
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Endogenous sex hormones and DNA methylation both play important roles in various diseases. However, their interplay is largely unknown. A deeper understanding of their interrelationships could provide new insights into the pathology of disease development. We, therefore, investigated associations between circulating sex hormones, sex hormone binding globulin (SHBG), and DNA methylation in blood, using samples from 77 men (65 with repeated samples), from the population-based Northern Sweden Health and Disease Study (NSHDS). DNA methylation was measured in buffy coat using the Infinium Methylation EPIC BeadChip (Illumina). Sex hormone (oestradiol, oestrone, testosterone, androstenedione, dehydroepiandrosterone, and progesterone) and SHBG concentrations were measured in plasma using a high-performance liquid chromatography tandem mass spectrometry (LC/MS-MS) method and an enzyme-linked immunoassay, respectively. Associations between sex hormones, SHBG, and DNA methylation were estimated using both linear regression and mixed-effects models. Additionally, we used the comb-p method to identify differentially methylated regions based on nearby P values. We identified one novel CpG site (cg14319657), at which DNA methylation was associated with dehydroepiandrosterone, surpassing a genome-wide significance level. In addition, more than 40 differentially methylated regions were associated with levels of sex hormones and SHBG and several of these mapped to genes involved in hormone-related diseases. Our findings support a relationship between circulating sex hormones and DNA methylation and suggest that further investigation is warranted, both for validation, further exploration and to gain a deeper understanding of the mechanisms and potential consequences for health and disease.
Introduction
Sex hormones are most commonly known for their role in sexual development and reproduction. For example, in men, the primary sex hormones are androgens, which typically associate with male traits developing during puberty. However, sex hormones have also been associated with an increased risk of some types of cancer, including breast and prostate cancer [Citation1]. These cancers are highly dependent on sex hormones for cell proliferation, and hormone suppression is, therefore, an effective therapeutic tool.
DNA methylation is another mechanism involved in the aetiology and progression of many diseases, including cancer [Citation2]. The addition of a methyl group to the nucleotide cytosine, at positions where it is followed by guanine (so called CpG sites), can affect gene expression. In carcinogenesis, this is often characterized by hypermethylation at promoter regions of tumour suppressor genes, resulting in the gene being turned off [Citation3]. This type of DNA methylation can repress transcription both directly, through inhibition of transcription factor binding, and indirectly, by for example recruiting methyl-binding proteins which in turn can repress transcription [Citation4]. DNA methylation patterns change naturally with age but can also be affected by several environmental factors such as diet and exposures to toxins [Citation5]. In addition, evidence suggests that biological sex influences DNA methylation patterns across multiple CpG sites during ageing [Citation6,Citation7], possibly attributed to differences in sex hormone levels. An epigenome-wide association study of individuals undergoing gender-affirming hormone therapy provides further evidence for the relationship between sex hormone levels and DNA methylation, reporting a progressive change in blood DNA methylation throughout therapy [Citation8].
Given the well-established roles of sex hormones and DNA methylation in the aetiology of different cancers, studying the relationship between DNA methylation and endogenous sex hormone levels may give us further insights into possible mediating effects of DNA methylation on the association between sex hormones and sex hormone dependent diseases. Several studies also indicate that oestradiol, and possibly progesterone, may impact the expression of DNA methyltransferases (DNMTs) in oestrogen sensitive tissues such as breast or endometrium [Citation9–11]. Taken together, better knowledge about these associations is likely important in order to increase our understanding about the sex hormone regulation.
Previously, four studies have investigated the association between circulating levels of endogenous sex hormones and DNA methylation. However, three [Citation12–15] measured only global DNA methylation levels either at repeated elements (LINE-1 and/or Alu repeats [Citation12–14]) or through the luminometric methylation assay (LUMA) [Citation15], all in postmenopausal women. Two studies measured site-specific DNA methylation, one in children and adolescents [Citation16] and one in children before- and after pubertal onset [Citation17]. In general, previous findings are inconsistent, likely due to the different study populations and DNA methylation measures. Data on men are lacking, despite the established importance of adult male hormone levels in health and disease.
To expand the knowledge about the relationship between endogenous sex-hormone levels and DNA methylation in blood we analysed genome-wide DNA methylation and sex hormone levels in blood samples from 80 men included in a population-based cohort in northern Sweden, all with repeated samples, mostly taken 10 years apart.
