further hierarchical grouping into biological categories [Citation23,Citation24]. To identify which biological pathways were most differentially methylated, we ranked them based on a weighted percentage from the differentially methylated genes within a biological pathway. This weighing prevented overrepresentation of those biological pathways comprising exceptionally small or large number of genes. The weighted percentage of differentially methylated genes was calculated by multiplying the natural logarithm from the number of differentially methylated genes with the percentage of differentially methylated genes within a biological pathway. Based on this ranking, the 25 most differentially methylated pathways were visualized in a tabular format.
Next, the differentially methylated biological pathways within the KEGG pathways database were grouped, by three authors in consensus (GC, IV, GVdB), into three classes depending on whether the pathway could be involved in ‘physical and neurocognitive development,’ whether it could be related to ‘the critical illness phase or its intensive medical care,’ or whether it could be related to ‘differences that may have been present before PICU admission.’ Class assignment was based on literature. For pathways that could be categorized into multiple classes, we prioritized assignment to the class ‘physical and neurocognitive development.’ For those pathways that were not assigned to this first class, we prioritized the class ‘critical illness phase or its intensive medical care.’ Next, we calculated the number and percentage of differentially methylated pathways of every class.
In addition, an online interactive visualization tool was created with the D3 javascript package to illustrate all the differentially methylated KEGG pathways, in relation to KEGG categories and genes [Citation25].
Results
Differentially methylated positions and regions in former PICU patients as compared with healthy children
Buccal mucosal swabs were collected from 821 patients, 407 randomized to early-PN and 414 to late-PN, and from 392 healthy children (). Three patients needed to be excluded for further analysis, as DNA yield was insufficient for one early-PN patient, and two late-PN patients showed deviation from the typical bi-peak curve of the methylation value distribution. Participants’ demographics and medical characteristics are shown in .
Table 1. Demographics and medical characteristics of participants included in the 2-year epigenetic follow-up analysis.
The adjusted comparison between the former PICU patients and the matched healthy children identified 4047 DMPs and 494 DMRs. The 4047 CpG-sites corresponding to the DMPs, their location within the genome, and the direction of change (hypo- or hypermethylated in former patients as compared with healthy children) are listed in Supplementary Table S1. The absolute mean differences in DNA-methylation beta-values for these CpG-sites were 2.24% (SD 1.42%), ranging up to 8.20% (), with mostly hypomethylation in patients (3654/4047 (90.3%), , Supplementary Table S2). Of the 4047 DMPs, 1446 (35.7%) were intergenic and 2601 (64.3%) were located within 2186 unique genes (, Supplementary Table S2). The majority of the 2601 DMPs within genes were located within the gene body excluding the first exon (1488 (57.2%), of which 1373 (92.3%) hypomethylated in former patients as compared with healthy children), or the promoter region (774 (29.8%), of which 633 (81.8%) hypomethylated in former patients) (, Supplementary Table S2). The genomic locations and width for each of the 494 DMRs are shown in Supplementary Table S3, together with the number of CpG-sites within each DMR. The individual CpG-sites within each of the DMRs, their location within the genome, and the direction of change are shown in Supplementary Table S4. The 494 DMRs, spanning a total of 343,442 base pairs, contained a total of 4024 CpG-sites, with absolute mean differences in DNA-methylation beta-values of 1.98% (SD 1.31%), ranging up to 7.41% (), again mostly towards hypomethylation in patients (2943/4024 CpG-sites (73.1%), , Supplementary Table S5). Of the 494 DMRs and the embedded 4024 CpG-sites, 120 DMRs (24.3%) and 599 CpG-sites (14.9%) were (partly) intergenic, and 409 DMRs (82.8%) and 3425 CpG-sites (85.1%) were associated with 468 unique genes (, Supplementary Table S5). The majority of the DMRs and corresponding CpG-sites found within genes were (partly) located within the promoter region (285/409 (69.7%) DMRs; 2043/3425 (59.6%) CpG-sites) or the gene body excluding the first exon (209/409 (51.1%) regions; 1027/3425 (29.7%) CpG-sites, , Supplementary Table S5).
