
ABSTRACT
Previously, we showed that DNA methylation defects in spermatozoa from male partners of couples undergoing recurrent pregnancy loss (RPL) could be a contributing paternal factor. In the present study, we aimed to determine whether the methylation levels of selected imprinted genes can be used as diagnostic markers to identify epigenetically abnormal spermatozoa sample in these cases. The methylation levels of selected imprinted genes in spermatozoa, which were previously found to be differentially methylated, were combined into a probability score (between 0–1) using multiple logistic regression. Different combinations of these genes were investigated using Receiver Operating Characteristic analysis, and the threshold values were experimentally validated in an independent cohort of 38 control and 45 RPL spermatozoa samples. Among the different combinations investigated, a combination of five imprinted genes comprising IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, and PEG3 (AUC = 0.88) with a threshold value of 0.61 was selected with a specificity of 90.41% and sensitivity of 70%. The results from the validation study indicated that 97% of the control samples had probability scores below this threshold, whereas 40% of the RPL samples were above this threshold with a post-hoc power of 97.8%. Thus, this combination can correctly classify control samples and potentially identify epigenetically abnormal spermatozoa samples in the male partners of couples undergoing RPL. We propose that the combined DNA methylation levels of these imprinted genes can be used as a diagnostic tool to identify spermatozoa samples with epigenetic defects which could contribute to the pathophysiology of RPL and the couple could be counselled appropriately.
Introduction
DNA methylation plays a crucial role in the transmission of epigenetic information from spermatozoa to developing embryos. This epigenetic information carried by spermatozoa is important because nearly 26% of the 5mC residues are retained in the paternal genome during early embryo development [Citation1]. Many of these CpG sites lie in the imprinting control region, where they regulate the parental allele-specific expression of imprinted genes, which are essential for early embryo development [Citation2]. Hence, any aberrations in the methylation pattern of the imprinted genes in the spermatozoa could detrimentally affect the expression of these genes and subsequent embryo development.
Recurrent pregnancy loss (RPL) presents as a frustrating reproductive disorder, where a couple undergoes two or more consequent pregnancy losses before the 20th week of gestation. It affects nearly 7% of Indian women and 1%–2% of women globally [Citation3,Citation4]. Although several female causes (such as genetic, anatomical, endocrine, immunological, infectious, and haematological) have been identified, there is a dearth of paternal factors associated with this condition, as nearly 50% of cases are idiopathic [Citation5]. To address this lacuna, we previously investigated the DNA methylation status of several important genes implicated in embryonic development in spermatozoa in a large cohort of male partners of couples undergoing idiopathic RPL and fertile men [Citation6]. We found aberrant DNA methylation patterns at several CpG sites in imprinted genes, such as IGF2-H19 DMR, IG-DMR, MEST, ZAC, KvDMR, PEG3, and PEG10 by pyrosequencing. Receiver operating characteristic (ROC) analysis identified that some differentially methylated genes could be used as diagnostic markers to distinguish samples with abnormal DNA methylation patterns in imprinted genes. In the current study, we investigated the diagnostic potential of several combinations of imprinted genes using multiple logistic regression. We further validated the results in an independent cohort of control (N = 38) and RPL (N = 45) spermatozoa.
Materials and methods
Multiple logistic regression and ROC analysis
The average methylation levels of differentially methylated sites for the genes IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, PEG3, MEST, and PEG10 for the control (N = 73) and RPL (N = 80) samples analysed in our previous study [Citation6] were subjected to multiple logistic regression in different combinations to obtain a predicted probability score for each sample. The probability score was then subjected to ROC analysis to obtain an ROC curve. Probability scores with a maximum sensitivity at 90% specificity were selected as threshold values. Multiple logistic regression and ROC analyses were performed using STATA software version 17 (StataCorp, Texas, USA). Diagnostic potential was calculated as previously described [(number of true negatives + number of true positives) × 100/total number of samples] [Citation6]. The level of significance was set at p < 0.05. Graphical representations were made using GraphPad prism version 9.3.1 (GraphPad Software, San Diego, USA).