Materials and methods
Study population
We included participants from the largest cohort in the population-based Northern Sweden Health and Disease Study, namely the Västerbotten Intervention Programme (VIP) [Citation18]. In VIP, residents of Västerbotten County are invited to undergo a health examination and to fill out questionnaires about health and lifestyle at the ages of 40, 50, and 60 years. In addition, participants are encouraged to provide a blood sample for future research. Blood samples are collected in the morning after at least 8 hours of fasting, (with deviations from protocol recorded) and stored at −80°C at the regional health care biobank, Biobanken Norr, in Umeå, Sweden. All participants provided written informed consent, and this study was approved by the regional ethical review board at Umeå University (Dnr: 2017/441–31).
Study participants
Eighty men with blood samples and data collected at two time points were included. The vast majority of them had their samples collected 10 years apart. Seven participants had sampling occasions deviating from the 10-year interval, of which four had an interval within 9–11 years, one had 7 years and two had 20 years. The participants had previously been selected as part of a prospective study of biomarkers for colorectal cancer [Citation19], and half of them (n = 40) were diagnosed with colorectal cancer between 3 months and 5 years (M = 2.0, SD = 1.2) after blood sampling. The remainder were control participants who were matched pairwise to the colorectal cancer cases based on age (±12 months), sampling date (±12 months) and fasting status (all>8 hours). Controls also had to be free of cancer for at least five years after the colorectal cancer diagnosis of their corresponding case, or at the end of follow up.
DNA methylation analysis
DNA-methylation measurements were generated as part of a previous study [Citation20]. In short, buffy coat DNA samples were bisulphite treated using the EZ DNA Gold Methylation kit from Zymo Research (Cat No: D5006) and analysed for methylation using Infinium MethylationEPIC BeadChip (Illumina, Cat No; WG-317-1001). DNA quality control, pre-processing, processing, and output data quality control were performed at the SNP&SEQ Technology Platform, Uppsala, Sweden, part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory.
DNA methylation pre-processing
Raw DNA methylation data were pre-processed using the ENmix R package [Citation21]. Prior to pre-processing, probes were excluded (N = 2420) if they were either SNP related [Citation22], had a call-rate of P < 0.01, were outliers, or were missing in more than 20% of the samples. Other exclusions included two samples with a gender mismatch error, one with low quality in>5% of CpGs, and four with>20% of CpGs missing (after removing outliers). Next, we conducted background correction of methylation signal intensities, using out-of-band Infinium I intensities, and performed quantile normalization of methylation intensity values for Infinium I and Infinium II probes separately. Finally, the data were corrected for probe type bias using the Regression on Correlated Probes (RCP) method. Methylation data were expressed as beta values at each CpG site, ranging from 0 (unmethylated) to 1 (fully methylated). After exclusion of cross-reactive probes, as suggested by Pidsley et al. [Citation23], the final number of CpG sites used in downstream analysis was 819,902. As methylation patterns can vary across cell types, we estimated white blood cell distribution within the buffy coat fraction using a method based on a reference dataset, as proposed by Houseman et al. [Citation24]. Finally, we also estimated surrogate variables to account for batch effects and unknown experimental confounders. This was done using intensity data for non-negative internal control probes and setting minimum percentage of variation explained by surrogate variables to 95%, which resulted in seven surrogate variables.
Sex hormone analysis
Because some samples stored in the biobank had insufficient volume for sex hormone analysis (<320 uL), only 149 out of 160 plasma samples were sent for sex hormone analysis. Levels of sex hormones and sex hormone binding globulin (SHBG) were analysed as previously described [Citation25]. All analyses were conducted at the International Agency for Research on Cancer in Lyon, France.
In brief, sex hormones (oestradiol, oestrone, testosterone, androstenedione, dehydroepiandrosterone, and progesterone) were measured using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. SHBG was measured using a commercially available enzyme-linked immunoassays kit by DRG (DRG Instruments GmbH, Marburg, Germany).
Cases and matched controls were measured within the same analytical batch. In each batch, 4 quality control samples were measured in duplicate. Intra-batch coefficients of variation (CVs) ranged from 0.8% for SHBG to 9.2% for progesterone, and inter-batch CVs ranged from 0.8% for progesterone to 11.8% for SHBG.
Free levels of oestradiol and testosterone were estimated from total oestradiol and testosterone concentrations, SHBG concentrations and an assumed constant concentration of albumin of 43 g/l, using a previously validated algorithm [Citation26,Citation27].
One sample had a progesterone concentration below the lower limit of quantification (LLOQ<15 pg/ml). In downstream analyses this concentration was set to the LLOQ.