Figure 2. Locations of Differentially Methylated Positions and Regions. Bar chart depicting the distribution of DMPs (a), DMRs (b) and CpG sites within DMRs (c) over the gene regions. The blue and red area represent the proportion that is hypomethylated and hypermethylated respectively in the children who had been admitted to the PICU 2 y prior to the follow-up study compared to the healthy control children. For our calculations, we defined the promoter as 0 to 1500 base pairs upstream of the transcription start site and the first exon is reported separately and thus is not included into the gene body. Note: Abbreviations: DMP, differentially methylated position; DMR, differentially methylated region; PICU, Paediatric Intensive Care Unit, UTR: untranslated region
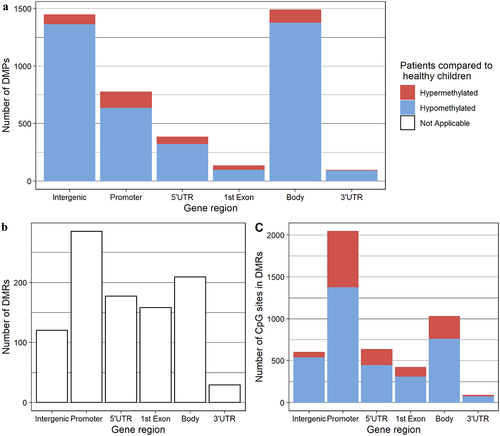
Table 2. Summary of absolute differences in methylation between former PICU patients and healthy children.
Out of the 159 CpG sites that were previously identified to become de novo abnormally methylated between PICU admission and discharge using DNA extracted from white blood cells [Citation8], we could assess 153 CpG sites in the 2-year follow-up buccal mucosa DNA sample after pre-processing. From these 153 CpG sites, 85 (55.5%) were abnormally methylated in former patients 2 y after PICU discharge.
Differentially methylated positions and regions in former late-PN versus early-PN PICU patients
Randomization to late-PN versus early-PN in the PICU did not significantly affect the methylation status of any of the 4047 DMPs or 494 DMRs found 2 y later in former PICU patients.
Functional annotation analysis
Combining the 2186 genes encompassing the identified DMPs and the 468 genes encompassing the identified DMRs yielded 2365 unique genes in which we found differential DNA-methylation between former PICU patients and matched healthy children. We subsequently functionally annotated these 2365 genes with use of the KEGG pathway database, which revealed that differential methylation between former PICU patients and matched healthy children affected genes in 323 KEGG biological pathways.
We subsequently grouped the 323 differentially methylated pathways into the 3 classes defined in function of whether they could be related to physical or neurocognitive development, to the critical illness phase or the intensive medical care, or to differences that may have been present before PICU admission (Supplementary Table S6). Most of the differentially methylated pathways could be attributed to physical and neurocognitive development (133 pathways, 41.2%), followed by the critical illness phase or the intensive medical care (106 pathways, 32.8%) and differences that may have been present before PICU admission (84 pathways, 26.0%).
The 25 most differentially methylated pathways are shown individually in and grouped into biological category and functional classes in Supplementary Table S7. For each of these pathways, at least 31.5% of the involved genes were methylated differently in former PICU patients than in healthy children. Twelve of these pathways (48%) could be related to physical or neurocognitive development with functions mostly in signal transduction and the endocrine and nervous system. Six pathways (24%) could be related to either the critical illness and its intensive medical care and comprised a wide range of systems, including the endocrine, circulatory, immune and sensory systems, as well as activities involved in environmental adaptation and substance dependence. The remaining 7 (28%) could be related to differences that may have been present before PICU admission, more specifically differences related to cancer and infectious diseases.
Table 3. Twenty-five most differentially methylated pathways in former patients 2 y after PICU-admission versus healthy children.
An interactive in-depth visualization of all KEGG biological categories, pathways with corresponding classifications and differentially methylated genes can be found at www.pepanic.com/2Y_epigenetics Instructions on how to use the interactive tool can be found in Supplementary Figure S2 [Citation26].