Experimental validation by pryosequencing
Participant recruitment and sample collection
To validate the probability score threshold for the combination of five imprinted genes to distinguish epigenetically abnormal spermatozoa, we performed pyrosequencing for IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, and PEG3 in 38 control and 45 RPL spermatozoa samples. This study was approved by the NIRRCH Ethics Committee for Human Studies (project no.250/2014). All protocols for participant recruitment, inclusion, and exclusion criteria were described previously [Citation6]. Briefly, the RPL group was comprised of male partners of couples undergoing more than two consecutive pregnancy losses before 20th weeks of gestation. Patients with known infectious, hormonal, haematological, and immunological causes of miscarriages or abnormal karyotypes were excluded, and only idiopathic cases were included in the RPL group. The control group comprised healthy fertile men who had fathered a child in the past year without any history of infertility or miscarriage. A total of 83 couples were recruited from September 2018 to March 2020. The male participants were in the age group 21–45 y and the female participants were between 18–35 y of age. All participants were recruited from the gynaecology outpatient departments of Nowrosjee Wadia Maternity Hospital (Parel, Mumbai, India) and Infertility Clinic at the ICMR-National Institute for Research in Reproductive and Child Health (ICMR-NIRRCH) after obtaining informed consent.
Semen analysis for validation cohort
Semen samples were obtained from male partners by masturbation after sexual abstinence for 3–5 d, and semen analysis was performed according to WHO guidelines (World Health Organization, 2010) as previously described [Citation6]. Sperm counts were measured on a haemocytometer and motility was assessed by computer assisted semen analysis after appropriate dilution. For assessing morphology, air-dried smears were fixed in 100% alcohol and subjected to Papanicolaou staining and the number of spermatozoa with normal head and tail morphology were counted. Chromomycin A3 (CMA3) (Sigma-Aldrich) staining was used to assess chromatin compaction as described by Bianchi et al. (detailed staining protocol provided in supplementary methods) [Citation7]. The Shapiro–Wilk test was used to determine the normality of the data. Since most of the data showed non-normal distribution, non-parametric Mann–Whitney test was used to ascertain the potential difference between the groups. A p-value less than 0.05 was considered significant.
DNA methylation analysis by pyrosequencing
After removal of seminal plasma, the sperm pellet was treated with somatic cell lysis buffer (0.1% sodium dodecyl sulphate, 0.5% Triton X-100 in diethyl pyrocarbonate water) treatment for 6 hours at room temperature on a shaker to remove any potential somatic cell contamination [Citation8]. The sperm samples were then washed twice with phosphate buffer saline (PBS) and stored at −80°C till genomic DNA extraction. Spermatozoa genomic DNA was extracted using a HiPurA Sperm Genomic DNA Purification Kit (HiMedia), and Genomic DNA was subjected to bisulphite conversion using the MethylCode Bisulphite Conversion Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Modified DNA was used for PCR amplification with primers specific for IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, and PEG3 using the Pyromark PCR Amplification Kit (Qiagen, Hilden, Germany). The PCR products were subjected to clean-up, followed by pyrosequencing on a PyroMark Q96 ID (Qiagen). All detailed protocols and primer sequences have been published previously [Citation6]. DNA methylation levels at differentially methylated sites in a gene were averaged to obtain the average methylation level at that locus. Thereafter, the average methylation levels for the five imprinted genes in the validation samples were subjected to multiple logistic regression to obtain the probability score, as mentioned above.
Unsupervised k-means clustering
Unsupervised k-means clustering of the probability score of the control and RPL samples in two clusters was performed using XLSTAT software (Data Analysis and Statistical Solution for Microsoft Excel, Addinsoft, Paris, France 2017).
Results
Multiple logistic regression and ROC analysis
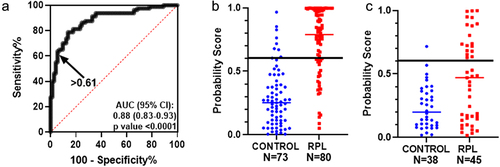
Ten combinations of genes for IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, PEG3, MEST, and PEG10 were tested. Since the imprinted gene IGF2-H19 DMR had the highest diagnostic potential (61.47) in our previous study [Citation6], it was included in all combinations. The probability scores obtained for the control and RPL samples in each combination were then subjected to ROC analysis, and the AUC and threshold values are presented in . The AUC for all combinations had a p-value of less than 0.0001.