Statistical analysis
Prior to multivariable analyses, we replaced missing data for BMI (N = 1), smoking status (N = 7), and alcohol consumption (N = 9) using values from the other sampling occasion. For alcohol consumption, an additional four participants lacked data also from the other sampling occasion. These missing values were replaced by the median of the colorectal cancer cases or controls as appropriate. Sex hormone and SHBG levels were log2 transformed to account for skewed distributions and to reduce the effect of outliers. Remaining outliers, or values greater or lower than Q1||Q3 ± 1.5 IQR (interquartile range) were excluded. To investigate associations between sex hormones, SHBG and CpG site-specific DNA methylation, we fitted linear regression models at each time point using the CpGassoc R package [Citation28] and mixed effects models using the lme4/lmerTest R packages [Citation29]. Prior to modelling, methylation beta values were transformed to M-values, , which are more suitable for this type of statistical modelling [Citation30]. All models included body mass index (BMI), smoking status, alcohol consumption, age, colorectal cancer case-control status, estimated cell-type composition, and the first seven surrogate variables as fixed effects in the mixed effects models. Participant identification number and case-control pair identification number were included as random effects. The model including SHBG was further adjusted for testosterone and oestradiol due to its regulatory role for these sex hormones. We chose not to adjust the testosterone and oestradiol models for SHBG levels, respectively, and considered instead free levels of these hormones, which are unbound and therefore independent of SHBG. Associations were tested using t-tests of regression coefficients equal to zero using Satterthwaite’s approximation of degrees of freedom. To control for multiple comparisons, we considered the genome-wide significance level of p < 9e-08 as proposed by Mansell et al. [Citation31] and the less conservative false discovery rate (FDR; q < 0.05). In addition, we plotted the observed P values (negative log10 transformed) against the expected P values (negative log10 transformed) in QQ-plots and calculated the genomic inflation factor (lambda).
Results from the epigenome-wide association study (EWAS) were further analysed to identify differentially methylated regions (DMR) affected by hormone levels. Although associations between sex hormones, sex hormone binding globulin, and methylation at individual CpG sites might not exceed a significance level corrected for multiple testing, regions comprising multiple neighbouring CpG sites may exert a joint significant effect. To identify such DMRs, we used the comb-p method [Citation32] as implemented in the ENmix (v. 1.30.01) [Citation21] R Bioconductor package. The method is based on estimating auto-correlation of adjacent P values from an EWAS. Once that is done, P values are weighed, using the Stouffer–Liptok–Kechris correction (slk), based on the previously calculated auto-correlations. Additionally, FDR correction is used, and regions surpassing the cut-off (q < 0.05) are returned. Finally, the P value of each region is further adjusted using Sidak correction, which takes into account the size of the region and the total number of regions. We applied the default maximum distance threshold of 500 base pairs between sites and an FDR threshold of 0.01. Furthermore, we only considered DMRs with a Sidak P value<0.05 and that consisted of at least 2 CpG sites. We repeated DMR analyses for linear regression and mixed effects models and noted overlapping significant DMRs for the first and second sampling time point, as well as in the linear regression models and mixed effects models. Significant findings were further tested for interaction with case-control status to determine disease influence on the associations. We applied FDR control to address multiple testing of interaction effects.
All statistical analyses were conducted in R v.4.1.1 (R Foundation for Statistical Computing, Vienna, Austria). All statistical tests for significance were two-sided and a P value of below 0.05 (or in the case of FDR, q < 0.05) was considered statistically significant, unless otherwise noted.
Gene enrichment, pathways, and associated diseases
We performed gene enrichment and pathway analysis using the gometh function in the missMethyl R package [Citation33]. Analysis was conducted by searching for significant CpGs from the EWAS and DMR analyses in the Gene Ontology (GO) databases as well as the Kyoto Encyclopedia of Genes and Genomes (KEGG). The gometh function specifically mapped the CpGs to the corresponding genes and tested GO enrichment and KEGG pathways using a Wallenius’ non central hypergeometric test. This method accounts for the number of CpGs per gene as well as CpGs annotated to multiple genes. P values were corrected using the false discovery rate (FDR). Finally, we searched DisGeNET database for the genes annotated to the significant CpGs [Citation34]. We used the gene2disease function from the disgenet2r R package to find diseases related to those genes with a minimum score of 0.5.
Results
Study participants and characteristics
The final study population consisted of 142 samples from 77 men (Supplemental Figure S1). Participant characteristics are presented in . In general, the men were 50 years old at the first sample time point and 60 years old at the second time point. Both BMI and alcohol consumption increased over time (median BMI increased from 25.7 to 26.5 kg/m2 and alcohol consumption by about one gram/day). For smoking status, most participants were non-smokers (never or former) at both time points, with a trend towards smoking cessation, as the proportion of current smokers decreased (from 26.5% to 13.5%) while former smokers increased (from 26.5% to 32.5%). Finally, as expected, endogenous sex hormone concentrations decreased between sample time points, with androgens and progesterone decreasing more than oestrogens. In contrast, SHBG increased slightly over time. Participant characteristics stratified by case-control status are found in Supplemental Table S1.