Discussion
As compared with matched healthy children, former PICU-patients who were assessed 2 y after critical illness revealed significantly different methylation of DNA extracted from buccal mucosa at 4047 CpG-sites (2186 genes) and 494 DNA-regions (468 genes). Most of these CpG-sites were hypomethylated (90.3%) in former PICU patients with an average absolute 2.06% effect size, irrespective of timing of PN initiation. Of the differentially methylated KEGG-pathways, 41.2% were related to physical/neurocognitive development, 32.8% to critically illness and the intensive medical care and 26.0% to pre-PICU-admission disorders. These data indicate that former PICU patients, assessed 2 y after critical illness, reveal aberrant DNA-methylation in genes and pathways that may explain at least part of their long-term developmental impairments.
We found that the methylome of DNA extracted from buccal mucosa of children who, 2 y earlier, had been critically ill and required intensive medical care in a PICU, was substantially different from that of matched healthy children, comprising many differentially methylated sites as well as regions. As compared with the previously published genome-wide DNA-methylation analysis, that was performed on DNA from peripheral blood samples harvested from patients while in the PICU and from healthy children [Citation8], ±25-fold more differentially methylated sites were identified in the current study. This difference in numbers is likely explained by differences in tissue type and by differences in the used methodology, which in the earlier study of peripheral blood DNA, identified only the de novo alterations occurring between PICU admission and discharge, after omitting all differences that were already present upon PICU admission, including differences related to cell type composition. This earlier methodology had the advantage of allowing identification only of de novo changes and linking these with the long-term physical and neurocognitive impairments, but the disadvantage of omitting assessment of many other genes and pathways that may have played a role. For example, assessing any role of altered methylation occurring in inflammation pathways was hereby not possible in the earlier study whereas these pathways turned out to be differentially methylated in the current study that used buccal swab DNA harvested 2 y after critical illness. Interestingly, in the current study, the functional annotation analysis also allowed to relate a large proportion of the DNA-methylation differences to physical and neurocognitive impairments. The current study could not assess whether these differences were pre-existing or occurred de novo during or after critical illness. However, 55.5% of the CpG sites that were previously found to become abnormally methylated between PICU admission and discharge in DNA extracted from peripheral blood cell, were also found to be abnormally methylated 2 y later in buccal mucosa DNA. Also, the direction of methylation changes, predominantly hypomethylation in patients, and the average effect size of the changes were comparable between both studies that used two different tissues.
Our findings are in agreement with other epigenome-wide methylation studies that have linked changes induced by other environmental exposures, for example early life adversity such as child abuse, parental smoking or diet-induced obesity, to physical growth and/or neurocognitive development or brain disorders [Citation27–33]. Other epigenome-wide studies have also identified a role of altered DNA-methylation in causing CNS disorders such Parkinson’s and Alzheimer’s disease [Citation34–38]. Furthermore, even earlier studies, that mostly used a target gene approach, also suggested such a role for altered DNA-methylation in neurodevelopment and in endocrine pathways controlling onset of puberty and physical growth [Citation39–44]. The current finding that the differences in DNA-methylation between former PICU patients, compared 2 y after critical illness, and healthy children were mostly revealing hypomethylation rather than hypermethylation was remarkable. This was also the case in the study of the de novo changes occurring during stay in the PICU [Citation8]. Also, in studies of neurodegenerative diseases, hypomethylation of DNA was found to be dominant [Citation10,Citation38,Citation45].
In contrast with the earlier study that assessed de novo alterations in DNA-methylation occurring while patients were in the PICU, and which revealed that the early use of PN, as compared with late PN, explained about a quarter of these changes, the current study did not detect such a difference. This could be a true finding or, alternatively, could be explained by differences in methodology. Indeed, the earlier study only assessed the role of early-PN versus late-PN in bringing about part of 159 DMPs that occurred in the PICU [Citation8]. The methodology used in the current study, which compared the entire methylome between former PICU patients and healthy children and identified 4047 DMPs, reduced the possibility to detect small differences related to the nutritional strategy in the PICU, as any such effect may have been diluted in more differences and may thus have remained hidden after adjusting for multiple testing.