Table 1. Diagnostic characteristics of combinations of genes tested.
From the ten combinations tested, one combination of five genes, IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, and PEG3, was selected, as this combination had a diagnostic potential of above 80 with the least number of genes. The ROC curve for this combination and the probability values for the control and RPL groups are shown in . A threshold of 0.61 was selected, with a specificity of 90.41% and sensitivity of 70%. Upon further experimental validation in additional controls (N = 38) and RPL (N = 45), 97.36% of controls had predicted probability scores below (specificity), and 40% of RPL samples had a score above this threshold (sensitivity) (). For the validation cohort, the mean predicted probability of the RPL group 0.4594 (±0.04) was significantly higher than the control group 0.2367 (±0.02) with a p-value of 0.0002 and post-hoc power of 97.8% (calculated using https://clincalc.com/). For the validation cohort, all semen parameters were within the normal range (Suplementary Table S1), as prescribed by the WHO 2010 reference values [Citation9].
Figure 1. Multiple regression and ROC analysis: (a) ROC curve for the probability score of average methylation levels of differentially methylated CpGs in IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, and PEG3 for the samples analysed in the previous study. Arrow indicates the threshold value of 0.61 probability score. Scatter plot with median for the probability score for the control (N = 73) and RPL samples (N = 80) in the previous study (b) and for the probability score for the control (N = 38) and RPL (N = 45) samples in the current validation study (c). The horizontal line indicates the threshold value of 0.61.
Unsupervised k-means clustering
Unsupervised k-means clustering was performed to cluster the predicted probability values of the combination of five imprinted genes in control and RPL samples in the validation study into two groups (). The majority of the control samples were in cluster 1 (86.84%), whereas 53.33% of the RPL samples were in cluster 2.
Table 2. Unsupervised k-means clustering of predicted probability values of combination of five imprinted genes into two clusters in validation cohort. Cluster 1 comprised of most of the control samples and some RPL samples with predicted probability values comparable to control; while Cluster 2 comprised of a sub-set of RPL samples with a predicted probability value different than the controls.
Discussion
Currently, there are limited paternal factors implicated in the pathophysiology of RPL, and most of the investigations and interventions are centred on the female partner [Citation3,Citation5]. There is still a lack of consensus regarding the association of general semen parameters such as spermatozoa count, motility, and morphology with RPL [Citation10]. Our earlier studies found normal semen parameters in male partners of couples undergoing RPL [Citation6,Citation11]. Thus, there is a need to investigate paternal factors beyond the routine semen parameters. Some studies have suggested that spermatozoa DNA methylation in imprinted genes (assessed by pyrosequencing) can be used as a diagnostic marker in andrological workup for idiopathic infertility and is significantly associated with oligospermia [Citation12]. Indeed, DNA methylation panels comprising a combination of candidate biomarker genes have been used for the screening and diagnosis of different types of cancers, such as colorectal, lung, and breast cancer [Citation13–16]
It is now widely recognized that the paternal epigenome contributes significantly to embryo development and pregnancy outcomes [Citation17]. In our previous study, we found that paternally imprinted (methylated) loci such as IGF2-H19 DMR and IG-DMR were hypomethylated at specific CpG sites in the spermatozoa in the RPL group as compared to the control. We also found that maternally imprinted genes such as ZAC, KvDMR, PEG3, MEST, and PEG10, which are usually unmethylated in spermatozoa, were hypermethylated in the RPL group. ROC analysis of these genes individually yielded AUC ranging from 0.64 to 0.59 and diagnostic potentials in the range of 61.47 to 54.63, indicated that they can be used to identify spermatozoa samples with epigenetic defects [Citation6]. Therefore, in this study, we investigated whether a combination of these genes could provide a better diagnostic performance than individual genes. We tested 10 combinations of genes that were found to be differentially methylated in our previous study [Citation6]. Using multiple logistic regression, we found that the combination of all seven differentially methylated genes, namely IGF2-H19, IG-DMR, ZAC, KvDMR, PEG3, MEST, and PEG10, provided much higher AUC and diagnostic potential values than the genes individually. For feasible use as a diagnostic marker, we aimed to have a combination of the least number of genes with a diagnostic potential of at least 80. Thus, we tested the AUC and diagnostic potential by eliminating the genes with lower individual AUC and diagnostic potential values, such as PEG10 and MEST. The combination of genes without PEG10 and MEST (i.e., with five genes, IGF2-H19, IG-DMR, ZAC, KvDMR, and PEG3) yielded similar AUC (above 0.8) and diagnostic potential (above 80). Further combinations of the four and three genes tested in had lower AUC and diagnostic potential values. Hence, a combination of five genes, IGF2-H19, IG-DMR, ZAC, KvDMR, and PEG3, was selected.