Table 1. Study participant characteristics.
Linear regression models
In linear regression models of single CpG sites and DNA methylation (), higher levels of free oestradiol were associated with DNA hyper-methylation at one CpG site, cg06070446 (q < 0.05), though only at the first measurement. An association of borderline statistical significance (q = 0.055) was observed between higher levels of dehydroepiandrosterone at the second measurement and DNA hypomethylation at the CpG site cg02327694. No other significant associations were found for single CpG sites in the linear regression models, and no association was statistically significant across both sampling time points.
Table 2. List of all most significant association between DNA methylation and each sex hormone and SHBG at each sampling time point, respectively.
Mixed effects models
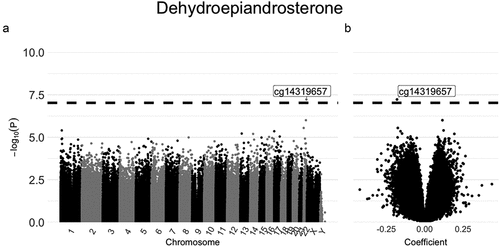
Results from mixed effects models of single CpG sites are shown in Manhattan and Volcano plots ( and Supplemental Figure S2, respectively). We identified one association that surpassed the proposed genome-wide significance level of<9e-08 [Citation31], between higher circulating levels of dehydroepiandrosterone and hypomethylation at cg14319657 in chromosome 22 (). This CpG site is located upstream of the LINC00898 gene. As apparent from the Volcano plot (), the regression coefficient is negative (approximately −0.18), suggesting an inverse relationship between levels of dehydroepiandrosterone and DNA methylation at that specific site. For the remaining sex hormones, there were no statistically significant associations (Supplemental Figure S2).
Figure 1. Manhattan plot (a) and Volcano plot (b) for the associations between DNA methylation and dehydroepiandrosterone estimated using mixed effects models.
In terms of P-value inflation, as indicated by the genomic inflation factor lambda, three exposures deviated from 1 by more than 0.1 (Supplemental Figure S3): dehydroepiandrosterone , testosterone
, and SHGB
.
Differentially methylated regions
Regions in which DNA was differentially methylated were identified using the comb-p method in two different ways. First, we combined P values from linear regression models at the different sampling time points. Here we identified 82 and 128 DMRs significantly (Sidak P < 0.05) associated with sex hormones and/or sex hormone binding globulin at the first and second sampling time point, respectively. As these are based on samples from the same individuals, we were also interested in seeing how many of the significant DMRs regions overlapped between time points. Four DMRs overlapped between the two time-points (). Furthermore, using P values from mixed effects models, 44 significant DMRs were identified (), 11 of which were also identified in the linear models at any sampling time point. Also, as seen in , the majority of DMRs overlap with androgen- and oestrogen response elements. Detailed information on significant DMRs is found in Supplemental Tables S2 & S3. Finally, none of the associations were significant for interaction by case-control status (all qinteraction>0.05).
Table 3. Differentially methylated regions calculated using the Comb-p method based on linear regression models. Restricted to regions overlapping between timepoints.
Table 4. Differentially methylated regions calculated using the Comb-p method based on mixed effects models.
Gene enrichment, pathways and associated diseases
We entered 28 and 281 unique CpGs from linear regression models and mixed models, respectively, into gene enrichment and pathway analyses. None of the CpGs were significantly associated with any GO terms or KEGG pathways (FDR<0.05). The corresponding top 5 hits for each sex hormone and SHBG are presented in Supplemental Tables S4-S7. Looking at associations between annotated genes and diseases in the DisGeNET database, genes were commonly associated with mental disorders (BLHE40, DGKH, FOXG1, and FOXP2) and nervous system diseases (FOXG1, FOXP2, and PLXND1), as illustrated in Supplemental Figure S4.
Discussion
Using samples from 77 men, 65 of which had repeated samples, we estimated associations between levels of circulating sex hormones, sex hormone binding globulin (SHBG) and DNA methylation in white blood cells. Assessing genome-wide DNA methylation levels, we identified three CpG-sites significantly and borderline significantly associated with levels of dehydroepiandrosterone (DHEA) and free oestradiol. We also identified multiple differentially methylated regions (DMRs) significantly associated with sex hormones and SHBG.