Strengths of this study were the large sample size and the multicentre, prospective, randomized controlled trial design with predefined follow-up assessments of patients and matched healthy children and the use of the 850 K Human MethylationEPIC BeadChip. Another strength was the development of a unique interactive tool (www.pepanic.com/2Y_epigenetics that allows readers to explore in detail every change in DNA-methylation in relation to genes, KEGG pathways and categories [Citation26]. Our study also has some limitations. First, we used buccal mucosa to extract DNA from, due to obvious practical and ethical limitations, which does not allow extrapolation to other tissues of interest. Second, due to the large number of DMPs that were identified, a statistical association analysis with physical and neurocognitive development was not possible. Instead we performed a functional annotation analysis. The assessment of the relevance of the identified pathways for physical and neurocognitive development comprised manual classification and class prioritization which hold a certain risk of bias.
In conclusion, 2 y after critical illness in children, buccal mucosa DNA showed abnormal methylation of CpG-sites and DNA-regions within genes located in pathways that are known to be important for physical and neurocognitive development. Future research should investigate whether the observed aberrant DNA-methylation in former PICU patients is permanent or could be restored when studied at a later stage of development.
Supplemental Material
Download Zip (1.4 MB)Acknowledgments
The resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation – Flanders (FWO) and the Flemish Government. The authors acknowledge the research team members involved in the study for their help with the technical and administrative support. Furthermore, they thank the children and their parents for their willingness to participate in the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is offered under the format of collaborative projects. Proposals can be directed to the corresponding author.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2022.2146966
Additional information
Funding
References
- Mesotten D, Gielen M, Sterken C, et al. Neurocognitive development of children 4 years after critical illness and treatment with tight glucose control: a randomized controlled trial. JAMA. 2012;308(16):1641–12.
- Mammen C, Al Abbas A, Skippen P, et al. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. Am J Kidney Dis. 2012;59:523–530.
- Banwell BL, Mildner RJ, Hassall AC, et al. Muscle weakness in critically ill children. Neurology. 2003;61:1779–1782.
- Verstraete S, Verbruggen SC, Hordijk JA, et al. Long-term developmental effects of withholding parenteral nutrition for 1 week in the paediatric intensive care unit: a 2-year follow-up of the PEPaNIC international, randomised, controlled trial. Lancet Respir Med. 2019;7:141–153.
- Lee JH, Choong K. Time to focus on paediatric critical care survivorship. Lancet Respir Med. 2019;7:103–105.
- Jacobs A, Dulfer K, Eveleens RD, et al. Long-term developmental effect of withholding parenteral nutrition in paediatric intensive care units: a 4-year follow-up of the PEPaNIC randomised controlled trial. Lancet Child Adolesc Health. 2020;4:503–514.
- Fivez T, Kerklaan D, Mesotten D, et al. Early versus Late parenteral nutrition in critically ill children. N Engl J Med. 2016;374:1111–1122.
- Guiza F, Vanhorebeek I, Verstraete S, et al. Effect of early parenteral nutrition during paediatric critical illness on DNA methylation as a potential mediator of impaired neurocognitive development: a pre-planned secondary analysis of the PEPaNIC international randomised controlled trial. Lancet Respir Med. 2020;8:288–303.
- Vandiver AR, Idrizi A, Rizzardi L, et al. DNA methylation is stable during replication and cell cycle arrest. Sci Rep. 2015;5:17911.
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775(1):138–162.
- Petryk N, Bultmann S, Bartke T, et al. Staying true to yourself: mechanisms of DNA methylation maintenance in mammals. Nucleic Acids Res. 2021;49:3020–3032.
- Team RC. R: a language and environment for statistical computing R foundation for statistical computing. 2019; Accessed 13 09, 2022. https://www.r-project.org
- LICMEpigenetics Package GitHub. 2022 Accessed 06 04, 2022. https://github.com/LICMLeuven/LICMEpigenetics
- PEPanic 2Y Epigenetic analysis GitHub. 2022; Accessed 13 09, 2022. https://github.com/LICMLeuven/PEPaNIC_2Y_Epigenetics
- Wu MC, Kuan P-F. A Guide to Illumina BeadChip Data Analysis. In: Tost J, editor. DNA Methylation Protocols. New York: Springer New York; 2018. p. 303–330.
- Fortin JP, Triche TJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33:558–560.