The aim of the ROC analysis was to select a threshold value for the probability score of the combination of five imprinted genes where the majority of the controls were correctly classified (i.e., having a high specificity of at least 90%). Accordingly, a threshold of 0.61 was selected for the probability score at which the specificity was 90.41% and the maximum sensitivity was 70%. In other words, the values for most of the control samples (90.41%) were below this threshold, and a subset of RPL cases (70%) was found to have higher values, indicating an abnormal DNA methylation pattern in these five imprinted genes. To experimentally validate this threshold, we recruited further study subjects in the control and RPL groups and performed pyrosequencing for the five imprinted genes in spermatozoa DNA. In this validation study, we found that 97.36% of the controls were correctly classified (i.e., below the threshold value of 0.61) and a subset of RPL cases (40%) above this threshold. Additionally, the mean predicted probability of the RPL group was higher than that of the control group. A post hoc power of 97.8% indicated that the sample size of this cohort was adequate. Thus, most of the control samples were reproducibly below the threshold value in our previous study as well as in this validation study. At this threshold, a subset of RPL cases was identified to have higher values, indicating abnormal DNA methylation patterns in the five imprinted genes.
To remove any potential bias from the data analysis, unsupervised k-means clustering was performed for the validation cohort, where the unlabelled predicted probability values for five imprinted genes were submitted for segregating into two clusters such that the data points in one group were more similar to each other. After labelling the sorted values, it was found that 86.84% of the control values were in Cluster 1, indicating that the majority of the control values were clustered together. However, more than half (53.33%) of the RPL samples were grouped in Cluster 2, indicating that their predicted probability values were different from those of the controls.
It is important to note that RPL can be a multifactorial condition, with more than one unknown cause contributing to it. Epigenetic aberrations in spermatozoa could be one of the contributing factors to RPL. Hence, not all the samples in the RPL group may have a predicted probability above the threshold level, and some could be clustered with most of the controls in cluster 1. This indicated that these samples may not have DNA methylation defects in these genes. Thus, it is more important to have a threshold with high specificity where most of the control samples are correctly classified. A threshold of 0.61 of the predicted probability of the combination of five imprinted genes could correctly classify 90.41% of the control samples in the earlier study and 97.36% of the control samples in the validation study, while identifying a subset of samples in the RPL group with DNA methylation defects.
Conclusions
To summarize, our results suggest that aberrant spermatozoa DNA methylation is a contributing paternal factor in a subset of RPL cases. The threshold value of the predicted probability score set in this study can be used to identify abnormal DNA methylation patterns in five imprinted genes in the spermatozoa of male partners. To the best of our knowledge, this is the first study to investigate the use of a combination of imprinted genes to detect paternal epigenetic factors contributing to RPL. We propose that the use of this marker should be explored in clinical settings.
Authors Role
NHB conceptualized the study. KK performed methylation analysis. SB and SR were involved in the statistical analyses. SM and DI were involved in sample collection. DS, VB, and AP were involved in the participant recruitment. KK drafted the manuscript. SR and NHB have made critical revisions. All authors agree with and approved the final manuscript.