To our knowledge, this study is the first to analyse associations between genome-wide DNA methylation and levels of sex hormones and SHBG in men (aged between 40 and 50 years at first sampling and with a repeat sample taken ten years later). The two previous studies of CpG specific DNA methylation and endogenous sex hormones included were restricted to children and adolescents [Citation16,Citation17]. The first study [Citation16] identified, despite not finding any individual significant CpG sites, eleven DMRs that were associated with total and bioavailable testosterone, as well as with SHBG in child and adolescent males. In adolescent females, levels of SHBG were significantly associated with DNA methylation at three individual CpG sites as well as two DMRs. None of the DMRs or individual CpG sites were replicated in our study, most likely due to the differently aged populations. The other previous study [Citation17], which had longitudinal data, found 999 CpGs significantly associated with levels of testosterone in boys. In girls, there were no significant associations. However, models were not adjusted for important confounding factors including BMI and not modelled longitudinally using mixed effects models despite the longitudinal study design. In addition to the previously described studies on DNA methylation and sex hormone levels in human samples, several studies have shown that sex hormones are involved in the regulation of epigenetic programs in multiple different cell types [Citation35], including endometrial cells [Citation36] and adipocytes [Citation37]. Although not addressed in this study (as it only included male subjects), there appear to be significant differences in the potential of sex hormones to illicit downstream responses in men and women, a concept that should be further investigated, especially in the context of colon cancer [Citation25].
In the EWAS, we identified one CpG site (cg14319657) at which DNA methylation was significantly associated with levels of dehydroepiandrosterone in mixed models. Cg14319657 is located in a CpG island upstream of the long intergenic non-protein coding RNA 898 (LINC00898). Long noncoding RNAs (lncRNAs) are involved in various diseases, including cancer [Citation38,Citation39], and upregulation of LINC00898 has been observed in bladder cancer [Citation40], oesophageal squamous cell carcinoma [Citation41], and lung adenocarcinoma [Citation42]. However, as for the majority of lncRNAs, the exact function of LINC00898 remains unknown.
In linear regression models based on specific sampling time points, we identified two CpG sites of interest. One site (cg06070446) was significantly associated with free oestradiol levels. It mapped to the gene body of GALNT3, previously found to be correlated with osteoporosis [Citation43], a disease in which oestrogen levels have an important role [Citation44]. The other CpG site (cg02327694) was borderline significantly associated with dehydroepiandrosterone. Cg02327694 mapped to an lncRNA called LINC01340, which is not functionally well characterized. However, as the relationship for baseline and repeated sampling measures was modelled separately, we conducted more statistical tests, and thus the risk that these two CpGs are false positives is larger. Additionally, the low number of samples at each time point could have led to insufficient statistical power to confirm these associations. Another explanation could be that sex hormone levels and DNA methylation at these CpG sites followed different time trajectories.
Aside from CpG site specific analyses, we also identified more than 40 DMRs that were differentially methylated and associated with sex hormones and/or SHBG. The combined effect of multiple methylated CpG sites, as it is the case in DMRs, is more likely to impact gene expression compared to methylation at individual sites. One DMR (CHR5:110062342–110062837) was associated with both dehydroepiandrosterone and oestrone levels, which is interesting as the former decreases and the latter increases in men over time. The DMR is annotated to the promoter region of the gene TMEM32 in chromosome 5 and includes 14 CpG sites, all of which were hypomethylated with median beta values ranging from 0.16 to 0.42 and a negative direction of effect. The gene TMEM32 has been associated with various diseases, including mild cognitive impairment [Citation45], in which a region within the gene was differentially methylated, and atopic dermatitis [Citation46,Citation47], associated with genetic variants of TMEM32. The latter is associated with both sex hormones and DNA methylation, mostly of genes regulating immune responses and inflammatory processes [Citation48,Citation49]. Another interesting DMR (CHR17:37123637–37123949), located within the promoter region of FBXO47 in chromosome 17, was associated with both progesterone and androstenedione. Median beta values ranged between 0.05 and 0.42 across both sampling time points, and methylation was positively associated with progesterone and androstenedione levels across all CpGs. FBXO47 belongs to a family of genes, F-Box only genes, of which many have oncogenic or tumour suppressive functions [Citation50]. FBXO47 in particular, has been suggested to have a tumour-suppressor role in kidney, liver, pancreas and gastric cancer [Citation51,Citation52]. However, it is unclear what, if any, role sex hormones have in the aetiology of these cancers.