- Hansen KD IlluminaHumanMethylationEPICanno.ilm10b4.hg19: annotation for Illumina’s EPIC methylation arrays 2017; Accessed 06 04, 2022. https://bitbucket.com/kasperdanielhansen/Illumina_EPIC
- Teschendorff A. Computational and Statistical Epigenomics. Computational Statistical Epigenomics. 2015;7:1–217.
- Lehne B, Drong AW, Loh M, et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015;16:12.
- Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375.
- Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
- Peters TJ, Buckley MJ, Statham AL, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:16.
- Kanehisa M, Furumichi M, Sato Y, et al. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–D51.
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
- Bostock M, Ogievetsky V, Heer J. D3: data-Driven Documents. IEEE Trans Vis Comput Graph. 2011;17:2301–2309.
- Coppens G PEPaNIC 2Y Epigenetics interactive visualisation tool. 2022; Accessed 03 03, 2022. https://www.pepanic.com/2Y_epigenetics
- Merrill SM, Moore SR, Gladish N, et al. Paternal adverse childhood experiences: associations with infant DNA methylation. Dev Psychobiol. 2021;63:e22174.
- Rzehak P, Saffery R, Reischl E, et al. Maternal smoking during pregnancy and DNA-Methylation in children at age 5.5 years: epigenome-wide-analysis in the European childhood obesity project (CHOP)-Study. PLoS One. 2016;11:e0155554.
- Xu R, Hong X, Zhang B, et al. DNA methylation mediates the effect of maternal smoking on offspring birthweight: a birth cohort study of multi-ethnic US mother-newborn pairs. Clin Epigenetics. 2021;13:47.
- van Dijk SJ, Tellam RL, Morrison JL, et al. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin Epigenetics. 2015;7:66.
- Nikpay M, Ravati S, Dent R, et al. Epigenome-wide study identified methylation sites associated with the risk of obesity. Nutrients. 2021;13: 1984.
- Meijer M, Klein M, Hannon E, et al. Genome-Wide DNA Methylation Patterns in Persistent Attention-Deficit/Hyperactivity Disorder and in Association With Impulsive and Callous Traits. Front Genet. 2020;11:16.
- Walton E, Relton CL, Caramaschi D. Using openly accessible resources to strengthen causal inference in epigenetic epidemiology of neurodevelopment and mental health. Genes (Basel). 2019;10:193.
- Hoffmann A, Sportelli V, Ziller M, et al. Epigenomics of major depressive disorders and schizophrenia: early life decides. Int J Mol Sci. 2017;18:1711.
- Chuang YH, Lu AT, Paul KC, et al. Longitudinal Epigenome-Wide Methylation Study of Cognitive Decline and Motor Progression in Parkinson’s Disease. J Parkinsons Dis. 2019;9:389–400.
- Moore K, McKnight AJ, Craig D, et al. Epigenome-wide association study for Parkinson’s disease. Neuromolecular Med. 2014;16:845–855.
- Li QS, Vasanthakumar A, Davis JW, et al. Association of peripheral blood DNA methylation level with Alzheimer’s disease progression. Clin Epigenetics. 2021;13:191.
- Chuang YH, Paul KC, Bronstein JM, et al. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017;9:76.
- Manotas MC, González DM, Céspedes C, et al. Genetic and Epigenetic Control of Puberty. Sex Dev. 2022;16:1–10.
- Bouwland-Both MI, van Mil NH, Stolk L, et al. DNA methylation of IGF2DMR and H19 is associated with fetal and infant growth: the generation R study. PLoS One. 2013;8:e81731.
- Fasolino M, Zhou Z. The Crucial Role of DNA Methylation and MeCP2 in Neuronal Function. Genes (Basel). 2017;8:141.
- Mattonet K, Nowack-Weyers N, Vogel V, et al. Prenatal exposure to endocrine disrupting chemicals is associated with altered DNA methylation in cord blood. Epigenetics. 2022;17:935–952.
- Leroy JL, Frongillo EA, Dewan P, et al. Can children catch up from the consequences of undernourishment? evidence from child linear growth, developmental epigenetics, and brain and neurocognitive development. Adv Nutr. 2020;11:1032–1041.
- Qureshi IA, Mehler MF. Understanding neurological disease mechanisms in the era of epigenetics. JAMA Neurol. 2013;70:703–710.
- Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–1204.