Supplemental Material
Download Zip (21.7 KB)Acknowledgments
The authors acknowledge the technical help provided by Mrs Tejashree Sontakke for the recruitment of participants in this study. The authors are grateful for the technical assistance provided by Mr Suryakant Mandavkar and Mr Deepak Shelar. We are thankful to all the couples participating in this study.
Disclosure statement
The authors have no potential conflict of interest to report.
Data availability statement
All the data contained in the manuscript can be available on request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2252244
Additional information
Funding
References
- Wang L, Zhang J, Duan J, et al. Programming and Inheritance of parental DNA methylomes in mammals. Cell. 2014;157(4):979–7. doi: 10.1016/j.cell.2014.04.017
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2(1):21–32. doi: 10.1038/35047554
- Bender Atik R, Christiansen OB, Elson J, et al. ESHRE guideline: recurrent pregnancy loss. Human Reproduction Open. 2018;2018(2):1–12. doi: 10.1093/hropen/hoy004
- Patki A, Chauhan N. An Epidemiology study to determine the prevalence and risk factors associated with recurrent spontaneous miscarriage in India. J Obstet Gynecol India. 2016;66(5):310–315. doi: 10.1007/s13224-015-0682-0
- Ford H, Schust D. Recurrent pregnancy loss: etiology, diagnosis, and therapy. Rev Obstet Gynecol. 2009;2(2):76–83. doi: 10.3390/jcm10215040
- Khambata K, Raut S, Deshpande S, et al. DNA methylation defects in spermatozoa of male partners from couples Experiencing recurrent pregnancy loss. Hum Reprod. 2021;36:48–60. doi: 10.1093/humrep/deaa278
- Bianchi PG, Manicardi GC, Bizzaro D, et al. Effect of deoxyribonucleic acid protamination on fluorochrome staining and in situ nick-translation of murine and human mature spermatozoa. Biol Reprod. 1993;49(5):1083–1088. doi: 10.1095/biolreprod49.5.1083
- Goodrich R, Johnson G, Krawetz SA. The preparation of human spermatozoal RNA for clinical analysis. Archives Of Andrology. 2007;53(3):161–167. doi: 10.1080/01485010701216526
- Cooper TG, Noonan E, von Eckardstein S, et al. World Health Organization reference values for human semen characteristics*‡. Human Reproduction Update. 2009;16(3):231–245. doi: 10.1093/humupd/dmp048
- Puscheck EE, Jeyendran RS. The impact of male factor on recurrent pregnancy loss. Curr Opin Obstet Gynecol. 2007;19:222–228. doi: 10.1097/GCO.0b013e32813e3ff0
- Ankolkar M, Patil A, Warke H, et al. Methylation analysis of idiopathic recurrent spontaneous miscarriage cases reveals aberrant imprinting at h19 icr in normozoospermic individuals. Fertil Sterility. 2012;98:1186–1192. doi: 10.1016/j.fertnstert.2012.07.1143
- Klaver R, Tuttelmann F, Bleiziffer A, et al. DNA methylation in spermatozoa as a prospective marker in andrology. Andrology. 2013;1(5):731–740. doi: 10.1111/j.2047-2927.2013.00118.x
- Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;370(14):1287–1297. doi: 10.1056/NEJMoa1311194
- Shirley M. Epi proColon® for colorectal cancer screening: A profile of its use in the USA. Mol Diagn Ther. 2020;24(4):497–503. doi: 10.1007/s40291-020-00473-8
- Shan M, Zhang L, Liu Y, et al. DNA methylation profiles and their diagnostic utility in BC. Dis Markers. 2019;2019:1–10. doi: 10.1155/2019/6328503
- Wei B, Wu F, Xing W, et al. A panel of DNA methylation biomarkers for detection and improving diagnostic efficiency of lung cancer. Sci Rep. 2021;11(1):16782. doi: 10.1038/s41598-021-96242-6
- Ibrahim Y, Hotaling J. Sperm epigenetics and its impact on male fertility. Semin Reprod Med. 2018;36(3/04):233–239. doi: 10.1055/s-0038-1677047