The link between circulating sex hormone levels and DNA methylation in white blood cells could be connected to inflammatory pathways. Inflammation is a complex process involving multiple different immune responses, and both oestrogens and androgens have been shown to have anti-inflammatory effects [Citation53]. Our lab previously investigated associations between circulating levels of inflammatory markers and DNA methylation in samples from the same cohort as in this study [Citation20], and were able to validate previous findings showing e.g., how CRP levels relate to DNA methylation levels.
Despite our findings, we cannot draw conclusions about the possible mediating aspect of DNA methylation on the association between sex hormones and sex hormone dependent diseases. Additionally, sex hormone levels are regulated through negative feedback loops involving, for example, expression of receptors. However, we found no statistically significant association between DNA methylation and sex hormones in regions coding for these receptors. Thus, we deem it more likely that the direction of the association is that sex hormones, and SHBG, can potentially alter DNA methylation in men.
A major strength of our study is the use of repeated samples, making it possible for us to conduct longitudinal assessments of the relationship between circulating sex hormones, sex hormone binding globulin, and DNA methylation. Although the two previous studies on males [Citation16,Citation17] also had data at different ages, these were either not repeat measurements of the same individuals or not modelled using mixed effects models. Further major strengths of our study are the use of a validated liquid chromatography tandem mass spectrometry (HPLC-MS/MS) method to measure sex hormones, as well as the use of the Illumina Infinium MethylationEPIC array, covering more than 850,000 CpG sites, to measure DNA methylation. In addition, we included a more comprehensive panel of sex hormones, also containing testosterone precursors (dehydroepiandrosterone and androstenedione), oestrogens (oestrone and oestradiol), and progesterone. All sex hormones and SHBG were measured in plasma collected after at least 8 hours of fasting and during the morning when concentrations of many sex hormones peak [Citation54]. Another strength was the adjustment for potentially important confounders such as BMI, which was measured by a health professional, alcohol consumption and physical activity, aside from age and estimated cell-type composition.
One limitation of our study is the modest sample size. However, this was partially compensated by the use of repeated samples. Another potential limitation was the inclusion of participants from a previous nested case-control study of colorectal cancer. Although the prospective cohort is population-based, the participants are not fully representative of the cohort, and the inclusion of cases could potentially distort associations between circulating sex hormones, sex hormone binding globulin, and DNA methylation in peripheral white blood cells. To account for this, we adjusted the analyses for colorectal cancer case status and tested for interaction by case-control status. However, we acknowledge that future studies with adequate statistical power should consider stratifying their data by case-control status to elucidate any differences caused by disease influence. Furthermore, as reflected by inflation factors values, some unexplained variation remains, despite the addition of case-control status and surrogate variables. Therefore, to adequately control the false-positive rate in analyses using EPIC array data, we adopted the proposed epigenome-wide significance level p < 9e-08 [Citation31]. Another potential limitation is the fact that DNA methylation was measured in buffy coat and not in other tissues (that might, in some cases, be more relevant for disease development). In the current study, however, investigating associations between buffy coat DNA methylation and circulating hormone levels is of particular interest as oestrogen has documented anti-inflammatory effects and have been shown to resolve inflammation in white blood cells such as macrophages [Citation55]. Despite this, future studies considering mediating effects of DNA methylation might consider measuring levels in the target tissue instead of blood [Citation56–58] or try to validate key findings from epidemiological studies in vitro e.g., in hormone-receptor positive breast cancer cells. Finally, our approach to impute for missing data assumed that BMI, alcohol intake and smoking did not change between sampling time points, which could have led to some bias.
Conclusion
In summary, we identified one novel CpG site that met a genome-wide significance level and more than 40 DMRs, associated with different sex hormones, and sex hormone binding globulin, supporting the relationship between sex hormones and DNA methylation. As these individually already play important roles in different diseases, we deem it likely that either the epigenetic regulation of sex hormone levels or the effect mediation of DNA methylation can be involved in disease initiation/progression. Nonetheless, mechanisms through which sex hormones may alter DNA methylation remain mostly unknown. Therefore, future studies focusing on this area, are needed.
Disclaimer
Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/World Health Organization.
Supplemental Material
Download Zip (1.1 MB)Acknowledgements
We would like to thank the participants and staff of the VIP cohort for their valuable contribution to this research. We thank the Biobank Research Unit at Umeå University, VIP, and the County Council of Västerbotten for providing data and samples and acknowledge the contribution from Biobank Sweden, supported by the Swedish Research Council (VR 2017-00650). Special thanks to Robert Johansson, Åsa Ågren, and their colleagues at the Biobank Research Unit, Umeå University, and former colleague Robin Myte for helpful assistance. Methylation profiling was performed by the SNP&SEQ Technology Platform in Uppsala (www.genotyping.se). The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory. The SNP&SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The datasets generated and/or analysed during the current study are considered personal data, which prohibits us from storing them in a public depository. However, all data are archived at the Biobank Research Unit at Umeå University (https://www.umu.se/en/biobank-research-unit/research/access-to-samples-and-data/), and access for secondary use can be granted conditional upon meeting Swedish requirements for human research.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2196759
Additional information
Funding
References
- Liu WJ, Zhao G, Zhang CY, et al. Comparison of the roles of estrogens and androgens in breast cancer and prostate cancer. J Cell Biochem. 2020;121(4):2756–13. DOI:10.1002/jcb.29515
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492.
- Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021;37:1012–1027.
- Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610.
- Feinberg AP, Longo DL. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. 2018;378:1323–1334.
- Yusipov I, Bacalini MG, Kalyakulina A, et al. Age-related DNA methylation changes are sex-specific: a comprehen-sive assessment. Aging (Albany NY. Aging. 2020;12(23):24057–24080. DOI:10.18632/aging.202251
- Vershinina O, Bacalini MG, Zaikin A, et al. Disentangling age-dependent DNA methylation: deterministic, stochastic, and nonlinear. Sci Rep. 2021;11:9201.
- Shepherd R, Bretherton I, Pang K, et al. Gender-affirming hormone therapy induces specific DNA methylation changes in blood. Clin Epigenetics. 2022;14:24.
- Yamagata Y, Asada H, Tamura I, et al. DNA methyltransferase expression in the human endometrium: down-regulation by progesterone and estrogen. Hum Reprod. 2009;24:1126–1132.
- Si X, Liu Y, Lv J, et al. ERα propelled aberrant global DNA hypermethylation by activating the DNMT1 gene to enhance anticancer drug resistance in human breast cancer cells. Oncotarget. 2016;7:20966–20980.
- Cui M, Wen Z, Yang Z, et al. Estrogen regulates DNA methyltransferase 3B expression in Ishikawa endometrial adenocarcinoma cells. Mol Biol Rep. 2009;36:2201–2207.
- Ulrich CM, Toriola AT, Koepl LM, et al. Metabolic, hormonal and immunological associations with global DNA methylation among postmenopausal women. Epigenetics. 2012;7:1020–1028.
- Boyne DJ, Friedenreich CM, McIntyre JB, et al. Endogenous sex hormone exposure and repetitive element DNA methylation in healthy postmenopausal women. Cancer Causes Control. 2017;28:1369–1379.
- Huen K, Harley K, Kogut K, et al. DNA methylation of LINE-1 and Alu repetitive elements in relation to sex hormones and pubertal timing in Mexican-American children. Pediatr Res. 2016;79:855–862.
- Iwasaki M, Ono H, Kuchiba A, et al. Association of postmenopausal endogenous sex hormones with global methylation level of leukocyte DNA among Japanese women. BMC Cancer. 2012;12:323.
- Arathimos R, Sharp GC, Granell R, et al. Associations of sex hormone-binding globulin and testosterone with genome-wide DNA methylation. BMC Genet. 2018;19:113.
- Almstrup K, Lindhardt Johansen M, Busch AS, et al. Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood. Sci Rep. 2016;6:28657.
- Norberg M, Wall S, Boman K, et al. The Västerbotten Intervention Programme: background, design and implications. Glob Health Action. 2010;3:4643.
- Harlid S, Harbs J, Myte R, et al. A two-tiered targeted proteomics approach to identify pre-diagnostic biomarkers of colorectal cancer risk. Sci Rep. 2021;11:5151.
- Myte R, Sundkvist A, Van Guelpen B, et al. Circulating levels of inflammatory markers and DNA methylation, an analysis of repeated samples from a population based cohort. Epigenetics. 2019;14:649–659.
- Xu Z, Niu L, Li L, et al. Enmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44:e20.
- Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017;45:e22.
- Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 2012;13:86.
- Harbs J, Rinaldi S, Gicquiau A, et al. Circulating Sex hormone levels and colon cancer risk in men: a nested case-control study and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2022;31:793–803.
- Rinaldi S, Geay A, Déchaud H, et al. Validity of free testosterone and free estradiol determinations in serum samples from postmenopausal women by theoretical calculations. Cancer Epidemiol Biomarkers Prev. 2002;11:1065–1071.
- Suzuki R, Allen NE, Appleby PN, et al. Lifestyle factors and serum androgens among 636 middle aged men from seven countries in the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Causes Control. 2009;20:811–821.
- Barfield RT, Kilaru V, Smith AK, et al. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics. 2012;28:1280–1281.
- Bates D, Mächler M, Bolker B, et al. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48.
- Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 2010;11:587.
- Mansell G, Gorrie-Stone TJ, Bao Y, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 2019;20:366.
- Pedersen BS, Schwartz DA, Yang IV, et al. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–2988.
- Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s Human Methylation 450 platform. Bioinformatics. 2016;32:286–288.
- Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020;48:D845–55.
- Kovács T, Szabó-Meleg E, Ábrahám IM. Estradiol-induced epigenetically mediated mechanisms and regulation of gene expression. Int J Mol Sci. 2020;21:3177.
- Houshdaran S, Oke AB, Fung JC, et al. Steroid hormones regulate genome-wide epigenetic programming and gene transcription in human endometrial cells with marked aberrancies in endometriosis. PLoS Genet. 2020;16:e1008601.
- Bjune JI, Strømland PP, Jersin R, et al. Metabolic and epigenetic regulation by estrogen in adipocytes. Front Endocrinol. 2022;13:828780.
- Huarte M. The emerging role of lncRnas in cancer. Nat Med. 2015;21:1253–1261.
- Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–463.
- Zhang S, Cao H, Ye L, et al. Cancer-associated methylated lncRnas in patients with bladder cancer. Am J Transl Res. 2019;11:3790–3800.
- Shen JF, Ge JF, Zheng SY, et al. Integrative analysis of differential circular RNA and long non-coding RNA profiles and associated competing endogenous RNA networks in esophageal squamous cell carcinoma. Funct Integr Genomics. 2021;21:125–138.
- Shi X, Tan H, Le X, et al. An expression signature model to predict lung adenocarcinoma-specific survival. Cancer Manag Res. 2018;10:3717–3732.
- Wang XY, Yang B, Liu CS, et al. Research on correlation between GALNT3 gene and osteoporosis. Eur Rev Med Pharmacol Sci. 2018;22:69–75.
- Ji MX, Yu Q. Primary osteoporosis in postmenopausal women. Chronic Dis Transl Med. 2015;1:9–13.
- Pathak GA, Silzer TK, Sun J, et al. Genome-wide methylation of mild cognitive impairment in Mexican Americans highlights genes involved in synaptic transport, alzheimer’s disease-precursor phenotypes, and metabolic morbidities. J Alzheimers Dis. 2019;72:733–749.
- Kim J-H, Lee S-Y, Kang M-J, et al. Association of genetic polymorphisms with atopic dermatitis, clinical severity and total IgE: a Replication and extended study. Allergy Asthma Immunol Res. 2018;10(4):397–405. DOI:10.4168/aair.2018.10.4.397
- Zheng J, Wu YY, Fang WL, et al. Confirming the TMEM232 gene associated with atopic dermatitis through targeted capture sequencing. Sci Rep. 2021;11:21830.
- Kanda N, Hoashi T, Saeki H. The roles of sex hormones in the course of atopic dermatitis. Int J Mol Sci. 2019;20:20.
- Nedoszytko B, Reszka E, Gutowska-Owsiak D, et al. Genetic and epigenetic aspects of atopic dermatitis. Int J Mol Sci. 2020;21:6484.
- Guardavaccaro D, Pagano M. Oncogenic aberrations of cullin-dependent ubiquitin ligases. Oncogene. 2004;23:2037–2049.
- Simon-Kayser B, Scoul C, Renaudin K, et al. Molecular cloning and characterization of FBXO47, a novel gene containing an F-box domain, located in the 17q12 band deleted in papillary renal cell carcinoma. Genes Chromosomes Cancer. 2005;43:83–94.
- Zhang F, Shi J, Wu Z, et al. A 3’-tRNA-derived fragment enhances cell proliferation, migration and invasion in gastric cancer by targeting FBXO47. Arch Biochem Biophys. 2020;690:108467.
- Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–574.
- Parikh TP, Stolze B, Ozarda Y, et al. Diurnal variation of steroid hormones and their reference intervals using mass spectrometric analysis. Endocr Connect. 2018;7:1354–1361.
- Villa A, Rizzi N, Vegeto E, et al. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci Rep. 2015;5:15224.
- Hannon E, Mansell G, Walker E, et al. Assessing the co-variability of DNA methylation across peripheral cells and tissues: implications for the interpretation of findings in epigenetic epidemiology. PLoS Genet. 2021;17:e1009443.
- Lowe R, Slodkowicz G, Goldman N, et al. The human blood DNA methylome displays a highly distinctive profile compared with other somatic tissues. Epigenetics. 2015;10:274–281.
- Huang YT, Chu S, Loucks EB, et al. Epigenome-wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics. 2016;11:227–236.